
Abstract
Rhodococcus opacus PD630 is a biotechnologically important bacterium with metabolic capability for bioremediation, metal recovery, and storage of triacylglycerols. Genome editing by homologous recombination in R. opacus is hampered by a very low combined frequency of DNA transfer and recombination. To improve recombination in the species, a conjugative, conditional suicide plasmid based on the replicon derived from the Corynebacterium glutamicum plasmid pGA1 was constructed and evaluated in R. opacus. The replication of this plasmid is controlled by a dual inducible and repressible promoter system originally developed for Mycobacterium spp. Next, we demonstrated that a derivative of this plasmid containing sacB as a counterselection marker and homologous regions of R. opacus could be used for homologous recombination, and that the problem of obtaining recombinants had been solved. Like for other Corynebacteriales, the cell wall of Rhodococcus spp. contains mycolic acids which form a hydrophobic and impermeable outer layer. Mycolic acids are essential for Mycobacterium smegmatis, but not for Corynebacterium glutamicum, and the new vector was used to study if mycolic acid is essential for R. opacus. We found that accD3 that is necessary for mycolic acid synthesis could only be deleted from the chromosome in strains containing a plasmid-encoded copy of accD3. This indicates that mycolic acid is important for R. opacus viability. The conditional suicide vector should be useful for homologous recombination or for delivering gene products like recombinases or Cas proteins and gRNA to Rhodococcus and related genera, while the approach should be applicable for any plasmid needing a plasmid-encoded protein for replication.
Key points
• Improved vector for homologous recombination in R. opacus.
• Mycolic acid is important for survival of R. opacus like it is for Mycobacterium.
• Similar conditional suicide plasmids may be constructed for other bacteria.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rhodococcus opacus PD630 is a Gram-positive, oleaginous actinobacterium that mainly occurs in soil. It stores surplus carbon as triacylglycerols under nitrogen-limiting conditions (Alvarez et al. 2000). R. opacus has recently attracted great interest because of its potential industrial applications such as production of large quantities of lipids, high value products from lignocellulosic biomass, production of carotenoids, tolerance towards the aromatic compounds that are generally produced from lignin, and bioremediation by breaking down different organic compounds (DeLorenzo et al. 2018; Anthony et al. 2019; Cappelletti et al. 2020).
Methods for homologous recombination in R. opacus are well described, but we found the combined frequencies of plasmid transfer and recombination to be rather low, necessitating several parallel conjugations in order to obtain the desired strains. In this study, we aimed to overcome this bottleneck by developing a conjugative conditional suicide plasmid where the replication in R. opacus is dependent on an externally added inducer. When a conditional suicide plasmid is applied for recombination, conjugation can be performed using conditions where the plasmid replicates. Then, recombinants can be selected for by cultivating a transconjugant under conditions where the plasmid does not replicate while simultaneously selecting for a positive selection marker encoded by the plasmid. Our hypothesis was that if the expression of a protein necessary for replication of a plasmid could be controlled with the lowest expression level being below that needed for each daughter cell to always receive a copy of the plasmid, then a conditional suicide plasmid could be constructed. Earlier, similar approaches have been developed and tested in Gram-negative bacteria, achieving a higher success rate for mutagenesis by homologous recombination when gene transfer and recombination were separated in time (Karunakaran et al. 1999; Gimmestad et al. 2009; Naorem et al. 2018).
The order Corynebacteriales to which R. opacus belongs is known for a unique cell wall structure containing mycolic acids as the major lipid species (Lechevalier et al. 1971; Goodfellow et al. 1982). Mycolic acids are α-branched β-hydroxy fatty acids containing a common mycolic acid motif (2-alkyl 3-hydroxy). The length varies between different species and can be up to 100 carbon atoms long (Marrakchi et al. 2014). The mycolyl-arabinogalactan-peptidoglycan complex forms a unique impermeable and hydrophobic outer layer (Minnikin 1991; de Carvalho et al. 2016; Houssin et al. 2020), which appears to be analogous to the outer membrane of Gram-negative bacteria.
Mycolic acid is formed when two molecules are condensed by the protein Pks13. One of these is a long fatty acyl CoA carboxylated by a specific acyl-CoA carboxylase encoded by accD3 in Corynebacterium glutamicum. The other is the long meromycolic acid which is AMP-activated by Fad32 (Marrakchi et al. 2014). In R. opacus, ten genes form the mycolic acid biosynthesis gene cluster (Supplemental Fig. S1); the last of these are three genes homologous to fad32, pks13, and accD3 from C. glutamicum (Gande et al. 2007). Portevin et al. (2004, 2005) have constructed three different C. glutamicum mutant strains that do not produce mycolic acid by deleting fad32, pks13, and accD3, respectively, although the mutants grew slower than the wild type. However, the corresponding genes could not be deleted in Mycobacterium smegmatis, suggesting that mycolic acids are essential for Mycobacterium but not for Corynebacterium (Portevin et al. 2004, 2005).
The objectives of this study were twofold. Firstly, we addressed the bottleneck for mutagenesis by homologous recombination by constructing a conditional suicide plasmid based on a cryptic plasmid pGA1 found in C. glutamicum (Nesvera et al. 1997). pGA1 encodes the Rep protein that is needed for replication of the plasmid. The amount of Rep and hence the plasmid’s copy number is controlled by a ctRNA overlapping the start of rep (Tauch et al. 2002). Several promoters were tested in this study, and the most promising was used to replace the endogenous promoter for rep. Secondly, the studies mentioned above on mycolic acid mutants addressed Mycobacterium and Corynebacterium, but not other genera in the order. It has been shown that the long mycolic acids are not necessary for growth of Rhodococcus equii and that growth of this bacterium was much less inhibited by the FASII inhibitors triclosan and isoniazid compared to M. smegmatis (Sydor et al. 2013). Still, this only indicates that the two genera might be different, while it still is not known if mycolic acid biosynthesis is necessary for R. opacus. Hence, we chose to test our new vector by trying to inactivate fad32, pks13, and accD3, using a similar approach as the one utilized by Portevin et al. (2004, 2005).
Methods
Strains, growth media, and culture conditions-
R. opacus PD630 (DSM 44193 renamed Rhodococcus wratislaviensis) was cultured at 30 °C and Escherichia coli at 37 °C, both at 225 rpm shaking in liquid Luria Bertani (LB) media (10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) or on LA plates (LB containing 15 g/L agar). Six percent sucrose was added to the solid media when needed for selection against SacB. For selection of transformants and conjugants, the concentrations of antibiotics used were kanamycin (Km) 25 μg/mL, chloramphenicol (Cm) 10 μg/mL, nalidixic acid (Nal) 40 μg/mL, and gentamycin (Gm) 20 μg/mL for R. opacus and Km 50 μg/mL, Gm 20 μg/mL, and Cm 20 μg/mL for E. coli. Gene expression was induced by 0.5 mM m-toluate or repressed by 100 ng/mL anhydrotetracycline (aTc) unless stated otherwise. E. coli DH5α was used for cloning and E. coli S17.1 (Simon et al. 1983) for conjugation.
Construction of plasmids
All plasmids and the details of their construction are described in Supplementary Table S1. All primers were designed using the Clone Manager 9 software from Sci Ed Software LLC, USA, and are listed in Supplementary Table S2. Plasmids constructed with PCR-amplified fragments were verified by Sanger sequencing (Eurofins Scientific, Luxembourg). Restriction enzymes, DNA modification enzymes, T4 DNA ligase, and Q5® High-Fidelity DNA Polymerase were obtained from New England Biolabs, USA. Zero Blunt™ TOPO™ PCR Cloning Kit (Invitrogen, USA) was used for cloning of some of the PCR fragments. SLIC cloning was performed as described earlier (Islam et al. 2017). For genomic DNA extraction, MasterPure™ Complete DNA and RNA Purification Kit (Lucigen, USA) was used, but cells from one 1 ml culture were treated with lysozyme (0.5 mg/ml) in 150 µl TE for 30 min prior to treatment with proteinase K. All construction of plasmids was performed using E. coli, and the final plasmids were then transferred to R. opacus by conjugation.
Construction of strains by conjugation and homologous recombination in R. opacus
Conjugation was performed as described earlier (Gimmestad et al. 2009), and the plasmid-encoded kanamycin or gentamycin resistances were used to select against R. opacus wild-type cells, while nalidixic acid was used to select against E. coli. For recombination studies using conditional suicide plasmids, the first crossover was selected for by omitting the inducer (m-toluate) and adding the repressor (aTc) to the medium. Double crossovers were selected on LA sucrose plates, and sucrose-resistant colonies were checked for sensitivity to kanamycin and further tested by PCR to identify clones with the desired mutant genotype. For colony PCR, some cell mass was transferred to the 1.5-mL tubes with 100 μL milli-Q water and pipetted up and down to mix the solution. The cells were then incubated at 100 °C for 15 min, and 0.5–1 µl of the boiled solution was used as DNA sample for PCR reactions. When necessary, isolated genomic DNA was used instead of boiled cells.
Assay of promoter strength
The Luciferase Assay System E1500 (Promega corporation, USA) was used to quantify the production of luciferase. Strains with plasmids were inoculated from precultures to an initial OD600 of 0.05, cultivated in LB Km and sampled after 24 and 48 h. R. opacus (pEC18Kmob2) was used as negative control for luciferase. All cultivations were performed in triplicates.
Results
Comparison of different promoters
Our first task was to find a promoter system for the rep gene that preferentially could be turned on and off, and with sufficient promoter strength to allow for good plasmid replication when the promoter was turned on. In order to compare different promoter systems, it is important that the vectors are as identical as possible. A vector backbone based on pEC18Kmob2 (Tauch et al. 2002) was constructed and denoted pRMG3 (Fig. 1a). pRMG3 was constructed by adding a NdeI-site downstream of the Plac and RBS found in the vector, cloning a luciferase-encoding open reading frame from the plasmid pUV15tetORm::luciferase into this NdeI-site, and finally add a NotI site upstream of the Plac promoter. The promoter is then flanked by NotI and NdeI restriction sites, and these were used to exchange this promoter with other promoters and control elements as depicted in Fig. 1a. Co-expression of lacI together with the Plac-promoter (pHE511) was tested since we were looking for an inducible system. In pHE518, production of luciferase was controlled by the strong Pmyc1 promoter from M. smegmatis combined with the tet-operator TetO (Ehrt et al. 2005). The TetR variant used, TetR#28, functions as a repressor for Pmyc1 with anhydrotetracycline as corepressor (Klotzsche et al. 2009). We also tested a double control system developed for Mycobacterium tuberculosis (Dragset et al. 2015) in which the TetR#28 repressor variant is used to repress expression of xylS from the Pmyc1/TetO promoter/operator. In this plasmid (pRMG4), the luciferase gene is expressed from the Pm promoter, which depends on XylS together with its coactivator m-toluate for efficient expression. For comparison, we included the constitutive promoter Pconst (pHE513) that has been analyzed earlier (DeLorenzo et al. 2018).

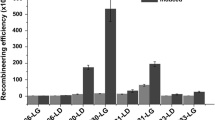
a Map of the plasmids used to test the expression systems. The NotI and NdeI restriction sites were used to exchange promoters and control elements. Promoters and operators are shown as thick arrows; the black boxes indicate the rrnBT1T2 transcriptional terminator. The intervening DNA is not shown in scale. b Promoter strength in R. opacus measured as luminescence/OD600. R. opacus cells containing these plasmids (label above bars) were cultivated in LB containing Km, and samples were collected after 24 and 48 h. The numbers are given as the average of three biological replicates. The first line in the horizontal axis shows the promoter used to express the luciferase gene, the second the regulatory protein, the third the regulatory protein used to regulate expression of the first regulatory protein, and the last line the addition of repressor or inducer. “–” is used when regulatory proteins or repressors/inducers are not relevant for that plasmid
The plasmids were transferred to R. opacus by conjugation, and the strains were cultivated in LB containing Km and the appropriate inducers and/or repressors, and growth and luciferase activity were measured. Measured luciferase activity per OD600 was then calculated and used to evaluate relative promoter strength (Fig. 1b). The results showed a difference of 1000 × in luciferase activity per cell. Pconst was the strongest of the tested promoters, followed by Plac while Pmyc1 was the weakest. Induction by IPTG or repression by aTc did not have any significant effect on Plac or Pmyc1, respectively. Still, co-expression of LacI resulted in a tenfold reduction in luciferase activity as compared to the cells containing the smaller plasmid pRMG3. In the absence of m-toluate, the luciferase activity in cells containing pRMG4 with the Pm promoter was five times less than for cells containing lacI and Plac (pHE511). However, when the inducer m-toluate was added, the promoter strength of the Pm-construct was similar to that of LacI-Plac (Fig. 1b). We did not observe any effect of using aTc in liquid media, but higher amounts of the repressor could have been tested.
Construction and verification of a conditional suicide plasmid
pEC18Kmob2 is based on plasmid pGA1 and encodes the Rep protein necessary for its own replication (Tauch et al. 2002). The plasmid replicates by a rolling circle mechanism, and the replication site, denoted “nic,” is found at the 3′-part of rep (Abrhámová et al. 2002). Since only the construct with a double control (Pmyc1/TetO and Pm) displayed inducible luciferase activity, we chose to use that system to control the protein necessary for replication in plasmid pEC18Kmob2. It should be noted that we did not know if the induced promoter strength would be sufficient for replication, or if the uninduced expression level was sufficiently low to make the plasmid unstable. However, this regulated promoter system seemed to be the most promising of those tested. The expression of rep is also controlled by a ctRNA overlapping the start codon and promoter of the gene (Venkova-Canova et al. 2003). This could be important for the stability of the plasmid, and two different versions were constructed: pHE524 containing the ctRNA and pHE523 containing only the ORF of rep.
The plasmids were transferred to R. opacus by conjugation, and many transconjugants were obtained for both plasmids when selected for on plates containing Km, Nal, and m-toluate. Transconjugants were then cultivated in LB with and without aTc and plated on LA Km with and without aTc, and on LA as a control for CFU. The results (Table 1) show that even without aTc in the medium, the plasmids were lost at a high frequency. There was no strong effect of the added aTc. Moreover, the cells did not grow well on plates with both Km and aTc, indicating some effect of the repressor on plasmid replication on the solid medium. The plasmid containing ctRNA, pHE524 (Fig. 2a), was lost ten times more frequently than the one without this element (Table 1) and was chosen as the best conditional suicide vector.

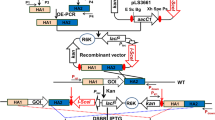
Maps of the a conjugative, conditional suicide plasmid pHE524. The elements used to control replication of the plasmid in R. opacus (nic, rep, TetR#28, and XylS) are described in the text. oripBR322 is used for replication in E. coli and oriT is necessary for conjugative transfer of the plasmids. aph(3,)-IIa encode resistance to kanamycin. b The pHE524 derivative pGJ1 used for homologous recombination in R. opacus, displaying the insertion of homologous arms flanking the chloramphenicol resistance gene cat and the SacB-encoding cassette used for negative selection of the second recombination step. Terminators are indicated as black boxes
The constructed conditional suicide plasmid can be used for homologous recombination
The next step would then be to demonstrate that this conditional suicide plasmid can be utilized for homologous recombination in R. opacus. As a first and simple test for the plasmid, we chose to inactivate gene PD630_RS00415, one of eleven genes in the genome that putatively encode fatty acyl CoA ligases. We have previously inactivated this gene in R. opacus using a conventional, non-replicative recombination plasmid, pMV11 (Supplemental Fig. S2), where PD630_RS00415 was inactivated by an inserted Cmr-gene and knew that this mutant grew similar to the wild-type strain (unpublished). The same inactivated copy of PD630_RS00415 as in pMV11 and the sacB gene encoding levan sucrase to enable selection of the second recombination step were inserted into pHE524 creating pGJ1 (Fig. 2b). The two plasmids were then conjugated to R. opacus. For each plasmid, three parallel conjugations were performed, and transconjugants were selected on plates containing Km for pGJ1 and Gm for pMV11; in both cases, the plates also contained Nal to select against E. coli. No transconjugants were found after conjugation using pMV11; we usually expect 0.1 to 0.2 transconjugants per experiment. However, transfer of pGJ1 resulted in several hundred transconjugants on selective medium and a conjugation frequency of around 3 per 106 recipient cells (Supplemental Fig. S3). We also tested the potential effects of enhancing replication by adding the inducer m-toluate to the conjugation plates and the effect of adding the replication inhibitor aTc to the selection plates. However, neither of these treatments resulted in any significant change in the conjugation frequency.
When a plasmid is used to transfer the homologous fragments flanking changed DNA, a double crossover is needed to obtain the mutant, and this necessitates a multi-step selection protocol (Fig. 3). In the first step, the plasmid will integrate through recombination with one of the homologous regions. In the second step, a second recombination event will result in either wild-type or mutant strains. After transferring the plasmid through conjugation in R. opacus, three transconjugant colonies were picked from a Nal, Km plate, and each was cultivated in LB containing Km, LB with aTc, LB with m-toluate, and LB with no additive. Dilutions were plated from each culture on LA, LA Km, and LA sucrose plates. After growth in liquid media without kanamycin, about 1% of the cells grew on LA Km. This is a relatively high number compared to those found for pHE524 (Table 1) indicating that some recombination had taken place at a previous step. Growth on sucrose plates could be caused by plasmid loss or by a successful double recombination, although it is also known that some cells might grow on sucrose despite the presence of sacB (Hashimoto et al. 2003). Therefore, cells from the sucrose plates were restreaked to plates with either Km or Cm since successful double recombinants should be Kms and Cmr. Four Cmr, Kms, sucroser colonies were confirmed by colony PCR to contain the inserted Cm-gene (Supplemental Fig. S4), demonstrating that the conditional suicide plasmid can be used for homologous recombination in R. opacus. The final protocol (Fig. 3) has the same number of steps as the one using a standard suicide plasmid, but the challenge of few or none transconjugants had been overcome by the new conditional suicide plasmid.

Conjugation scheme using the traditional suicide plasmid (example pMV11) and the conditional suicide plasmid (example pGJ1). The improved step is emphasized
Genes in the mycolic acid gene cluster could not be easily inactivated by homologous recombination
The hydrophobicity of the R. opacus cells results in properties like foaming in high-density cultures and adherence to each other and to some particles. Mycolic acids might be important for these properties, and mycolic acid–negative cells could be useful as control strains in some experiments exploring the surface properties of R. opacus. The second objective of this study was to see if it is possible to remove the genes necessary for mycolic acid biosynthesis in R. opacus like it has been done for C. glutamicum (Portevin et al. 2005). Plasmid pGJ6 (Supplemental Fig. S5) is similar to pGJ1, but that the target is the three genes fad32-pks-accD3 at the 3′ end of the mycolic acid gene cluster (Supplemental Fig. S1). At the end of the recombination protocol (Fig. 3), 17 sucroser, Kms colonies were tested by colony PCR using 6 different primer pairs. We found that although strains where the plasmid was incorporated into the genome had been obtained, only wild-type strains were obtained after the second cross over (Supplemental Fig. S6). The whole experiment was repeated, and this time, 30 sucroser, Kms colonies were tested after genomic DNA extraction, but again, we ended up with the similar negative results (not shown).
These results indicated that mycolic acid could be essential for the survival of R. opacus like it is for M. smegmatis (Portevin et al. 2004, 2005). In their study, this was demonstrated by showing that deletion of essential genes in mycolic acid biosynthesis in M. smegmatis was possible only if a copy of the same gene was expressed from a complementation plasmid (Portevin et al. 2005). Using their approach, a new recombination plasmid, pGJ7 (Supplemental Fig. S5), was constructed targeting accD3, the last gene in the operon. pGJ7 contains the homologous regions flanking both sides of a 533 nt partial accD3 deletion (Supplemental Fig. S7). pGJ7 was conjugated to R. opacus, and transconjugants were cultivated in the presence of aTc and kanamycin for recombination (Supplemental Fig. S8). We had found that cells with the suicide plasmid did not grow well on LA plates with Km and aTc (Table 1); hence, this step was included to select for the recombinants as depicted in Supplemental Fig S8. After selecting for cells that grew on the LA Km aTc plates (Supplementary Fig S8), 4.26 × 106 colonies/ml culture were obtained. These two extra steps might not have been necessary but were added to ensure that strains with an integrated copy of the plasmid were obtained. Colonies picked from the LA Km aTc plates were shown by colony PCR to be first recombinants. One such strain containing pGJ7 inserted into the genome was named GJ7(7) and chosen for the further studies (Supplemental Fig. S9). Similar to the recombination experiments using pGJ6, after selection on sucrose, only wild-type cells or cells that still retained the integrated plasmid were found (Supplemental Fig. S9). The latter probably resulted from inactivation of sacB; this selection system is known to give some false positives (Cianfanelli et al. 2020).
However, first recombinants like strain GJ7(7) could be used to see if accD3 could be deleted in the chromosome when a plasmid expressing accD3 was present in the cell. To do so, complementation plasmid pGJ8 was constructed. In this plasmid, accD3 is controlled by the strong Pconst. Plasmid pGJ8 was then transferred to strain GJ7(7) by conjugation and selected for by Gm (Supplemental Fig. S8). Then, double cross-over events were selected for by sucrose, and the strains were subsequently tested for sensitivity to Km. This is the same approach as used by Portevin et al. (2005) and is illustrated in Fig. 4 and Supplemental Fig. S8.

Overview of homologous recombination approach for deletion of accD3 in R. opacus using conditional suicide plasmid pGJ7 and complementation plasmid pGJ8. The gene of interest (accD3) with the partial deletion is depicted as a thick, black line
Genomic DNA was extracted from 15 sucroser, Kms, and Gmr colonies and tested by PCR. Five of the colonies showed the mutant band (0.192 kb), while ten showed bands corresponding GJ7(7) (0.192 and 0.719 kb) (Fig. 5). Surprisingly, none of the mutants had reverted to the wild type. The PCR products for two of the mutants (#2 and #8) were confirmed by sequencing to rule out the possibility of non-specific PCR products of the apparently correct length. The result strongly indicates that biosynthesis of mycolic acid is important for the viability of R. opacus.

Gel image showing R. opacus ΔaccD3 mutant strain resulting in presence of complementation plasmid, tested with primers delaccD3F/R. Expected bands for wild type: 0.719 kb, mutant: 0.192 kb and first recombinants: 12.4, 0.719, and 0.192 kb. R: R. opacus (wild type), F7: GJ7(7), P: pGJ7, M: standard (PstI-restricted phage λ DNA)
Discussion
R. opacus is among those bacteria where the combined rate of conjugation and rate of recombination is rather low. Therefore, it becomes laborious and time consuming to obtain strains where the plasmid has integrated into the chromosome when non-replicating plasmids are used. There is no reason why homologous recombination cannot take place even if a replicating plasmid were used; however, it would then be very difficult to select for the mutants or to get rid of the plasmid after a successful second recombination had removed it from the chromosome. The latter would be necessary to avoid new rounds of recombination events.
In this work, we developed a conditional suicide plasmid for R. opacus (Fig. 2). Obviously, a much higher number of transconjugants were obtained when this plasmid was used for homologous recombination compared to using a non-replicating plasmid. Since the process of plasmid transfer became decoupled from that of recombination, the plasmid solved the problem of the low number of transconjugants obtained when using non-replicating recombination plasmids.
The plasmid was then used to delete essential genes in the mycolic acid biosynthetic pathway of R. opacus. However, a chromosomal deletion mutant was only obtained in the presence of a complementing plasmid. Earlier, Portevin et al. used a similar approach to show that a mutant with deletions of pks13, fad32, or accD4 was viable in M. smegmatis only when a complementing plasmid expressing these genes was present. However, in their study, they used a thermosensitive plasmid as their conditional suicide plasmid (Portevin et al. 2004, 2005). Our results suggest that mycolic acid is essential for the viability of R. opacus like it is for Mycobacterium but not for Corynebacterium (Portevin et al. 2004, 2005; Raad et al. 2010). For the latter genus, one species lacking mycolic acid has also been described (Collins et al. 1988).
Measurements of relative promoter strengths (Fig. 1b) combined with the identification of pHE524 as a conditional suicide plasmid (Table 1) indicate that a promoter strength above the uninduced level of our dual control system (pRMG4) but below the induced level is necessary for the plasmid to be stably maintained. If the tested promoters work equally well for the rep gene as for the luciferase-expressing gene, it follows that if Pconst or Plac had been used to control rep, the plasmid would have been too stable. If Pmyc1 had been used, the plasmid would have been less stable since that promoter seemed weaker than the uninduced Pm dual control system. Still, it might have been sufficient for recombination purposes, especially if the ctRNA had been removed.
Similar conditional suicide plasmids to ours have earlier been developed for Gram-negative bacteria (Gimmestad et al. 2009; Naorem et al. 2018), and this suggests that the approach may be utilized in other species as well, if an inducible or repressible promoter is available for the species. The Pm-XylS expression system was chosen as one of our candidates because of its known broad host range (Gawin et al. 2017), and our result shows that the system constructed for M. tuberculosis (Dragset et al. 2015) provided inducible expression in R. opacus, too. Even though we did not observe a significant effect of aTc in liquid culture for R. opacus, the system still worked well. It would have been possible to test higher concentrations of the repressor if we had needed the effect. As it is, our results show that when a fairly weak promoter is used to express xylS, further repression of this promoter was not necessary for obtaining a suitably unstable delivery plasmid in R. opacus. For Gram-negative bacteria, it has been shown that Pm promoter mutants and 5′UTR mutants with a wide range of expression strengths may be found (Bakke et al. 2009; Lale et al. 2011), and this could be an alternative if the wild-type promoter has a too low induced activity or a too high uninduced activity in the bacterium of interest.
The conditional suicide vector pHE524 should also be useful as a vector for delivering, e.g., transposons or genome-editing systems like CRISPR-Cas or Cre-Lox, because the plasmid disappears with high frequency when it is not selected for by kanamycin. Moreover, since the replication system originates from C. glutamicum pGA1 (Tauch et al. 2002), while the promoter-regulator system controlling replication in the conditional suicide vector was developed for M. smegmatis (Dragset et al. 2015), the plasmid could potentially function in the genera Mycobacterium and Corynebacterium as well as for Rhodococcus.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. Plasmids are available upon request.
References
Abrhámová Z, Pátek M, Nesvera J (2002) Atypical location of double-strand origin of replication (nic site) on the plasmid pGA1 from Corynebacterium glutamicum. Folia Microbiol (praha) 47(4):307–310. https://doi.org/10.1007/bf02818687
Alvarez HM, Kalscheuer R, Steinbüchel A (2000) Accumulation and mobilization of storage lipids by Rhodococcus opacus PD630 and Rhodococcus ruber NCIMB 40126. Appl Microbiol Biotechnol 54(2):218–223
Anthony WE, Carr RR, DeLorenzo DM, Campbell TP, Shang Z, Foston M, Moon TS, Dantas G (2019) Development of Rhodococcus opacus as a chassis for lignin valorization and bioproduction of high-value compounds. Biotechnol Biofuels 12:192. https://doi.org/10.1186/s13068-019-1535-3
Bakke I, Berg L, Aune TE, Brautaset T, Sletta H, Tøndervik A, Valla S (2009) Random mutagenesis of the Pm promoter as a powerful strategy for improvement of recombinant-gene expression. Appl Environ Microbiol 75(7):2002–2011. https://doi.org/10.1128/AEM.02315-08
Cappelletti M, Presentato A, Piacenza E, Firrincieli A, Turner RJ, Zannoni D (2020) Biotechnology of Rhodococcus for the production of valuable compounds. Appl Microbiol Biotechnol 104(20):8567–8594. https://doi.org/10.1007/s00253-020-10861-z
Cianfanelli FR, Cunrath O, Bumann D (2020) Efficient dual-negative selection for bacterial genome editing. BMC Microbiol 20(1):129. https://doi.org/10.1186/s12866-020-01819-2
Collins MD, Burton RA, Jones D (1988) Corynebacterium amycolatum sp. nov. a new mycolic acid-less Corynebacterium species from human skin. FEMS Microbiol Lett 49(3):349–352. https://doi.org/10.1111/j.1574-6968.1988.tb02755.x
de Carvalho CCCR, Fischer MA, Kirsten S, Würz B, Wick LY, Heipieper HJ (2016) Adaptive response of Rhodococcus opacus PWD4 to salt and phenolic stress on the level of mycolic acids. AMB Express 6(1):66. https://doi.org/10.1186/s13568-016-0241-9
DeLorenzo DM, Rottinghaus AG, Henson WR, Moon TS (2018) Molecular toolkit for gene expression control and genome modification in Rhodococcus opacus PD630. ACS Synth Biol 7(2):727–738. https://doi.org/10.1021/acssynbio.7b00416
Dragset MS, Barczak AK, Kannan N, Maerk M, Flo TH, Valla S, Rubin EJ, Steigedal M (2015) Benzoic acid-inducible gene expression in Mycobacteria. PLoS One 10(9):e0134544. https://doi.org/10.1371/journal.pone.0134544
Ehrt S, Guo XV, Hickey CM, Ryou M, Monteleone M, Riley LW, Schnappinger D (2005) Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res 33(2):e21. https://doi.org/10.1093/nar/gni013
Gande R, Dover LG, Krumbach K, Besra GS, Sahm H, Oikawa T, Eggeling L (2007) The two carboxylases of Corynebacterium glutamicum essential for fatty acid and mycolic acid synthesis. J Bacteriol 189(14):5257–5264. https://doi.org/10.1128/JB.00254-07
Gawin A, Valla S, Brautaset T (2017) The XylS/Pm regulator/promoter system and its use in fundamental studies of bacterial gene expression, recombinant protein production and metabolic engineering. Microb Biotechnol. https://doi.org/10.1111/1751-7915.12701
Gimmestad M, Ertesvåg H, Heggeset TMB, Aarstad O, Svanem BIG, Valla S (2009) Characterization of three new Azotobacter vinelandii alginate lyases, one of which is involved in cyst germination. J Bacteriol 191(15):4845–4853
Goodfellow M, Weaver CR, Minnikin DE (1982) Numerical classification of some Rhodococci, Corynebacteria and Related Organisms. Microbiol 128(4):731–745. https://doi.org/10.1099/00221287-128-4-731
Hashimoto JG, Stevenson BS, Schmidt TM (2003) Rates and consequences of recombination between rRNA operons. J Bacteriol 185(3):966–972. https://doi.org/10.1128/jb.185.3.966-972.2003
Houssin C, de Sousa DC, Constantinesco F, Dietrich C, Labarre C, Bayan N (2020) Architecture and biogenesis of the cell envelope of Corynebacterium glutamicum. In: Inui M, Toyoda K (eds) Corynebacterium glutamicum: biology and biotechnology. Springer International Publishing, Cham, pp 25–60
Islam MN, Lee KW, Yim HS, Lee SH, Jung HC, Lee JH, Jeong JY (2017) Optimizing T4 DNA polymerase conditions enhances the efficiency of one-step sequence- and ligation-independent cloning. Biotechniques 63(3):125–130. https://doi.org/10.2144/000114588
Karunakaran P, Endresen DT, Ertesvåg H, Blatny JM, Valla S (1999) A small derivative of the broad-host-range plasmid RK2 which can be switched from an replicating to a non-replicating state as a response to an externally added inducer. FEMS Microbiol Lett 180:221–227
Klotzsche M, Ehrt S, Schnappinger D (2009) Improved tetracycline repressors for gene silencing in mycobacteria. Nucleic Acids Res 37(6):1778–1788. https://doi.org/10.1093/nar/gkp015
Lale R, Berg L, Stüttgen F, Netzer R, Stafsnes M, Vee Aune TE, Valla S (2011) Continuous control of the flow in metabolic pathways through 5’-UTR sequence modifications in mRNA expressed from the broad-host-range Pm promoter. Appl Env Microbiol 77:2648–2655
Lechevalier MP, Horan AC, Lechevalier H (1971) Lipid composition in the classification of Nocardiae and Mycobacteria. J Bacteriol 105(1):313–318. https://doi.org/10.1128/jb.105.1.313-318.1971
Marrakchi H, Lanéelle M-A, Daffé M (2014) Mycolic acids: structures, biosynthesis, and beyond. Chem Biol 21(1):67–85. https://doi.org/10.1016/j.chembiol.2013.11.011
Minnikin DE (1991) Chemical principles in the organization of lipid components in the mycobacterial cell envelope. Res Microbiol 142(4):423–427. https://doi.org/10.1016/0923-2508(91)90114-p
Naorem SS, Han J, Zhang SY, Zhang J, Graham LB, Song A, Smith CV, Rashid F, Guo H (2018) Efficient transposon mutagenesis mediated by an IPTG-controlled conditional suicide plasmid. BMC Microbiol 18(1):158. https://doi.org/10.1186/s12866-018-1319-0
Nesvera J, Pátek M, Hochmannová J, Abrhámová Z, Becvárová V, Jelínkova M, Vohradský J (1997) Plasmid pGA1 from Corynebacterium glutamicum codes for a gene product that positively influences plasmid copy number. J Bacteriol 179(5):1525–1532. https://doi.org/10.1128/jb.179.5.1525-1532.1997
Portevin D, Sousa-D’Auria CC, Houssin C, Grimaldi C, Chami M, Daffe M, Guilhot C (2004) A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. PNAS 101(1):314–319. https://doi.org/10.1073/pnas.0305439101
Portevin D, D’Auria LD, Montrozier H, Houssin C, Stella A, Laneelle MA, Bardou F, Guilhot C, Daffe M (2005) The acyl-AMP ligase FadD32 and AccD4-containing acyl-CoA carboxylase are required for the synthesis of mycolic acids and essential for mycobacterial growth - Identification of the carboxylation product and determination of the acyl-CoA carboxylase components. J Biol Chem 280(10):8862–8874. https://doi.org/10.1074/jbc.M408578200
Raad RB, Méniche X, de Sousa-d’Auria C, Chami M, Salmeron C, Tropis M, Labarre C, Daffé M, Houssin C, Bayan N (2010) A deficiency in arabinogalactan biosynthesis affects Corynebacterium glutamicum mycolate muter membrane mtability. J Bacteriol 192(11):2691–2700. https://doi.org/10.1128/JB.00009-10
Simon R, Priefer U, Pühler A (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Biotechnology (n y) 1:784–791
Sydor T, von Bargen K, Hsu FF, Huth G, Holst O, Wohlmann J, Becken U, Dykstra T, Sohl K, Lindner B, Prescott JF, Schaible UE, Utermohlen O, Haas A (2013) Diversion of phagosome trafficking by pathogenic Rhodococcus equi depends on mycolic acid chain length. Cell Microbiol 15(3):458–473. https://doi.org/10.1111/cmi.12050
Tauch A, Kirchner O, Löffler B, Götker S, Pühler A, Kalinowski J (2002) Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr Microbiol 45(5):362–367. https://doi.org/10.1007/s00284-002-3728-3
Venkova-Canova T, Patek M, Nesvera J (2003) Control of rep gene expression in plasmid pGA1 from Corynebacterium glutamicum. J Bacteriol 185(8):2402–2409
Acknowledgements
We thank Marte Vilnes for creating pMV11 and the original PD630_RS00415 mutant and Monika Wikarská and Rocio Maciel Gómez for constructing some of the plasmids. Plasmids pDD112 and pDD120 were gifts from Professor Moon, Department of Energy, Environmental and Chemical Engineering, Washington University in St. Louis, Missouri, USA.
Funding
Open access funding provided by NTNU Norwegian University of Science and Technology (incl St. Olavs Hospital - Trondheim University Hospital) This work was funded by the Norwegian Research council grants 239001 and 274691.
Author information
Authors and Affiliations
Contributions
GJ and HE conceived, designed, and performed the experiments and interpreted the results. Both authors wrote the manuscript and read and approved the final version.
Corresponding author
Ethics declarations
Compliance with ethical standards
This article does not contain any studies with animals or with human participants.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jain, G., Ertesvåg, H. Improved site-specific mutagenesis in Rhodococcus opacus using a novel conditional suicide plasmid. Appl Microbiol Biotechnol 106, 7129–7138 (2022). https://doi.org/10.1007/s00253-022-12204-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-022-12204-6