
Abstract
Smokeless tobacco products (STP) contain diverse microbial communities that contribute to the formation of harmful chemical byproducts. This is concerning since 300 million individuals around the globe are users of smokeless tobacco. Significant evidence has shown that microbial metabolic activities mediate the formation of carcinogens during manufacturing. In recent years, studies have revealed a series of additional health impacts that include lesions and inflammation of the oral mucosa and the gastrointestinal tract, as well as alterations of the endogenous microbiota. These findings are due to recent developments in molecular technologies that allowed researchers to better examine the microbial component of these products. This new information illustrates the scale of the STP microbiota and its diversity in the finished product that is sold for consumption. Additionally, the application of metagenomics and metatranscriptomics has provided the tools to look at phylogenies across bacterial, viral, and eukaryotic groups, their functional capacities, and viability. Here we present key examples of tobacco microbiology research that utilizes newer approaches and strategies to define the microbial component of smokeless tobacco products. We also highlight challenges in these approaches, the knowledge gaps being filled, and those gaps that warrant further study. A better understanding of the microbiology of STP brings vast public health benefits. It will provide important information for the product consumer, impact manufacturing practices, and provide support for the development of attainable and more meaningful regulatory goals.
Key points
Newer technologies allowed quicker and more comprehensive identification of microbes in tobacco samples, encapsulating microorganisms difficult or impossible to culture.
Current research in smokeless tobacco microbiology is filling knowledge gaps previously unfilled due to the lack of suitable approaches.
The microbial ecology of smokeless tobacco presents a clearer picture of diversity and variability not considered before.
Similar content being viewed by others

Introduction
The use of tobacco products is considered one of the biggest public health threats in the world according to the World Health Organization (WHO 2020). Worldwide marketplaces consist of a vast array of tobacco products including smokeless tobacco products (STP), which are suspected to have reached every inhabited continent (Siddiqi et al. 2020). Global estimates indicate 300 million people in at least 70 countries use STP, with an estimated 5.9 million in the USA alone (Creamer et al. 2019; Hatsukami et al. 2014; Siddiqi et al. 2020). A fraction of this population will develop cancers and other health complications associated with the use of these products (IARC 2007; Sinha et al. 2016; Timberlake et al. 2017). The term smokeless tobacco indicates the use of unburned tobacco, referring to a large variety of products for oral consumption and nasal insufflation (Delnevo et al. 2020; WHO, FCOTC 2020). These products are highly addictive and differ greatly in manufacturing methods, composition, and associated health risks, making them a global health concern (Mutti et al. 2016; Siddiqi et al. 2020).
A considerable amount of effort has been placed on investigating the chemical composition of cured and fermented tobacco, to identify chemical constituents that may pose significant health risks (Hearn et al. 2013; Lawler et al. 2013; Richter et al. 2008; Stanfill et al. 2011; Stepanov et al. 2008). In STP, an estimated 4000 compounds have been found in chemical characterizations of these products, with over 30 of these linked to cancer (Hatsukami et al. 2014). Many of these characterizations have investigated the mechanism of tobacco-specific nitrosamines (TSNAs) formation, their in-product concentrations, and associations to disease. Interestingly, some of these studies have suggested microbial metabolic activities in tobacco play a role in the formation of TSNAs. For instance, it is understood that TSNAs form during the air-curing of burley tobacco as a result of the microbial-mediated conversion of nitrate to nitrite, and the subsequent reaction of nitrite with alkaloids (i.e., nicotine) in tobacco (IARC 2007).
As early as the 1970s, a substantial amount of research was steered towards addressing the “tobacco microbiology” aspects of different tobacco products in the market (Mitchell and Stauber 1972). A large portion of the initial scientific contributions came from major tobacco companies (Pauly and Paszkiewicz 2011). Many of these studies were disadvantaged by the technology available at the time the research was done, consequently reaching limited conclusions. However, in recent years, several studies have applied newer molecular-based technologies to study microbial compositions in STP (Al-Hebshi et al. 2017; Han et al. 2016; Monika et al. 2020; Rivera et al. 2020; Smyth et al. 2017; Tyx et al. 2020; Tyx et al. 2016). Their observations suggest that STP microbiota are variable and diverse, in contrast to consumable products outside the tobacco industry, which normally possess reduced diversities with fewer species in high abundance.
As the microbial ecology of STP is further explored, a clearer picture of diversity and variability informs regulators of additional potential harmful constituency not considered before. Areas of concern like pathogenic species or toxin-carrying microorganisms are described in more detail by recent work (Al-Hebshi et al. 2017; Han et al. 2016; Monika et al. 2020; Rivera et al. 2020; Tyx et al. 2020; Tyx et al. 2016). Microbial contributions outside the canonical chemistry found in smokeless tobacco are beginning to emerge in the literature. It is also recognized that a compendium of the latest microbial studies on these products is lacking. Therefore, in this review, we aim to summarize current scientific works pertaining to American-made STP microbiology and the impact the latest findings have had on the microbiology knowledge of these products.
Smokeless tobacco harbors diverse microbial communities
Early reports on tobacco for cigar production identified microbial species with functional roles during fermentation in the early stages of production (Di Giacomo et al. 2007). For instance, the fungi Debaryomyces hansenii utilizes lactic acid as a main carbon source and produces ammonia, leading to pH increases that stimulate growth of other microorganisms. Among these microorganisms are Bacillus species (i.e., B. licheniformis, B. subtills), evidenced to reduce NO3 without producing N2 gas, and species like Corynebacterium ammoniagenes, reported to accumulate nitrite during subsequent stages of tobacco maturation (Di Giacomo et al. 2007).
Similarly, other groups have identified a vast diversity of bacterial species present in cigars, cigarettes, and smokeless tobacco products (Chattopadhyay et al. 2019; Chopyk et al. 2017a; Sapkota et al. 2010). These studies have shown that several genera, such as Bacillus, Pseudomonas, Pantoea, and Staphylococcus in particular, tend to be present in most or all tobacco products. However, the dominant organisms for each product category are mostly different. Cigars have been found to be dominated by the Family Enterobacteriaceae, and the genera Bacillus, Pantoea, Pseudomonas, and Staphylococcus (Di Giacomo et al. 2007; Smyth et al. 2017). Cigarette tobacco tends to be dominated by Pseudomonas (Chopyk et al. 2017b), while smokeless (snuff) tobacco has been found to be dominated by a variety of different genera including Bacillus, Corynebacterium, Lactobacillus, Marinilactibacillus, Oceanobacillus, Paenibacillus, Staphylococcus, and Tetragenococcus (Al-Hebshi et al. 2017; Han et al. 2016; Rivera et al. 2020; Smyth et al. 2017; Tyx et al. 2020; Tyx et al. 2016). Consequently, the microbial factor of these products remains to some extent, enigmatic.
Microbial life has been long understood to drive reactions that make smokeless tobacco more palatable and has been studied using numerous technologies throughout the years. Earlier studies into tobacco microbiology involved culturing microbes (bacteria and fungi) from tobacco itself, or from products (Pauly et al. 2008). Most older studies that identified various organisms focused on those that grew in common culture media used for clinical isolations. These studies identified taxa of note corresponding to genera Micrococcus, Bacillus, Phytomonas, Staphylococcus, Alternaria, Aspergillus, and Penicillium among a few others (Jensen and Parmele 1950; Pauly et al. 2008; Peiser et al. 1982; Verweij et al. 2000; Welty 1972; Welty et al. 1968). Later studies used classical phenotypic and metabolic assays to classify bacteria and fungi. More recently, studies backed up or supplemented culturing methods by attempting to measure total bacterial load, often using quantitative PCR (qPCR). Law et al. measured nitrogen-reducing bacteria using qPCR on napA and narG genes while Al-Hebshi et al. obtained total bacterial load by qPCR on the 16S gene itself (Al-Hebshi et al. 2017; Law et al. 2016). However, a more robust complementation came with the implementation of Sanger sequencing, usually of the 16S/18S ribosomal RNA sequences commonly used for taxonomic identification (Huang et al. 2010; Su et al. 2011).
Newer technologies allowed quicker and more comprehensive identification of microbes in samples, encapsulating difficult or impossible to culture microorganism. At least a few studies published prior to the availability of high-throughput sequencing (HTS) used denaturing gradient gel electrophoresis (DGGE) technology with Sanger sequencing and restriction fragment length polymorphism (RFLP) analysis (Di Giacomo et al. 2007; Zhao et al. 2007). Though Di Giacomo et al. focused on cigar tobacco, this oft-cited study is of particular note because of the breadth and detail used, including chemical measurements and the use of novel media to grow isolates from tobacco.
With the development of microarrays and more advanced HTS technologies like semiconductor-based sequencing (IonTorrent™), sequence by synthesis (Pyrosequencing™, Illumina™), and nanopore sequencing (Oxford Nanopore™), used in combination with 16S rRNA gene, amplification allowed for much greater sample depth as well as faster and easier identification. Several publications were released in succession using some of these technologies. One of the first reports on microbial communities in tobacco was from Sapkota et al. who used microarray and HTS technologies to characterize cigarette microbial communities (Sapkota et al. 2010). Smokeless tobacco products were not investigated using these technologies until Tyx et al. released a 16S HTS study using semiconductor-based sequencing technology (Tyx et al. 2016). Shortly thereafter, this study was followed by several others along the same lines, demonstrating the diversity of bacterial life in STP, while also showing the breadth of life observed between products and product types (Al-Hebshi et al. 2017; Han et al. 2016; Law et al. 2016; Smyth et al. 2017; Tyx et al. 2016).
It should be noted that a few of the more recent studies (Al-Hebshi et al. 2017; Mehra et al. 2020; Zhang et al. 2020; Zhou et al. 2020) focus mainly on products or aging tobacco not widely available in the USA. While “international” products are quite interesting, especially in the capacity that some products such as Sudanese toombak, Yemeni Shammah, and Asian betel quid contain higher amounts of carcinogenic TSNAs as compared with US domestic products, we will forego discussing them in this review in order to focus on US domestic products. For the aforementioned reasons, we suggest that a follow-up review on international products would be of great benefit to the community.
The most prominent difference between the culturing methods and the culture-independent methods is the much-increased diversity observed in most products interrogated with molecular approaches. While some products were still found to have low diversity using these methods, mainly some moist snuff products, it is clear that newer technologies like HTS allow researchers to observe the microbial community in much greater depth than had previously been reported. Prior to the advent of HTS and microarray studies, most noted bacteria were classified within three phyla. Among the most abundant are Firmicutes, specifically the genera Bacillus and Staphylococcus. Proteobacteria, with an abundant Class of Gammaproteobacteria, mainly represented by genera Acinetobacter, Proteus, and Pseudomonas. And finally, Actinobacteria for which abundances were observed in the genus Micrococcus (Cockrell et al. 1989; Dygert 1957; Jensen and Parmele 1950; Peiser et al. 1982). High throughput sequencing methods revealed the presence of several additional taxa not previously reported, including additional families in the phylum Actinobacteria. In phylum Bacteroidetes, Classes Flavobacteriia, Sphingobacteriia, and Bacteroidia were identified, while in phylum Proteobacteria, Family Leuconostocaceae was also identified (Han et al. 2016; Law et al. 2016; Smyth et al. 2017; Tyx et al. 2016).
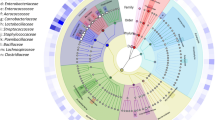
There are some disadvantages to using high-throughput amplicon sequencing. The main drawback of short amplicon 16S v-region sequencing is the inability to distinguish individual species or strains, as is possible with full-length 16S rRNA sequences (Wang et al. 2018). Another disadvantage is the apparent differences in microbiomes based on the 16S region used. Some regions can differentiate specific taxonomic groups better than others, and since many different regions are used in the literature, it can be difficult to compare studies directly. This is demonstrated in the notable differences in the core microbiomes between the various HTS studies. We have compiled data from several studies, based on reported results, where we highlight overlapping groups which may make up the “core” microbiome genera present in STP (Fig. 1, Table S1).

Venn diagram of the shared taxa found in five STP microbiota studies. These studies determined microbial composition using different variable (V) regions 16S rRNA gene. (A) Al-Hebshi et al., used V1–V3; (B) Han et al., V6; (C) Law et al., V4; (D) Smyth et al., V1–V2; and (E) Tyx et al., V4. Notable differences in the core microbiomes between the various studies are shown as the numbers in each overlapping region. The overlapping groups at the center represent the potential “core” microbiome of genera present in STP. Shared taxa names are illustrated in supplemental table 1 (Table S1)
One advantage of HTS capabilities is the ability to characterize the microbiota in STP in much greater depth than ever before. This is because HTS enables the use of shotgun sequencing, where every piece of DNA from a sample can be sequenced without amplification and then classified, provided the database is sufficiently comprehensive. Sequencing without amplification eliminates some of the bias that may be present in 16S studies that use PCR amplification. Thus, sequencing every piece of DNA enables the detailing of an entire metagenome — all genomes of every organism in the sample — a powerful tool for observing functional capabilities of the species present. Functional information of this type can be imputed using 16S studies (i.e., Phylogenetic Investigation of Communities by Reconstruction of Unobserved States or PICRUSt) but may be prone to the aforementioned PCR bias and further limitations of the databases involved (Langille et al. 2013). Two recent studies using shotgun metagenomic sequencing were able to look in depth at both the phylogeny and the functional capacities of microbial communities in snuff products (Rivera et al. 2020; Tyx et al. 2020). These studies were able to highlight the genomic content of all microbes found in smokeless tobacco.
Another criticism of using 16S rRNA gene-based studies for microbial community characterization is that there is no way to identify whether the bacteria detected are viable or if their DNA is persisting in the product. Tyx et al. attempted to address this issue, where the authors used both DNA and RNA (as reverse-transcribed cDNA) for analysis using HTS (Rivera et al. 2020; Tyx et al. 2020). The study found differences in phylogenetic abundances between the two approaches, but it was also demonstrated that phylogeny and abundances identified using the transcriptome (RNA transcribed to cDNA) closely resembled that of the phylogeny and abundance using a metagenome (DNA sample) for the same product. This suggests that organisms identified in DNA-only studies are giving fairly accurate representations of what is truly present in products. It should also be noted that in that study, in addition to 16S RNA, total RNA was characterized and functionality of the total genome was presented (Tyx et al. 2020).
Bacteria and fungi in smokeless tobacco
The microbial life found in smokeless tobacco products is derived as a result of a long procession of changing environments, starting with tobacco itself. Plants have their own microbiotas, which vary from leaf, to stem, to roots (Bulgarelli et al. 2013). Endophytic bacteria are commonly associated with plants and are helpful to the plant by providing nutrients, combatting disease, etc. (Kandel et al. 2017). Rhizobial-associated bacteria are sometimes considered separately but are normally called endophytic as well; many of these types fix nitrogen into the soil, allowing for uptake of nitrogen by the plant. Additional sources of bacteria to the freshly harvested tobacco include those from the soil and those contributed through fertilizers, handling, and animal contamination. Many of the genera commonly identified in smokeless tobacco, such as Bacillus, Corynebacterium, Microbacterium, and Pseudomonas, contain species commonly known as plant endophytes (Kandel et al. 2017).
In the early stages of smokeless tobacco manufacturing, curing and fermentation play key roles in toxicant formation, presumably mediated by microbial metabolic activity (Di Giacomo et al. 2007; Vigliotta et al. 2007; Zhou et al. 2021). Curing methods are aimed at creating dried tobacco leaves with suitable chemical composition for subsequent fermentation, aging, and consumption. During this process, the amount of reducing sugars increases, leaving the bulk content of the leaf consisting of sugars and proteins prior to fermentation (IARC 2007). In fermentation, cured tobacco carbohydrates and polyphenols in the leaves decrease. This reduction is due to controlled conditions facilitating chemical reactions in the tobacco mixture that produce Maillard reactions in the natural tobacco sugars resulting in darkened tobacco and producing snuff flavor precursors and other compounds (Sensabaugh Jr et al. 1985).
As tobacco is cured, a progression begins where specific groups of microbial species change in their relative abundances. Communities increase or decrease as various nutrients are depleted, pH changes, moisture evaporates, and temperature rises. Overall, this progression produces desirable characteristics sought by manufacturers. However, in STP, this process has not been completely characterized to date, most likely because these are manufacturing trade secrets. An effort to detail at least one specific type — cigar tobacco — has been conducted by Di Giacomo et al. (2007). This study succeeded in demonstrating changes in the microbial community over time during tobacco preparations which correspond with chemical changes taking place simultaneously. Tobacco pH changes gradually, presumably as a result of the several acid-producing species excreting metabolic byproducts like lactic acid.
The authors also found that reducing sugars were depleted over 17 days, while malic and citric acids were reduced substantially during the first 100 h. Additionally, ammonium concentrations were found to initially increase but subsequently decrease. During this fermentation process, nitrate was found to decrease, while nitrite and TSNAs increased. Nitrite, once in the vicinity of alkaloids (i.e., nicotine), tends to form TSNAs through chemical reactions, without the need of biochemical catalysis processes by bacteria in the tobacco milieu.
Fungal species in tobacco have not been a well-explored aspect of tobacco microbiology, yet some studies suggest these microorganisms are present. Notable fungi commonly identified in cured tobacco (but not necessarily smokeless products) included species in the genera Alternaria, Aspergillus, Candida, Cladosporium, Debaryomyces, Epicoccum, Penicillium, and Trichosporon (Di Giacomo et al. 2007; Welty 1971; Welty 1972; Welty and Lucas 1969; Welty et al. 1968). For instance, yeast species have been reported to persist during the early stages of fermentation in tobacco for Italian cigars (Di Giacomo et al. 2007). From these cigar tobaccos, a yeast strain has been identified (Debaryomyces hansenii) and its growth and biochemical properties studied, revealing its potential role in the formation of TSNAs (Vigliotta et al. 2007). Similarly, Larsson et al. detected fungi in cigarette tobacco by quantifying ergosterol, a fungal cell wall component, in fresh and fermented tobacco leaves. They also found that cigarettes stored in high humidity increase the measure of fungi or fungal particulate detected in cigarettes and cigarette smoke (Larsson et al. 2008). Although these are important findings, results did not yield mycobial community structure information in tobacco products. In smokeless tobacco, a limited number of studies involving the identification of fungal species have been published in recent years. Saleem and others reported a more comprehensive isolation of different species of Aspergillus as the predominant genera in Pakistani smokeless tobacco (Saleem et al. 2018). Correspondingly, taxa belonging to the genera Aspergillus and Alternaria were found in metagenomic interrogations of American snuff products (Rivera et al. 2020). While comprehensive characterizations of the fungal component in STP microbiomes are scarce, these studies suggest the possibility of a relevant role for fungal species in the early stages of smokeless products manufacturing.
An element relatively overlooked in STP studies is the viral component of its microbial communities. These taxonomic groups have been casually passed over in some of the early community structure characterizations or studies favoring pathogens searches and species with more metabolically relevant potential (e.g., NO3 reduction). However, some studies do mention the importance of the viral component in smokeless tobacco. Rivera and colleagues found gene sequences for the bacteriophage families Myovirideae, Podovirideae, and Siphovirideae in moist snuff metagenomes. The authors underscore the exogenous bacteriophages as probable vehicles for undesirable genetic exchanges within the user’s microbiomes (Rivera et al. 2020). Tyx et al. presented metatranscriptomic results showing taxa of the family Virgaviridae, provenance of the ssRNA tobacco mosaic virus (TMV). The authors propose TMV presence as a likely contributing factor to chronic oral inflammation (Tyx et al. 2020). Other more benign roles for TMV have been proposed as a recent study submits that the virus may be an immunological mediator for resistance against the SARS-CoV-2 virus (de Bernardis and Busà 2020). Overall, these findings suggest that as tobacco microbiology research evolves, these taxonomic groups merit deeper and more careful examination.
Many species identified in the studies to date have been found to contain various nitrate reductases and transporters. Nitrate reduction may happen by two separate but important pathways, assimilatory and dissimilatory reduction. Assimilatory pathways usually involve the nas genes (nasAB), and couple nitrate to nitrite conversion to further reduction into ammonium. Dissimilatory pathways, usually associated with nitrate reductases encoded in the nar and nap operons (narGHJI and napABC) (Stewart et al. 2002), along with transporters narK, narT, and narU (Clegg et al. 2002), enable the respiratory conversion of nitrate to nitrite. This reaction is usually associated in hypoxic environments where an electron-accepting nitrate ion is used in lieu of an oxygen atom in an energy-generating electron transport chain reaction (Sparacino-Watkins et al. 2014).
An interesting aspect of the microbiota of tobacco is that some microbes have evolved to take advantage of the high levels of nicotine in tobacco, which can be found in levels up to 3% of the total mass of the plant matter (Armstrong et al. 1998; Armstrong et al. 1999). Thus far, Pseudomonas spp. and Arthrobacter spp. are examples of bacteria with nicotine degradation pathways (Brandsch 2006). However, in STP, these species have not been found in high abundance; hence, further research is warranted.
Smokeless tobacco impact on human microbiomes (microbiotas) and health
The use of smokeless tobacco products causes a wide range of pathologies within which disruption of the oral cavity environment is prevalent. It is well documented that STP causes cancer, predominantly in the oral cavity (i.e., cheek, gums, and lips) and pharynx (Boffetta et al. 2008; IARC 2007; Tomar et al. 2019). The harm is not restricted to these areas since esophageal and pancreatic cancers have also been linked to STP use (Boffetta et al. 2008; IARC 2007; Tomar et al. 2019). Apart from cancerous illness, there are other oral health outcomes resulting from the use of these products. These include gingival and periodontal inflammation, oral mucosal lesions, and outside the oral environment, cardiovascular disease which has been associated with hematogenous spread of oral bacteria (Critchley and Unal 2003; Grady et al. 1990; Greer 2011). Additionally, smokeless tobacco contained relatively high concentrations of bacterial lipopolysaccharide (LPS) that may induce inflammatory changes activating endothelium in ways that promote recruitment of leukocytes (Furie et al. 2000). Smokeless tobacco use is not confined to male populations; therefore, we add to this list adverse reproductive outcomes in female STP users (Jin et al. 2018; Nair et al. 2015).
Although STP have circulated the markets for decades, their impact on the user’s microbiome has not been extensively studied. As mentioned above, it is only recently that technology has allowed for a more precise exploration of the oral microbial community composition and alterations from STP use. It has been suggested that an unbalance oral microbiota can result from physiological changes caused by stresses affecting bacterial viability and metabolite levels within the bacterial community (Meurman and Bascones-Martinez 2011). Significant disruption of the oral microbiota can occur in response to STP exposure as it is observed in studies using Syrian Golden hamsters as an in vivo community dynamics model (Jin et al. 2018). Sun and others showed that STP extract enhances oxidative stress and perturbs the arginine-NO pathway in oral bacterial cells (Sun et al. 2016). Furthermore, they observed enrichment of specific species within the community, some classified as opportunistic pathogens (Sun et al. 2016). A similar enrichment phenomenon was observed in studies where Arabian snuff (Shammah) induced changes in the tongue microbiome. That study highlighted the enrichment of bacteria with high acetaldehyde production (i.e., Rothia and other Streptococcii), which were evidenced to ultimately be responsible for increased incidences of cancer for users of these products (Halboub et al. 2020).
Parallel to cancer is a myriad of long suspected STP-related health effects underlying the pathophysiology of many tobacco use–related diseases. In early research efforts, associations between smokeless tobacco use and periodontal disease were established in a survey of young STP users and disease symptoms (Weintraub and Burt 1987). Winn and Tomar underscored the use of STP as a risk factor in the development of root-surface and coronal caries. They argue these effects may be the result of high sugar content in STP, increased gingival recession, and enhanced collagenase activity (Tomar and Winn 1999). More recently, periodontal disease has been linked to enhance biofilm formation and metabolic activity in specific bacterial species mediated by nicotine exposure in a concentration dependent manner (El-Ezmerli and Gregory 2019; Huang et al. 2014; Wu et al. 2018). Moreover, important oral pathogens such as Scardovia wiggsiae, Porphyromonas gingivalis, Lactobacilus casei, Actynomyces viscosus, Rothia dentocariosa, Enterococcus faecalis, and Candida albicans have been associated with enhanced biofilm formation in studies in vitro (Balhaddad et al. 2019; DuBois et al. 2014; Wagenknecht et al. 2018).
While these pathogens play important roles in tooth decay and periodontal disease, we omit further details of those mechanisms but highlight their role as a collective, responding to nicotine exposure. It is noteworthy that mouth commensal streptococci like Streptococcus mutans and S. gordonii undergo gene expression dysregulation when exposed to nicotine (El-Ezmerli and Gregory 2019; Huang et al. 2014; Wagenknecht et al. 2018).
These regulatory effects may serve as modulating factors for bacterial-mediated metabolism, increasing pathogenicity potential for some commensals in the oral cavity (Zhang et al. 2019). For instance, some streptococci play key roles in the development of biofilms, and it is suggested that a species like S. gordonii is integral in initiating colonization by producing surfaces for other colonizers to adhere (El-Ezmerli and Gregory 2019; Huang et al. 2014). Thus, it is reasonable to envision colonization mechanisms whereby the nicotine delivered by STP induces upregulation of biofilm formation facilitating establishment and persistence of species introduced by smokeless products.
A similar dynamic has been observed in an in vitro microbial–mucosal interface model of oral commensals and pathogens. These two defined communities were simultaneously exposed to cigarette smoke, demonstrating the downregulation of essential metabolic gene functions in commensal biofilms while observing an upregulation of some metabolic functions and virulence factor genes in the pathogenic group biofilm. These regulatory responses occurred alongside host cell inflammatory responses causing early commensal death and preventing niche saturation by commensals while enriching for pathogenic species within the biofilm model (Shah et al. 2017). These dynamics may not be restricted to virulence factors but rather include the exchange of other gene functions like toxins and antibiotic resistance. Rivera and others use a metagenomic approach to describe microbial communities’ gene contributions in moist and dry snuff. The study found gene functions conferring genetic transference and antibiotic resistance mechanisms among other functions for species relatives of those found in the oral cavity. They propose that in the event of colonization, a permissive environment like a biofilm matrix may increase the likelihood for undesirable genetic exchanges between endogenous and exogenous species (Rivera et al. 2020).
These scenarios are plausible when considering smokeless tobacco use consists of placing tobacco in the mouth for extended periods of time while chemical (nicotine) and microbial content percolates into the oral environment. Hence, it is interesting to consider what proportion of STP’s microbial species and metabolites inputs into the oral cavity. To date, no study has been reported on these aspects and therefore this possibility warrants investigation.
As in buccal microbiotas, the gastrointestinal (GI) tract exhibits change in its microbiota as a consequence of tobacco products use. A large proportion of the work in the literature looks at effects of smoking and nicotine on the gut microbiome. For example, nicotine exposure has been observed to systematically affect the gut microbiota with diet and sex specificities (Chi et al. 2017; Wang et al. 2019). Associations between smoking and GI microbiota fluctuations have been observed in studies where the phylum Bacteroidetes was found dominant while Firmicutes and Proteobacteria decreased in smokers (Lee et al. 2018). Lin and others observed similar trends with additional smoking positive associations to a reduction in short chain fatty acids in the GI tract which can negatively impact a healthy gut microbiome (Lin et al. 2020).
Alternative explanations for the above described dysbiosis are the smoke-induced modulation of key inflammatory pathways and alterations on level of cytokines in the intestinal immune system (Savin et al. 2018). These altered levels of cytokines generate reactive oxygen species (ROS) in the blood stream, resulting in oxidative stress (Talukder et al. 2011). Furthermore, smoke-related molecular changes of the gut mucin composition can be considered as additional mechanisms where alterations to this protective barrier may lead to alterations in the bacterial composition of this protective layer (Tomoda et al. 2011). Interestingly, these mechanisms are discussed in the context of smoke exposure, yet studies evaluating similar effects from smokeless tobacco products have not been completed.
A clear overlapping aspect between combustion and smokeless tobacco products is their nicotine content and delivery similarities. Smokeless tobacco delivers concentrations of nicotine comparable to cigarettes. However, it does so at slower rates than smoking, and does not expose users to combustion gases and particles (Benowitz 2011). Due to the lack of studies examining these aspects in the context of smokeless tobacco, it is impossible to conjecture if similarities in products’ nicotine delivery affects smokers and STP users’ GI microbiotas in a similar manner.
Smoking toxicities appear to play a key role in the suppression of some species and enrichment of others creating dysbiosis in the GI tract and possible development of several systemic diseases in the host (Wilkins et al. 2019). A recent study examining electronic cigarettes effects over user microbiomes showed that the diversity of the buccal cavity and the gut microbiomes are comparable to that of never or non-smokers. This was in contrast to significant differences observed in the composition of active smokers (Stewart et al. 2018). Immunological alterations notwithstanding, this brings into question whether nicotine alone has the predominant effect on microbial communities or are there additional factors in tobacco affecting the microbial content of these microbiomes. Sapkota et al. proposed that cigarettes per se are the direct cause of exposure of specific bacteria to the GI tract causing changes to the microbiome composition (Sapkota et al. 2010). A similar question can be asked where we look at effects of smokeless tobacco users versus other nicotine delivery systems and the potential introduction of microbial species to an established niche. Remarkably STP studies of this nature are yet to be reported.
Concluding remarks
A long-standing history exists for investigations addressing the negative impact of tobacco use on health. The progression of this history entails an expansion of research questions ranging from chemical composition and toxicology to the microbial component of tobacco products. To date, we have ascertained nicotine as an important culprit for commensal species displacement, leading to microbiome compositional shifts (dysbiosis) and adverse effects on immunological responses. However, nicotine is not the only harmful constituent in STP. For instance, STP extract creates microbial shifting dynamics that decreases favorable bacteria while enriching species that promote disease in both mouth and host gut. In the mouth, STP use can create an ecological imbalance and promote the formation of biofilms which can be considered a conduit for opportunistic pathogens and genetic transference. We now know that STP bacterial communities carry the genes for horizontal gene transfer (HGT) and antimicrobial resistance mechanisms (AMR) among other undesirable traits (Rivera et al. 2020; Tyx et al. 2020). Although, species compatibilities for these mechanisms to function or the likelihood of a significant HGT frequency of events is unknown, the potential may be enhanced in a biofilm environment. Additionally, peripheral factors like sodium salicylate, commonly used as a flavoring and additive agent in STP, are known to induce antibiotic resistance in bacteria (Cohen et al. 1993; Hartog et al. 2010; Riordan et al. 2007). Some of these questions require further investigation and may define future research in smokeless tobacco microbiology.
We cited above a microbial succession phenomenon observed in the early stages of cigar tobacco fermentation during manufacturing (Di Giacomo et al. 2007). Similarly, Zhou and colleagues recently presented a longitudinal microbial dynamics analysis of STP during a 24-month storage (aging) (Zhou et al. 2021). They found significant spatiotemporal heterogeneity in the microbial community structure during aging and emphasized the potential influences species interactions and enzyme activity could have on diversity and composition over time (Zhou et al. 2021). Other studies describing STP microbial structure and function have done end point sampling of products available in the market and ready for consumption. However, smokeless tobacco samples from early stages of manufacturing may allow researchers to identify microbial dynamic events to help pinpoint key changes associated to the generation of TSNAs. Consequently, improved practices to help highlight the toxic agents and their sources in these products and the ability to evaluate industry claims of reduced toxicity, overall harm, and potential unintended consequences are a priority for the field.
Current research in smokeless tobacco microbiology is filling knowledge gaps previously vacant due to the lack of suitable approaches. The incorporation of more advanced technologies in studies of the STP microbial component is now a feasible proposition. DNA sequencing strategies including metagenomic and metatranscriptomic revealed not only species present and their diversity, but also genetic contributions carried as they are introduced into the different microbiotas of the STP user. Moreover, they provide an assessment of species viability and highlight the presence of viral component in STP previously overlooked. Ideally, a combined omics approach whereby transcriptomics and metabolomics are incorporated may be the path to defining species composition and metabolic activity at any given step in the manufacturing of STP.
References
Al-Hebshi NN, Alharbi FA, Mahri M, Chen T (2017) Differences in the bacteriome of smokeless tobacco products with different oral carcinogenicity: compositional and predicted functional analysis. Genes 8(4). https://doi.org/10.3390/genes8040106
Armstrong DW, Wang X, Ercal N (1998) Enantiomeric composition of nicotine in smokeless tobacco, medicinal products, and commercial reagents. Chirality 10(7):587–591. https://doi.org/10.1002/(SICI)1520-636X(1998)10:7<587::AID-CHIR6>3.0.CO;2-#
Armstrong DW, Wang X, Lee J-T, Liu Y-S (1999) Enantiomeric composition of nornicotine, anatabine, and anabasine in tobacco. Chirality 11(1):82–84. https://doi.org/10.1002/(SICI)1520-636X(1999)11:1<82::AID-CHIR14>3.0.CO;2-C
Balhaddad AA, Melo MAS, Gregory RL (2019) Inhibition of nicotine-induced Streptococcus mutans biofilm formation by salts solutions intended for mouthrinses. Restor Dent Endod 44(1):e4. https://doi.org/10.5395/rde.2019.44.e4
Benowitz NL (2011) Smokeless tobacco as a nicotine delivery device: harm or harm reduction? Clin Pharmacol Ther 90(4):491–493. https://doi.org/10.1038/clpt.2011.191
Boffetta P, Hecht S, Gray N, Gupta P, Straif K (2008) Smokeless tobacco and cancer. Lancet Oncol 9(7):667–675. https://doi.org/10.1016/s1470-2045(08)70173-6
Brandsch R (2006) Microbiology and biochemistry of nicotine degradation. Appl Microbiol Biotechnol 69(5):493–498. https://doi.org/10.1007/s00253-005-0226-0
Bulgarelli D, Schlaeppi K, Spaepen S, Ver Loren van Themaat E, Schulze-Lefert P (2013) Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol 64:807-838 doi:https://doi.org/10.1146/annurev-arplant-050312-120106
Chattopadhyay S, Smyth EM, Kulkarni P, Babik KR, Reid M, Hittle LE, Clark PI, Mongodin EF, Sapkota AR (2019) Little cigars and cigarillos harbor diverse bacterial communities that differ between the tobacco and the wrapper. PLoS One 14(2):e0211705. https://doi.org/10.1371/journal.pone.0211705
Chi L, Mahbub R, Gao B, Bian X, Tu P, Ru H, Lu K (2017) Nicotine alters the gut microbiome and metabolites of gut-brain interactions in a sex-specific manner. Chem Res Toxicol 30(12):2110–2119. https://doi.org/10.1021/acs.chemrestox.7b00162
Chopyk J, Chattopadhyay S, Kulkarni P, Claye E, Babik KR, Reid MC, Smyth EM, Hittle LE, Paulson JN, Cruz-Cano R, Pop M, Buehler SS, Clark PI, Sapkota AR, Mongodin EF (2017a) Mentholation affects the cigarette microbiota by selecting for bacteria resistant to harsh environmental conditions and selecting against potential bacterial pathogens. Microbiome 5(1):22. https://doi.org/10.1186/s40168-017-0235-0
Chopyk J, Chattopadhyay S, Kulkarni P, Smyth EM, Hittle LE, Paulson JN, Pop M, Buehler SS, Clark PI, Mongodin EF, Sapkota AR (2017b) Temporal variations in cigarette tobacco bacterial community composition and tobacco-specific nitrosamine content are influenced by brand and storage conditions. Front Microbiol 8:358. https://doi.org/10.3389/fmicb.2017.00358
Clegg S, Yu F, Griffiths L, Cole JA (2002) The roles of the polytopic membrane proteins NarK, NarU and NirC in Escherichia coli K-12: two nitrate and three nitrite transporters. Mol Microbiol 44(1):143–155. https://doi.org/10.1046/j.1365-2958.2002.02858.x
Cockrell WT, Roberts JS, Bernard KE, Robert SF (1989) Microbiology of oral smokeless tobacco products. Tob Sci 191(107):55–57
Cohen SP, Levy SB, Foulds J, Rosner JL (1993) Salicylate induction of antibiotic resistance in Escherichia coli: activation of the mar operon and a mar-independent pathway. J Bacteriol 175(24):7856–7862. https://doi.org/10.1128/jb.175.24.7856-7862.1993
Creamer MR, Wang TW, Babb S, Cullen KA, Day H, Willis G, Jamal A, Neff L (2019) Tobacco product use and cessation indicators among adults - United States, 2018. MMWR 68(45):1013–1019. https://doi.org/10.15585/mmwr.mm6845a2
Critchley JA, Unal B (2003) Health effects associated with smokeless tobacco: a systematic review. Thorax 58(5):435–443
de Bernardis E, Busà L (2020) A putative role for the tobacco mosaic virus in smokers’ resistance to COVID-19. Med Hypotheses 143:110153. https://doi.org/10.1016/j.mehy.2020.110153
Delnevo CD, Hrywna M, Miller Lo EJ, Wackowski OA (2020) Examining market trends in smokeless tobacco sales in the United States: 2011-2019. Nicotine Tob Res. https://doi.org/10.1093/ntr/ntaa239
Di Giacomo M, Paolino M, Silvestro D, Vigliotta G, Imperi F, Visca P, Alifano P, Parente D (2007) Microbial community structure and dynamics of dark fire-cured tobacco fermentation. Appl Environ Microbiol 73(3):825–837. https://doi.org/10.1128/AEM.02378-06
DuBois AE, Bennett ZC, Khalid U, Khalid A, Meece RA, Difiore GJ, Gregory RL (2014) Nicotine: its stimulating and inhibitory effects on oral microorganisms. J Fine Focus 1(1):63–75. https://doi.org/10.33043/FF.1.1.63-75
Dygert HP (1957) Snuff-a source of pathogenic bacteria in chronic bronchitis. N Engl J Med 257(7):311–313. https://doi.org/10.1056/NEJM195708152570704
El-Ezmerli NF, Gregory RL (2019) Effect of nicotine on biofilm formation of Streptococcus mutans isolates from smoking and non-smoking subjects. J Oral Microbiol 11(1):1662275. https://doi.org/10.1080/20002297.2019.1662275
Furie MB, Raffanello JA, Gergel EI, Lisinski TJ, Horb LD (2000) Extracts of smokeless tobacco induce pro-inflammatory changes in cultured human vascular endothelial cells. Immunopharmacology 47(1):13–23. https://doi.org/10.1016/S0162-3109(99)00181-2
Grady D, Greene J, Daniels TE, Ernster VL, Robertson PB, Hauck W, Greenspan D, Greenspan J, Silverman S (1990) Oral mucosal lesions found in smokeless tobacco users. J Am Dent Assoc 121(1):117–123. https://doi.org/10.14219/jada.archive.1990.0139
Greer R, O. Jr. (2011) Oral manifestations of smokeless tobacco use. Otolaryngol Clin N Am 44(1):31-56, v doi:https://doi.org/10.1016/j.otc.2010.09.002
Halboub E, Al-Ak’hali MS, Alamir AH, Homeida HE, Baraniya D, Chen T, Al-Hebshi NN (2020) Tongue microbiome of smokeless tobacco users. BMC Microbiol 20(1):201. https://doi.org/10.1186/s12866-020-01883-8
Han J, Sanad YM, Deck J, Sutherland JB, Li Z, Walters MJ, Duran N, Holman MR, Foley SL (2016) Bacterial populations associated with smokeless tobacco products. Appl Environ Microbiol 82(20):6273–6283. https://doi.org/10.1128/AEM.01612-16
Hartog E, Menashe O, Kler E, Yaron S (2010) Salicylate reduces the antimicrobial activity of ciprofloxacin against extracellular Salmonella enterica serovar Typhimurium, but not against Salmonella in macrophages. J Antimicrob Chemother 65(5):888–896. https://doi.org/10.1093/jac/dkq077
Hatsukami D, Zeller M, Gupta P, Parascandola M, Asma S (2014) Smokeless tobacco and public health: a global perspective. National Cancer Institute and Centers for Disease Control and Prevention. Bethesda, MD
Hearn BA, Renner CC, Ding YS, Vaughan-Watson C, Stanfill SB, Zhang L, Polzin GM, Ashley DL, Watson CH (2013) Chemical analysis of Alaskan Iq’mik smokeless tobacco. Nicotine Tob Res 15(7):1283–1288. https://doi.org/10.1093/ntr/nts270
Huang J, Yang J, Duan Y, Gu W, Gong X, Zhe W, Su C, Zhang KQ (2010) Bacterial diversities on unaged and aging flue-cured tobacco leaves estimated by 16S rRNA sequence analysis. Appl Microbiol Biotechnol 88(2):553–562. https://doi.org/10.1007/s00253-010-2763-4
Huang R, Li M, Ye M, Yang K, Xu X, Gregory RL (2014) Effects of nicotine on Streptococcus gordonii growth, biofilm formation, and cell aggregation. Appl Environ Microbiol 80(23):7212–7218. https://doi.org/10.1128/AEM.02395-14
IARC (2007) Smokeless tobacco and some tobacco-specific N-nitrosamines. International Agency for Research on Cancer. Lyon, France
Jensen CO, Parmele HB (1950) Fermentation of cigar-type tobacco. J Ind Eng Chem 42(3):519–522.
Jin J, Guo L, VonTungeln L, Vanlandingham M, Cerniglia CE, Chen H (2018) Smokeless tobacco impacts oral microbiota in a Syrian Golden hamster cheek pouch carcinogenesis model. Anaerobe 52:29–42. https://doi.org/10.1016/j.anaerobe.2018.05.010
Kandel SL, Joubert PM, Doty SL (2017) Bacterial endophyte colonization and distribution within plants. Microorganisms 5(4):77. https://doi.org/10.3390/microorganisms5040077
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31(9):814–821. https://doi.org/10.1038/nbt.2676
Larsson L, Szponar B, Ridha B, Pehrson C, Dutkiewicz J, Krysinska-Traczyk E, Sitkowska J (2008) Identification of bacterial and fungal components in tobacco and tobacco smoke. Tob Induc Dis 4:4. https://doi.org/10.1186/1617-9625-4-4
Law AD, Fisher C, Jack A, Moe LA (2016) Tobacco, microbes, and carcinogens: correlation between tobacco cure conditions, tobacco-specific nitrosamine content, and cured leaf microbial community. Microb Ecol 72(1):120–129. https://doi.org/10.1007/s00248-016-0754-4
Lawler TS, Stanfill SB, Zhang L, Ashley DL, Watson CH (2013) Chemical characterization of domestic oral tobacco products: total nicotine, pH, unprotonated nicotine and tobacco-specific N-nitrosamines. Food Chem Toxicol 57:380–386. https://doi.org/10.1016/j.fct.2013.03.011
Lee SH, Yun Y, Kim SJ, Lee EJ, Chang Y, Ryu S, Shin H, Kim HL, Kim HN, Lee JH (2018) Association between cigarette smoking status and composition of gut microbiota: population-based cross-sectional study. J Clin Med 7(9). https://doi.org/10.3390/jcm7090282
Lin R, Zhang Y, Chen L, Qi Y, He J, Hu M, Zhang Y, Fan L, Yang T, Wang L, Si M, Chen S (2020) The effects of cigarettes and alcohol on intestinal microbiota in healthy men. J Microbiol 58(11):926–937. https://doi.org/10.1007/s12275-020-0006-7
Mehra R, Mohanty V, Balappanavar AY, Kapoor S (2020) Bacterial contamination of packaged smokeless tobacco sold in India. Tob Prev Cessat 6:11. https://doi.org/10.18332/tpc/115064
Meurman JH, Bascones-Martinez A (2011) Are oral and dental diseases linked to cancer? Oral Dis 17(8):779–784. https://doi.org/10.1111/j.1601-0825.2011.01837.x
Mitchell TG, Stauber PC (1972) Methods for the microbiological examination of tobacco and tobacco products. British American Tobacco Records, p 82
Monika S, Dineshkumar T, Priyadharini S, Niveditha T, Sk P, Rajkumar K (2020) Smokeless tobacco products (STPs) harbour bacterial populations with potential for oral carcinogenicity. Asian Pac J Cancer Prev 21(3):815–824. https://doi.org/10.31557/APJCP.2020.21.3.815
Mutti S, Reid JL, Gupta PC, Pednekar MS, Dhumal G, Nargis N, Hussain AG, Hammond DJTc (2016) Perceived effectiveness of text and pictorial health warnings for smokeless tobacco packages in Navi Mumbai, India, and Dhaka, Bangladesh: findings from an experimental study. Tob Control 25(4):437–443. https://doi.org/10.1136/tobaccocontrol-2015-052315
Nair S, Schensul JJ, Begum S, Pednekar MS, Oncken C, Bilgi SM, Pasi AR, Donta B (2015) Use of smokeless tobacco by Indian women aged 18-40 years during pregnancy and reproductive years. PLoS One 10(3):e0119814. https://doi.org/10.1371/journal.pone.0119814
Pauly JL, Paszkiewicz G (2011) Cigarette smoke, bacteria, mold, microbial toxins, and chronic lung inflammation. J. Oncol 2011:13. https://doi.org/10.1155/2011/819129
Pauly JL, Waight JD, Paszkiewicz GM (2008) Tobacco flakes on cigarette filters grow bacteria: a potential health risk to the smoker? Tob Control 17(Suppl 1):i49–i52. https://doi.org/10.1136/tc.2007.022772
Peiser AJ, Nocella DE, Gray RJH (1982) Microbiological safety and stability of chewing tobacco. J Food Prot 45(5):462–465. https://doi.org/10.4315/0362-028x-45.5.462
Richter P, Hodge K, Stanfill S, Zhang L, Watson C (2008) Surveillance of moist snuff: total nicotine, moisture, pH, un-ionized nicotine, and tobacco-specific nitrosamines. Nicotine Tob Res 10(11):1645–1652. https://doi.org/10.1080/14622200802412937
Riordan JT, Muthaiyan A, Van Voorhies W, Price CT, Graham JE, Wilkinson BJ, Gustafson JE (2007) Response of Staphylococcus aureus to salicylate challenge. J Bacteriol 189(1):220–227. https://doi.org/10.1128/jb.01149-06
Rivera AJ, Tyx RE, Stanfill SB, Watson CH (2020) Microbial communities and gene contributions in smokeless tobacco products. Appl Microbiol Biotechnol 104(24):1–17. https://doi.org/10.1007/s00253-020-10999-w
Saleem S, Naz SA, Shafique M, Jabeen N, Ahsan SW (2018) Fungal contamination in smokeless tobacco products traditionally consumed in Pakistan. J Pak Med Assoc 68(10):1471–1477
Sapkota AR, Berger S, Vogel TM (2010) Human pathogens abundant in the bacterial metagenome of cigarettes. Environ Health Perspect 118(3):351–356. https://doi.org/10.1289/ehp.0901201
Savin Z, Kivity S, Yonath H, Yehuda S (2018) Smoking and the intestinal microbiome. Arch Microbiol 200(5):677–684. https://doi.org/10.1007/s00203-018-1506-2
Sensabaugh Jr AJ, Parks RL, Marsh Jr AC (1985) Process for producing moist snuff. US4528993A
Shah SA, Ganesan SM, Varadharaj S, Dabdoub SM, Walters JD, Kumar PS (2017) The making of a miscreant: tobacco smoke and the creation of pathogen-rich biofilms. npj. Biofilms Microbiomes 3(1):1–9. https://doi.org/10.1038/s41522-017-0033-2
Siddiqi K, Husain S, Vidyasagaran A, Readshaw A, Mishu MP, Sheikh A (2020) Global burden of disease due to smokeless tobacco consumption in adults: an updated analysis of data from 127 countries. BMC Med 18(1):222. https://doi.org/10.1186/s12916-020-01677-9
Sinha DN, Abdulkader RS, Gupta PC (2016) Smokeless tobacco-associated cancers: a systematic review and meta-analysis of Indian studies. Int J Cancer 138(6):1368–1379. https://doi.org/10.1002/ijc.29884
Smyth EM, Kulkarni P, Claye E, Stanfill S, Tyx R, Maddox C, Mongodin EF, Sapkota AR (2017) Smokeless tobacco products harbor diverse bacterial microbiota that differ across products and brands. Appl Microbiol Biotechnol 101(13):5391–5403. https://doi.org/10.1007/s00253-017-8282-9
Sparacino-Watkins C, Stolz JF, Basu P (2014) Nitrate and periplasmic nitrate reductases. Chem Soc Rev 43(2):676–706. https://doi.org/10.1039/c3cs60249d
Stanfill SB, Connolly GN, Zhang L, Jia LT, Henningfield JE, Richter P, Lawler TS, Ayo-Yusuf OA, Ashley DL, Watson CH (2011) Global surveillance of oral tobacco products: total nicotine, unionised nicotine and tobacco-specific N-nitrosamines. Tob Control 20(3):e2. https://doi.org/10.1136/tc.2010.037465
Stepanov I, Jensen J, Hatsukami D, Hecht SS (2008) New and traditional smokeless tobacco: comparison of toxicant and carcinogen levels. Nicotine Tob Res 10(12):1773–1782. https://doi.org/10.1080/14622200802443544
Stewart V, Lu Y, Darwin AJ (2002) Periplasmic nitrate reductase (NapABC enzyme) supports anaerobic respiration by Escherichia coli K-12. J Bacteriol 184(5):1314–1323. https://doi.org/10.1128/jb.184.5.1314-1323.2002
Stewart CJ, Auchtung TA, Ajami NJ, Velasquez K, Smith DP, De La Garza RII, Salas R, Petrosino JF (2018) Effects of tobacco smoke and electronic cigarette vapor exposure on the oral and gut microbiota in humans: a pilot study. PeerJ 6:e4693. https://doi.org/10.7717/peerj.4693
Su C, Gu W, Zhe W, Zhang KQ, Duan Y, Yang J (2011) Diversity and phylogeny of bacteria on Zimbabwe tobacco leaves estimated by 16S rRNA sequence analysis. Appl Microbiol Biotechnol 92(5):1033–1044. https://doi.org/10.1007/s00253-011-3367-3
Sun J, Jin J, Beger RD, Cerniglia CE, Yang M, Chen H (2016) Metabolomics evaluation of the impact of smokeless tobacco exposure on the oral bacterium Capnocytophaga sputigena. Toxicol in Vitro 36:133–141. https://doi.org/10.1016/j.tiv.2016.07.020
Talukder MA, Johnson WM, Varadharaj S, Lian J, Kearns PN, El-Mahdy MA, Liu X, Zweier JL (2011) Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice. Am J Physiol Heart Circ Physiol 300(1):H388–H396. https://doi.org/10.1152/ajpheart.00868.2010
Timberlake DS, Nikitin D, Johnson NJ, Altekruse SF (2017) A longitudinal study of smokeless tobacco use and mortality in the United States. Int J Cancer 141(2):264–270. https://doi.org/10.1002/ijc.30736
Tomar SL, Winn DM (1999) Chewing tobacco use and dental caries among U.S. men. J Am Dent Assoc 130(11):1601–1610. https://doi.org/10.14219/jada.archive.1999.0099
Tomar SL, Hecht SS, Jaspers I, Gregory RL, Stepanov I (2019) Oral health effects of combusted and smokeless tobacco products. Adv Dent Res 30(1):4–10. https://doi.org/10.1177/0022034519872480
Tomoda K, Kubo K, Asahara T, Andoh A, Nomoto K, Nishii Y, Yamamoto Y, Yoshikawa M, Kimura H (2011) Cigarette smoke decreases organic acids levels and population of Bifidobacterium in the caecum of rats. J Toxicol Sci 36(3):261–266. https://doi.org/10.2131/jts.36.261
Tyx RE, Stanfill SB, Keong LM, Rivera AJ, Satten GA, Watson CH (2016) Characterization of bacterial communities in selected smokeless tobacco products using 16S rDNA analysis. PLoS One 11(1):e0146939. https://doi.org/10.1371/journal.pone.0146939
Tyx RE, Rivera AJ, Keong LM, Stanfill SB (2020) An exploration of smokeless tobacco product nucleic acids: a combined metagenome and metatranscriptome analysis. Appl Microbiol Biotechnol 104(2):751–763. https://doi.org/10.1007/s00253-019-10232-3
Verweij PE, Kerremans JJ, Voss A, Meis JF (2000) Fungal contamination of tobacco and marijuana. JAMA 284(22):2875. https://doi.org/10.1001/jama.284.22.2875
Vigliotta G, Di Giacomo M, Carata E, Massardo DR, Tredici SM, Silvestro D, Paolino M, Pontieri P, Del Giudice L, Parente D (2007) Nitrite metabolism in Debaryomyces hansenii TOB-Y7, a yeast strain involved in tobacco fermentation. Appl Microbiol Biotechnol 75(3):633–645. https://doi.org/10.1007/s00253-007-0867-2
Wagenknecht DR, BalHaddad AA, Gregory RL (2018) Effects of nicotine on oral microorganisms, human tissues, and the interactions between them. Curr Oral Health Rep 5(1):78–87. https://doi.org/10.1007/s40496-018-0173-3
Wang F, Men X, Zhang G, Liang K, Xin Y, Wang J, Li A, Zhang H, Liu H, Wu L (2018) Assessment of 16S rRNA gene primers for studying bacterial community structure and function of aging flue-cured tobaccos. AMB Express 8(1):182. https://doi.org/10.1186/s13568-018-0713-1
Wang R, Li S, Jin L, Zhang W, Liu N, Wang H, Wang Z, Wei P, Li F, Yu J, Lu S, Chen Y, Li Z, Wu C (2019) Four-week administration of nicotinemoderately impacts blood metabolic profile and gut microbiota in a diet-dependent manner. Biomed Pharmacother 115:108945. https://doi.org/10.1016/j.biopha.2019.108945
Weintraub JA, Burt BA (1987) Periodontal effects and dental caries associated with smokeless tobacco use. Public Health Rep 102(1):30–35
Welty RE (1971) Fungi isolated from flue-cured tobacco inoculated in the field with storage fungi. Appl Microbiol 21(3):552–554. https://doi.org/10.1128/am.21.3.552-554.1971
Welty RE (1972) Fungi isolated from flue-cured tobacco sold in Southeast United States, 1968-1970. Appl Microbiol 24(3):518–520. https://doi.org/10.1128/am.24.3.518-520.1972
Welty RE, Lucas GB (1969) Fungi isolated from flue-cured tobacco at time of sale and after storage. Appl Microbiol 17(3):360–365. https://doi.org/10.1128/am.17.3.360-365.1969
Welty RE, Lucas GB, Fletcher JT, Yang H (1968) Fungi isolated from tobacco leaves and brown-spot lesions before and after flue-curing. Appl Microbiol 16(9):1309–1313. https://doi.org/10.1128/am.16.9.1309-1313.1968
World Health Organization (2020) Tobacco. Publisher. https://www.who.int/news-room/factsheets/detail/tobacco#:~:text=The%20tobacco%20epidemic%20is%20one,exposed%20to%20second%2Dhand%20smoke. Accessed 23 Oct 2020
WHO FCOTC (2020) Commonly used smokeless tobacco products around the globe. Publisher. https://untobaccocontrol.org/kh/smokeless-tobacco/paan-betelquid-tobacco/. Accessed 23 Oct 2020
Wilkins LJ, Monga M, Miller AW (2019) Defining dysbiosis for a cluster of chronic diseases. Sci Rep 9(1):12918. https://doi.org/10.1038/s41598-019-49452-y
Wu Y, Ma Y, Xu T, Zhang Q-Z, Bai J, Wang J, Zhu T, Lou Q, Götz F, Qu D, Zheng C-Q, Zhao K-Q (2018) Nicotine enhances Staphylococcus epidermidis biofilm formation by altering the bacterial autolysis, extracellular DNA releasing, and polysaccharide intercellular adhesin production. Front Microbiol 9:2575–2575. https://doi.org/10.3389/fmicb.2018.02575
Zhang Y, He J, He B, Huang R, Li M (2019) Effect of tobacco on periodontal disease and oral cancer. Tob Induc Dis 17:40. https://doi.org/10.18332/tid/106187
Zhang Q, Geng Z, Li D, Ding Z (2020) Characterization and discrimination of microbial community and co-occurrence patterns in fresh and strong flavor style flue-cured tobacco leaves. Microbiology open 9(2):e965. https://doi.org/10.1002/mbo3.965
Zhao M, Wang B, Li F, Qiu L, Li F, Wang S, Cui J (2007) Analysis of bacterial communities on aging flue-cured tobacco leaves by 16S rDNA PCR-DGGE technology. Appl Microbiol Biotechnol 73(6):1435–1440. https://doi.org/10.1007/s00253-006-0625-x
Zhou J, Yu L, Zhang J, Zhang X, Xue Y, Liu J, Zou X (2020) Characterization of the core microbiome in tobacco leaves during aging. MicrobiologyOpen 9(3):e984. https://doi.org/10.1002/mbo3.984
Zhou J, Yu L, Zhang J, Liu J, Zou X (2021) Dynamic characteristics and co-occurrence patterns of microbial community in tobacco leaves during the 24-month aging process. Ann Microbiol 71(1):1–13. https://doi.org/10.1186/s13213-021-01620-0
Funding
Funding for this study was provided by the Centers for Disease Control and Prevention, National Center for Environmental Health, Division of Laboratory Sciences. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
AJR conceived concept and delineated manuscript. AJR and RET worked on structure, compiled, and summarized literature. AJR and RET wrote manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention. Use of trade names is for identification only and does not imply endorsement by the Centers for Disease Control and Prevention, the Public Health Service, or the US Department of Health and Human Services.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 229 kb)
Rights and permissions
About this article

Cite this article
Rivera, A.J., Tyx, R.E. Microbiology of the American Smokeless Tobacco. Appl Microbiol Biotechnol 105, 4843–4853 (2021). https://doi.org/10.1007/s00253-021-11382-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-021-11382-z