
Abstract
A hydrophobic gadolinium-based magnetic ionic liquid (MIL) was investigated for the first time as an extraction solvent in dispersive liquid–liquid microextraction (DLLME). The tested MIL was composed of trihexyl(tetradecyl)phosphonium cations and paramagnetic gadolinium chloride anions. The prepared MIL showed low water miscibility, reasonable viscosity, markedly high magnetic susceptibility, adequate chemical stability, low UV background, and compatibility with reversed-phase HPLC solvents. These features resulted in a more efficient extraction than the corresponding iron or manganese analogues. Accordingly, the overall method sensitivity and reproducibility were improved, and the analysis time was reduced. The applicability of the proposed MIL was examined through the microextraction of four sartan antihypertensive drugs from aqueous samples followed by reversed-phase HPLC with UV detection at 240 nm. The DLLME procedures were optimized for disperser solvent type, MIL mass, disperser solvent volume, as well as acid, base, and salt addition. The limits of quantitation (LOQs) obtained with the analysis of 1.2-mL samples after DLLME and HPLC were 80, 30, 40, and 160 ng/mL for azilsartan medoxomil, irbesartan, telmisartan, and valsartan, respectively. Correlation coefficients were greater than 0.9988 and RSD values were in the range of 2.48–4.07%. Under the optimized microextraction conditions and using a 5-mL sample volume, enrichment factors were raised from about 40 for all sartans using a 1.2-mL sample to 175, 176, 169, and 103 for azilsartan medoxomil, irbesartan, valsartan, and telmisartan, respectively. The relative extraction recoveries for the studied sartans in river water varied from 82.5 to 101.48% at a spiked concentration of 0.5 μg/mL for telmisartan and irbesartan and 1 μg/mL for azilsartan medoxomil and valsartan.

Graphical abstract
Similar content being viewed by others

Introduction
Despite significant technological advances, a limited number of analytical techniques are specific and sensitive enough to allow for direct sample analysis. Therefore, sample preparation prior to analysis is still an essential step in most analytical protocols to remove potentially interfering substances, preconcentrate the sample, and/or make it readily compatible with the analytical instrument.
Dispersive liquid–liquid microextraction (DLLME) [1] is undoubtedly a successful sample preparation technique as evident by its impressive number of applications reported in the literature [2]. The technique has become very popular because of its simplicity, rapidness, high extraction efficiency (rate and recovery), and minimal consumption of solvents [3]. However, its reliance on volatile toxic organic extractants, such as chloroform and carbon tetrachloride, is inconsistent with modern trends toward green analytical methods and compromises the method reproducibility. Therefore, the investigation into alternative extraction solvents is indispensable to address the limitations of traditional organic solvents.
Ionic liquids (ILs) are organic salts with melting points at or below 100 °C. Their interesting properties, such as high thermal stability, negligible vapor pressure, and non-flammability [4], make them attractive alternatives and help improve the precision of the analytical measurement. Additionally, their physical and chemical characteristics can be fine-tuned by structural changes in their cationic and anionic parts. This synthesis could be exploited to enhance the extraction recovery and selectivity. ILs have been used in a wide variety of extraction modes [5] including single-drop microextraction (SDME) and solid-phase extraction (SPE) as well as DLLME [6, 7]. The application of ILs as extraction solvents for DLLME first appeared in 2008 [8, 9].
The development of fully automated extractive and measurement procedures is highly desirable because it helps reduce the analysis time, increase sample throughput, and improve the overall method precision through the reduction of human errors [7]. Although ILs have been proven to be excellent extractants in DLLME, the technique still requires centrifugation for phase separation, which is time-consuming and bothersome to automate. In response to this limitation, an innovative class of ionic liquids bearing magnetic properties, i.e., magnetic ionic liquids (MILs) which often involve an imidazolium [10], choline [11], or phosphonium [12] cation and a paramagnetic metal (Fe, Co, Mn, or Gd) chloride anion, has been synthesized. They have been applied not only for analytical applications such as density magnetic levitation [13] but also as both liquid extraction solvents [14] and for SPE [15]. Comparison of headspace single-drop microextraction and DLLME, both using the same MILs, showed the former technique was better for aromatic compounds of higher vapor pressure and the latter better for compounds with lower vapor pressure [16]. In situ formation of hydrophobic MILs has been applied using DLLME for the extraction of PAHs and alkylphenols with HPLC detection [17] and with stir bar DLLME for extraction of fused ring compounds with headspace GC detection [18]. Extractions of asphaltenes, phenols, and herbicides show their broad range of solvation [19]. A comparison of MILs with trichloroferrate anions but differing hydrophobic cations was used for extraction of PAHs from water and tea infusions [20]. Extraction of DNA with in situ DLLME [21] and sequence-specific DNA extraction from biological samples using ion-tagged oligonucleotides bound to MILs [22] have recently been reported.
One class of MILs, with the phosphonium cation trihexyl(tetradecyl)phosphonium (P6,6,6,14) and the metal chloride or hexafluoroacetylacetonate (hfacac) anion, has received considerable attention for DLLME. A comparison of P6,6,6,14-metal(hfacac)x MILs where the metal is Ni, Co, or Dy for the stir bar DLLME of salicylates [23] or preconcentration of bacteria [24] has been made. The [P6,6,6,14+]2 [MnCl42−] MIL overcame the hydrolysis instability and strong UV absorbance of the FeCl4− analogue [25, 26] and was effective for the extraction of selected drugs, phenolics, insecticides, and PAHs [27]. Estrogens have been extracted using the [P6,6,6,14+]2[MnCl42−] MIL from urine [28]. The [P6,6,6,14+]2[CoCl42−] species showed advantages over the Ni, Mn, and Fe tetrachloride analogues for DLLME of estrogens from milk and cosmetics [29]. The [P6,6,6,14+] [Dy(III)(hfacac)4−] MIL has been utilized to introduce an enhanced variant of MIL-based DLLME for microextraction of triazines and sulfonamides through a one-pot, pH-modulated approach [30].
The current study introduces, we believe for the first time, the application of a gadolinium-based MIL (Fig. 1) as an extractant in DLLME and discusses the main characteristics that make it superior to the currently employed iron- and manganese-based [P6,6,6,14+] MIL extractants. The applicability of the proposed MIL was evaluated through the DLLME of four antihypertensive drugs of the sartan class (azilsartan medoxomil, AZI; irbesartan, IRB; telmisartan, TEL; and valsartan, VAL) (Fig. 1) followed by HPLC/UV analysis. Sartans are usually considered one of the first-line medications for the management of hypertension [31]. Recently, these drugs have gained much attention after a retrospective study showed that they could be beneficial in hospitalized COVID-19 patients with hypertension; inpatient treatment with sartans was associated with a lower risk of mortality [32]. These drugs are administered at low doses and are present at low concentration levels in various matrices indicating preconcentration is desirable. Previously, losartan and valsartan have been extracted from plasma using a stir bar approach [33] but to the best of our knowledge, previous application of MIL-based DLLME to this class of compounds has not been reported. In general, pharmaceuticals are rarely considered as analytes for DLLME, particularly using MILs, and therefore the development of effective sample preconcentration procedures for such a class of drugs is undoubtedly important.

Chemical structures of the studied magnetic ionic liquids and the separated sartans
Experimental
Chemicals and materials
Trihexyl(tetradecyl)phosphonium chloride (≥ 95.0%), gadolinium(III) chloride hexahydrate (99.0%), acetone (≥ 99.9%), and phosphoric acid (≥ 99.999% trace metal basis) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dichloromethane (99.9%), manganese(II) chloride tetrahydrate (≥ 99.0%), and iron(III) chloride hexahydrate (≥ 99.0%) were purchased from Acros Organics (Pittsburgh, PA, USA). Acetonitrile (HPLC grade), methanol (HPLC grade), sodium chloride (≥ 99.0%), sodium hydroxide (≥ 97.0%), and hydrochloric acid (ACS, ≥ 38.0%) were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Azilsartan medoxomil (≥ 99.5%), irbesartan (99.6%), telmisartan (99.5%), and valsartan (99.5%) were generously donated by Sigma Pharmaceutical Industries (Quesna, Menofyia, Egypt).
Instrumentation and chromatographic conditions
Throughout this study, all chromatographic analyses were performed using an UltiMate 3000 UHPLC system (Thermo Fisher Scientific™, Sunnyvale, CA, USA). The instrument was equipped with a DGP-3600RS pump, a WPS-3000RS autosampler, a TCC-3000RS column thermostat, and an Ultimate multiwavelength 3000RS UV-VIS detector. Chromeleon 7.2.1 software was used for data acquisition and processing. Chromatographic separation was performed on a Waters Symmetry C18 column (5-μm particle size, 150 mm × 3.9 mm i.d.). A mobile phase consisting of a mixture of acetonitrile and water containing triethylamine (50 mM, pH was adjusted to 2.4 with orthophosphoric acid) in a ratio of 46:54% (v/v) was delivered at a flow rate of 0.8 mL/min. The injection volume was 5 μL, and the UV detector was set at 240 nm. A Sorvall™ Legend™ Micro 21 Microcentrifuge (Thermo Fisher Scientific™, Sunnyvale, CA, USA) was used for centrifugation.
Synthesis of magnetic ionic liquids
In the present work, three hydrophobic MILs based on the trihexyl(tetradecyl)phosphonium (P6,6,6,14) cation and containing different paramagnetic metal-halide anions (FeCl4−, MnCl42−, and GdCl63−) (Fig. 1) were synthesized according to previously published procedures [10, 12]. Iron(III) chloride (1 equiv.), manganese(II) chloride (0.5 equiv.), or gadolinium(III) chloride (0.3 equiv.) was added to a solution of [P6,6,6,14+][Cl−] (1 equiv.) in dichloromethane and the reaction mixture was stirred for 24 h at room temperature. The solvent was removed, and the product was dried under vacuum at 60 °C overnight.
The synthesized [P6,6,6,14+]3[GdCl63−] MIL was characterized by elemental analysis, Fourier transform infrared spectroscopy (FT-IR), and UV/Vis spectroscopy. FT-IR spectra were acquired on a Nicolet iS10 FT-IR spectrometer (Thermo Fisher Scientific™, Waltham, MA, USA) in a KBr pellet. The UV/Vis spectra were recorded in acetonitrile using a PG T80+ spectrophotometer (PG Instruments Limited, Leicestershire, UK).
The water content of the [P6,6,6,14+]3[GdCl63−] MIL as measured by the Karl Fischer titration method was 0.35% (w/w). Elemental analysis for C96H204Cl6GdP3 showed that the %found of C:H equals 63.19:11.43%, respectively (calculated: C, 63.30%; H, 11.29%), which is typical for the formed [P6,6,6,14+]3[GdCl63−] MIL. The FT-IR spectrum of the synthesized [GdCl63−]-based MIL showed a combination of the peaks of [P6,6,6,14+][Cl−] and GdCl3 (Electronic Supplementary Material (ESM) Fig. S1) and is almost identical with the spectra reported for analogous transition metal-based MILs [29], emphasizing that they all contain the same cationic structure. In terms of characterization by UV/Vis, shown as spectra in ESM Figs. S2 and S3, the sharp and relatively narrow absorption peak at 273 nm is characteristic for the GdCl63− anion [34].
Standard solutions and dispersive liquid–liquid microextraction procedure
Stock standard solutions of AZI, IRB, TEL, and VAL were separately prepared at 1 mg/mL in methanol. Appropriate aliquots were transferred from these stock solutions to daily prepare an aqueous standard mixture of the four sartans containing 1 μg/mL for IRB and TEL, and 2 μg/mL for AZI and VAL.
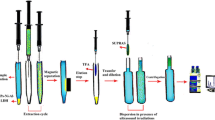
The DLLME procedure (Fig. 2) was performed by transferring 1.2 mL of the aqueous sartan mixture into a 2-mL Eppendorf tube. The premixed solution of the MIL extractant and disperser solvent was injected rapidly into the sample solution through a chromatographic syringe. After manual shaking for 2 min, the cloudy solution produced was then separated using a strong neodymium magnet (B = 1.4 T) or by centrifugation for 6 min at 6000 rpm. Afterward, about 15 μL of the floating extract layer was cautiously collected and transferred into HPLC vials for analysis. Collection of larger volumes of the extract layer increased the chance of mistakenly withdrawing some of the aqueous phase which significantly affected the method reproducibility and, therefore, was avoided.

Schematic representation of the employed DLLME procedure
Extraction optimization, method performance, and recovery
The DLLME procedure was optimized to achieve the highest possible enrichment factors (EFs) with the studied sartans. Experimental conditions, such as disperser solvent type, MIL mass, disperser solvent volume, as well as the effect of acid, base, and salt addition, were studied. Measurements were made in duplicate for error bar calculations. EFs were calculated according to the following equation: EF = Ce/Ci where Ce is the concentration of the analyte in the extract and Ci is the initial concentration of the analyte in the aqueous sample.
The analytical performance of the proposed method was evaluated in ultrapure water by determining the concentration limits of quantitation (LOQs) (defined as the lowest concentration yielding a signal-to-noise ratio of 10), linear ranges, and relative standard deviation values (RSDs). The relative recovery was studied in a river water matrix, collected locally (Cairo, Egypt) in glass amber bottles. All water samples were filtered through a 0.45-μm nylon membrane before extraction to remove particulate matter and analysis was performed within a few hours after sample collection.
Results and discussion
Advantages of [P6,6,6,14 +]3[GdCl6 3−] MIL as an extraction solvent in DLLME
In recent years, growing attention has been paid to the use of MILs as extraction solvents in DLLME to make the technique faster and easier to automate. In this context, several hydrophobic P6,6,6,14-based MILs with different paramagnetic metal-halide anions (e.g., FeCl4−, MnCl42−) were considered for use in DLLME [27, 35, 36]. These tetraalkylphosphonium salts favorably showed compatibility with common reversed-phase HPLC solvents and low miscibility with aqueous samples during extraction. Nevertheless, they suffered from some serious limitations that can affect the extraction efficiency and analytical method performance (e.g., accuracy and precision). The [P6,6,6,14+][FeCl4−] MIL was found to be chemically unstable, as it was susceptible to rapid hydrolysis in aqueous sample solutions [26, 37], and exhibits a strong UV absorbance [27, 35] which limits its compatibility with HPLC when coupled to UV detection. The [P6,6,6,14+]2[MnCl4−2] MIL showed better chemical stability, but it has markedly high viscosity (112,300 cP at 293 K [12]), which influences the mixing and mass transfer processes in DLLME. Accordingly, analyte partitioning and phase separation are slowed down. Also, the high viscosity complicates extract collection and injection into the analytical instrument. Furthermore, both [P6,6,6,14+][FeCl4−] and [P6,6,6,14+]2[MnCl4−2] MILs have only fair magnetic susceptibility (4.34 and 4.22 emu K mol−1, respectively [10, 35]) which reduces their response to an applied external magnetic field during phase separation.
As shown in Table 1, the prepared [P6,6,6,14+]3[GdCl63−] MIL has reasonable viscosity, markedly higher magnetic susceptibility, and is less prone to undergo hydrolysis in aqueous samples. The UV/Vis absorption spectrum of the investigated [P6,6,6,14+]3[GdCl63−] MIL is shown in ESM Fig. S4. Compared to the spectra reported for Fe, Co, and Ni analogues [27, 29], the tested MIL showed considerably lower UV absorption. These features should result in a more efficient extraction (in terms of rate and recovery), faster phase separation, and easier handling of extract during collection and subsequent analysis leading to an improvement in the overall method sensitivity and reproducibility, reducing the analysis time, and allowing for better automation of DLLME. In addition, an aquatic ecotoxicological study of MILs showed those with Fe and Gd metal chloride anions when coupled with certain choline cations could have lower toxicity although overall MILs with the MnCl42− anion were considered the least toxic [38]. However, a cytotoxicity comparison of MILs with two different cations showed a higher human cell toxicity with Co and Mn metal chloride anions as compared to those with Fe and Gd [39].
To evaluate the performance of [P6,6,6,14+]3[GdCl63−] MIL, an aqueous sample of the selected sartans is subject to DLLME followed by HPLC analysis (according to the procedures under the “Experimental” section) and enrichment factors (EFs) were determined. Results were compared with those obtained with [P6,6,6,14+][FeCl4−] and [P6,6,6,14+]2[MnCl4−2] MILs. In agreement with our hypothesis, the [P6,6,6,14+]3[GdCl63−] MIL provided the best enrichment with all sartans. EFs obtained for three sartans with [P6,6,6,14+]3[GdCl63−] were roughly twice those obtained with [P6,6,6,14+]2[MnCl4−2]) (Fig. 3), with the only exception being TEL.

Effect of the MIL type on the enrichment factors of sartans. Extraction conditions: sample volume, 1.2 mL; extraction solvent mass, 30 mg; dispersive solvent, 70 μL acetonitrile
Considering the abovementioned characteristics, [P6,6,6,14+]3[GdCl63−] MIL could be a superior alternative to current MILs as an extractant in DLLME and represents an important advance toward faster and better automation of the extraction technique.
Optimization of the DLLME of sartans using [P6,6,6,14 +]3[GdCl6 3−] MIL extractant
The prepared [P6,6,6,14+]3[GdCl63−] MIL extractant was applied for the extraction and simultaneous determination of the selected sartans in aqueous samples combining DLLME with HPLC/UV analysis. To achieve high sensitivity, the effect of disperser solvent type, MIL mass, and disperser solvent volume, and the effect of acid, base, and salt addition were experimentally investigated.
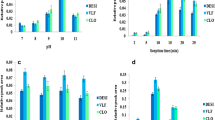
Dispersion of the MIL extractant into very fine droplets through the aqueous sample is a crucial step for effective extraction as it increases the sample/extractant interfacial area and, consequently, enhances the mass transfer yield and kinetics. In order to study the effect of the disperser solvent type, solvents that are miscible with both water and [P6,6,6,14+]3[GdCl63−] MIL extractant, including acetone, acetonitrile, ethanol, and methanol, were studied. EF values for all sartans were highest with both acetone and methanol (Fig. 4); however, acetone has a high UV cut-off point, and the chromatographic peaks obtained with methanol showed better shape and symmetry. For this reason, methanol was chosen as the disperser solvent for the rest of the study.

Effect of dispersive solvent type on the enrichment factor of sartans. Extraction solvent, 30 mg [P6,6,6,14+]3[GdCl63−]. Other conditions as in Fig. 3
The MIL extractant mass was secondly optimized using different amounts of the MIL ranging from 25 to 45 mg. The enrichment for all analytes increased as the mass of the MIL reduced (Fig. 5). A MIL mass of 25 mg provided the best EF for all analytes, but the upper extract layer was relatively thin, complicating its collection and noticeably affecting the precision of the results. On the other hand, a good level of enrichment and reproducibility was obtained with 30 mg of the MIL extractant and, therefore, this amount was employed in the following optimization experiments.

Effect of extraction solvent ([P6,6,6,14+]3[GdCl63−]) mass on the enrichment factor of sartans. Extraction conditions: sample volume, 1.2 mL; dispersive solvent volume, 70 μL MeOH
The effect of the disperser solvent volume was also studied using different volumes of methanol: 30, 50, 70, 100, and 200 μL. For most of the analytes, the EF did not significantly change as the disperser solvent volume varied (ESM Fig. S5). A disperser solvent volume of 70 μL was selected for use in subsequent optimization studies because it is in the middle of a plateau which supports the robustness of the method against small changes in the disperser solvent volume.
The pH of the sample solution usually has a significant effect on the extraction efficiency, particularly when analytes have acidic or basic functional groups. To study the effect of acid or base addition on the extraction efficiency, a 50 μL volume of 0.05 M HCL, 0.05 M NaOH, or water was added to the aqueous sample mixture (1.2 mL) before microextraction, and EFs were compared (ESM Fig. S6). Comparable enrichment was observed with all analytes after DLLME of samples with the acid or water added. The sartan drugs tend to be charged when in solution because of the COOH and amine groups (see Fig. 1 for structures). VAL has two acidic substituents, the COOH group and the tetrazole ring having pKa values of 3.60 and 4.70, respectively [40]. IRB has the acidic tetrazole ring (pKa 3.69) [40] and AZI has one benzimidazole structure with a pKa of 6.1 [41]. TEL has two benzimidazole groups with pKa values of 4.1 and 6.0 and an acidic COOH group with a pKa of 3.5 [40]. As expected based on the pKa values, extraction efficiencies of the sartans should be about the same at pH 7 and alkaline pH conditions. AZI, being neutral in charge, should extract better at alkaline pH than acid pH; the opposite should be true for VALand IRB. Neither trend was clearly observed, probably due to the sufficient remaining hydrophobic structure of the sartans. A marked increase (threefold) in EF was observed with TEL at basic pH compared to acid pH. Apparently, the neutralization of the two benzimidazole groups to reduce the high number of charged substituents in TEL was important. This sartan is considered the most lipophilic with the highest octanol/water partition coefficient [42].
The salting-out effect on the microextraction efficiency was studied by using different KCl concentrations (0, 1, 2.5, 5, and 10% (w/v)) in the aqueous sample solution before doing the DLLME. KCl concentrations above 10% (w/v) caused the precipitation of TEL from the aqueous sample solution and were excluded from this study. Results showed no significant change in the EF of the examined sartans as KCl concentration increased from 0 to 10% (w/v) (ESM Fig. S7), which could be because the maximum possible enrichment was already attained at the previously optimized conditions.
Analytical performance and recovery from real water samples
At the optimized extraction conditions for 1.2-mL sample volumes (30 mg of the [P6,6,6,14+]3[GdCl63−] MIL extractant, 70 μL of the disperser solvent methanol, and the medium rendered basic with 50 μL of 0.05 M NaOH), linearity ranges, LOQs, and RSD values were determined. Enrichment factors for all studied sartans were around 40. The employed chromatographic conditions allowed an adequate resolution of all analyte peaks (Fig. 6). Calibration curves (n = 6) of the studied sartans were constructed by plotting the peak area versus the concentration in μg/mL. Linearity was studied over the ranges 0.25–4.0, 0.125–2.0, 0.125–2.0, and 0.25–8.0 μg/mL for AZI, IRB, TEL, and VAL, respectively. The correlation coefficients were greater than 0.9988, indicating an excellent level of linearity (Table 2). The LOQ values were 80, 30, 40, and 160 ng/mL for AZI, IRB, TEL, and VAL, respectively. RSD values (n = 6) were in the range of 2.48–4.07%. Much lower quantitation limits could be achieved with larger 5-mL sample volumes since enrichment factors of 175, 176, 169, and 103 were obtained with azilsartan medoxomil, irbesartan, valsartan, and telmisartan, respectively.

Chromatograms of (a) blank sample after DLLME using [P6,6,6,14+]3[GdCl63−] MIL; (b) sample of four sartans without extraction; and (c) sample of four sartans after DLLME using [P6,6,6,14+]3[GdCl63−] MIL. Extraction conditions: sample, an aqueous mixture of TEL, IRB, AZI, and VAL at 1 μg/mL, 1 μg/mL, 2 μg/mL, and 2 μg/mL, respectively; sample volume, 1.2 mL; extraction solvent, 30 μg [P6,6,6,14+]3[GdCl63−] MIL; dispersive solvent volume, 70 μL MeOH; base volume, 50 μL 0.05 M NaOH. Chromatographic conditions: described under the “Experimental” section
The relative recovery was studied in river water samples. The studied analytes were below the detectable level in all samples. The water samples were then spiked with standards of the investigated sartans to contain 0.5 μg/mL for TEL and IRB, and 1 μg/mL for AZI and VAL. The chromatograms of the unspiked and spiked river water samples are shown in ESM Fig. S8. The recoveries (± RSD) obtained were 82.5% (± 6.1), 97.45% (± 3.9), 92.23% (± 4.8), and 101.48% (± 3.3) for TEL, IRB, AZI, and VAL, respectively, indicating that the [P6,6,6,14+]3[GdCl63−] MIL-DLLME HPLC/UV method is feasible for the determination of the examined sartans in river water samples.
Comparison with other reported methods
Table 3 compares LOQs, RSDs, extraction times, and sample volumes for SPE [43, 44], solid-phase microextraction (SPME) [45], IL-DLLME [46], and [P6,6,6,14+]3[GdCl63−] MIL-DLLME method for the extraction and determination of sartan drugs in various matrices. The results show that the extraction time in the proposed [P6,6,6,14+]3[GdCl63−] MIL–based DLLME method is very short (less than 2 min), whereas extraction time for SPE, SPME, and IL-DLLME methods ranged from 10 to greater than 30 min. In addition, RSDs for the DLLME are lower than those obtained with SPE and SPME. In contrast to SPE, SPME, and IL-DLLME, the proposed [P6,6,6,14+]3[GdCl63−] MIL-DLLME is potentially more easily automated. LOQ values were lower for the SPE methods as compared to the DLLME studies. It is worthy to note that much lower quantitation limits should be achieved if the proposed [P6,6,6,14+]3[GdCl63−] MIL-DLLME showing high extraction efficiency is coupled with highly sensitive MS/MS detection and/or applied to larger sample volumes.
Conclusion
To the best of our knowledge, this study explores for the first time the potential of the paramagnetic [GdCl63−] anion-based MILs as the microextraction solvent in DLLME. The prepared hydrophobic [P6,6,6,14+]3[GdCl63−] MIL was tested by the microextraction of four sartan drugs and their subsequent HPLC/UV determination. Compared to trihexyl(tetradecyl)phosphonium cation–based MILs containing [FeCl4−] and [MnCl4−2], the [P6,6,6,14+]3[GdCl63−] MIL showed the highest enrichment with all analytes. The prepared Gd(III)-based MIL showed resistance to hydrolysis in aqueous samples and low UV background signal. Additionally, the reasonable viscosity of the proposed MIL promotes analyte partitioning, accelerates phase separation, and facilitates extract handling and transfer into the analytical instrument. Furthermore, the introduced MIL exhibited substantially high magnetic susceptibility allowing for faster retrieval of the extraction solvent with a strong magnet. Therefore, the [P6,6,6,14+]3[GdCl63−] MIL is a promising extraction solvent that could permit faster and better automation of the microextraction technique. Future studies to be considered are use of this MIL and additional gadolinium-based ones for DLLME with potential application to selected pharmaceuticals often found in wastewater such as carbamazepine, atenolol, sulfamethoxazole, and ciprofloxacin.
Data availability
Not applicable
References
Rezaee M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadi F, Berijani S. Determination of organic compounds in water using dispersive liquid-liquid microextraction. J Chromatogr A. 2006;1116:1–9.
Mansour FR, Khairy MA. Pharmaceutical and biomedical applications of dispersive liquid–liquid microextraction. J Chromatogr B Anal Technol Biomed Life Sci. 2017;1061–1062:382–91.
Trujillo-Rodríguez MJ, Rocío-Bautista P, Pino V, Afonso AM. Ionic liquids in dispersive liquid-liquid microextraction. TrAC - Trends Anal Chem. 2013;51:87–106.
Ho TD, Zhang C, Hantao LW, Anderson JL. Ionic liquids in analytical chemistry: fundamentals, advances, and perspectives. Anal Chem. 2014;86:262–85.
Clark KD, Emaus MN, Varona M, Bowers AN, Anderson JL. Ionic liquids: solvents and sorbents in sample preparation. J Sep Sci. 2018;41:209–35.
Rykowska I, Ziemblińska J, Nowak I. Modern approaches in dispersive liquid-liquid microextraction (DLLME) based on ionic liquids: a review. J Mol Liq. 2018;259:319–39.
Mansour FR, Danielson ND. Solvent-terminated dispersive liquid-liquid microextraction: a tutorial. Anal Chim Acta. 2018;1016:1–11.
Zhou Q, Bai H, Xie G, Xiao J. Trace determination of organophosphorus pesticides in environmental samples by temperature-controlled ionic liquid dispersive liquid-phase microextraction. J Chromatogr A. 2008;1188:148–53.
Baghdadi M, Shemirani F. Cold-induced aggregation microextraction: a novel sample preparation technique based on ionic liquids. Anal Chim Acta. 2008;613:56–63.
Del Sesto RE, McCleskey TM, Burrell AK, Baker GA, Thompson JD, Scott BL, et al. Structure and magnetic behavior of transition metal based ionic liquids. Chem Commun. 2002;8:447–9.
Rosatella AA, Siopa F, Frade RFM, Afonso CAM. New low viscous cholinium-based magnetic ionic liquids. New J Chem. 2016;40:3124–9.
Santos E, Albo J, Rosatella A, Afonso CAM, Irabien Á. Synthesis and characterization of magnetic ionic liquids (MILs) for CO2 separation. J Chem Technol Biotechnol. 2014;89:866–71.
Bwambok DK, Thuo MM, Atkinson MBJ, Mirica KA, Shapiro ND, Whitesides GM. Paramagnetic ionic liquids for measurements of density using magnetic levitation. Anal Chem. 2013;85:8442–7.
Joseph A, Zyła G, Thomas VI, Nair PR, Padmanabhan AS, Mathew S. Paramagnetic ionic liquids for advanced applications: a review. J Mol Liq. 2016;218:319–31.
Capriotti AL, Cavaliere C, La Barbera G, Montone CM, Piovesana S, Laganà A. Recent applications of magnetic solid-phase extraction for sample preparation. Chromatographia. 2019;82:1251–74.
An J, Rahn KL, Anderson JL. Headspace single drop microextraction versus dispersive liquid-liquid microextraction using magnetic ionic liquid extraction solvents. Talanta. 2017;167:268–78.
Trujillo-Rodríguez MJ, Anderson JL. In situ formation of hydrophobic magnetic ionic liquids for dispersive liquid-liquid microextraction. J Chromatogr A. 2019;1588:8–16.
Trujillo-Rodríguez MJ, Anderson JL. In situ generation of hydrophobic magnetic ionic liquids in stir bar dispersive liquid-liquid microextraction coupled with headspace gas chromatography. Talanta. 2019;196:420–8.
Clark KD, Nacham O, Purslow JA, Pierson SA, Anderson JL. Magnetic ionic liquids in analytical chemistry: a review. Anal Chim Acta. 2016;934:9–21.
Trujillo-Rodríguez MJ, Nacham O, Clark KD, Pino V, Anderson JL, Ayala JH, et al. Magnetic ionic liquids as non-conventional extraction solvents for the determination of polycyclic aromatic hydrocarbons. Anal Chim Acta. 2016;934:106–13.
Bowers AN, Trujillo-Rodríguez MJ, Farooq MQ, Anderson JL. Extraction of DNA with magnetic ionic liquids using in situ dispersive liquid–liquid microextraction. Anal Bioanal Chem. 2019;411:7375–85.
Emaus MN, Zhu C, Anderson JL. Selective hybridization and capture of KRAS DNA from plasma and blood using ion-tagged oligonucleotide probes coupled to magnetic ionic liquids. Anal Chim Acta. 2020;1094:1–10.
Chisvert A, Benedé JL, Anderson JL, Pierson SA, Salvador A. Introducing a new and rapid microextraction approach based on magnetic ionic liquids: stir bar dispersive liquid microextraction. Anal Chim Acta. 2017;983:130–40.
Clark KD, Purslow JA, Pierson SA, Nacham O, Anderson JL. Rapid preconcentration of viable bacteria using magnetic ionic liquids for PCR amplification and culture-based diagnostics. Anal Bioanal Chem. 2017;409:4983–91.
Xie ZL, Taubert A. Thermomorphic behavior of the ionic liquids [C4mim][FeCl 4] and [C12mim][FeCl4]. ChemPhysChem. 2011;12:364–8.
Stefánsson A. Iron(III) hydrolysis and solubility at 25°C. Environ Sci Technol. 2007;41:6117–23.
Yu H, Merib J, Anderson JL. Faster dispersive liquid-liquid microextraction methods using magnetic ionic liquids as solvents. J Chromatogr A. 2016;1463:11–9.
Merib J, Spudeit DA, Corazza G, Carasek E, Anderson JL. Magnetic ionic liquids as versatile extraction phases for the rapid determination of estrogens in human urine by dispersive liquid-liquid microextraction coupled with high-performance liquid chromatography-diode array detection. Anal Bioanal Chem. 2018;410:4689–99.
Feng X, Xu X, Liu Z, Xue S, Zhang L. Novel functionalized magnetic ionic liquid green separation technology coupled with high performance liquid chromatography: a rapid approach for determination of estrogens in milk and cosmetics. Talanta. 2020;209:120542.
Chatzimitakos TG, Pierson SA, Anderson JL, Stalikas CD. Enhanced magnetic ionic liquid-based dispersive liquid-liquid microextraction of triazines and sulfonamides through a one-pot, pH-modulated approach. J Chromatogr A. 2018;1571:47–54.
Flack JM, Adekola B. Blood pressure and the new ACC/AHA hypertension guidelines. Trends Cardiovasc Med. 2020;30:160–4.
Zhang P, Zhu L, Cai J, Lei F, Qin J, Xie J, et al. Association of inpatient use of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res. 2020. https://doi.org/10.1161/CIRCRESAHA.120.317134.
Babarahimi V, Talebpour Z, Haghighi F, Adib N, Vahidi H. Validated determination of losartan and valsartan in human plasma by stir bar sorptive extraction based on acrylate monolithic polymer, liquid chromatographic analysis and experimental design methodology. J Pharm Biomed Anal. 2018;153:204–13.
Moeller T, Moss FAJ. Observations on the rare earths. LX. The ultraviolet absorption spectrum of the gadolinium ion in aqueous solutions. J Am Chem Soc. 1951;73:3149–51.
Deng N, Li M, Zhao L, Lu C, de Rooy SL, Warner IM. Highly efficient extraction of phenolic compounds by use of magnetic room temperature ionic liquids for environmental remediation. J Hazard Mater. 2011;192:1350–7.
Chatzimitakos T, Binellas C, Maidatsi K, Stalikas C. Magnetic ionic liquid in stirring-assisted drop-breakup microextraction: proof-of-concept extraction of phenolic endocrine disrupters and acidic pharmaceuticals. Anal Chim Acta. 2016;910:53–9.
Nordstrom DK, Plummer LN, Langmuir D, Busenberg E, May HM, Jones BF, Parkhurst DL. Chapter 31. Revised chemical equilibrium data for major water—mineral reactions and their limitations. In: Melchoir DC, Bassett RL, editors. Chemical Modeling of Aqueous Systems II. ACS Symposium Series, vol. 416. American Chemical Society; 1990. pp. 398–413.
Sintra TE, Nasirpour M, Siopa F, Rosatella AA, Gonçalves F, Coutinho JAP, et al. Ecotoxicological evaluation of magnetic ionic liquids. Ecotoxicol Environ Saf. 2017;143:315–21.
Frade RFM, Simeonov S, Rosatella AA, Siopa F, Afonso CAM. Toxicological evaluation of magnetic ionic liquids in human cell lines. Chemosphere. 2013;92:100–5.
Tosco P, Rolando B, Fruttero R, Henchoz Y, Martel S, Carrupt PA, et al. Physicochemical profiling of sartans: a detailed study of ionization constants and distribution coefficients. Helv Chim Acta. 2008;91:468–82.
Jassem NA, Rajab NA. Formulation and in vitro evaluation of azilsartan medoxomil nanosuspension. Int J Pharm Pharm Sci. 2017;9:110–9.
Wienen W, Entzeroth M, Van Meel JCA, Stangier J, Busch U, Ebner T, et al. A review on telmisartan: a novel, long-acting angiotensin II-receptor antagonist. Cardiovasc Drug Rev. 2000;18:127–54.
Gurke R, Rossmann J, Schubert S, Sandmann T, Rößler M, Oertel R, et al. Development of a SPE-HPLC-MS/MS method for the determination of most prescribed pharmaceuticals and related metabolites in urban sewage samples. J Chromatogr B Anal Technol Biomed Life Sci. 2015;990:23–30.
Tarcomnicu I, Van Nuijs ALN, Simons W, Bervoets L, Blust R, Jorens PG, et al. Simultaneous determination of 15 top-prescribed pharmaceuticals and their metabolites in influent wastewater by reversed-phase liquid chromatography coupled to tandem mass spectrometry. Talanta. 2011;83:795–803.
Unceta N, Sampedro MC, Bakar NKA, Gómez-Caballero A, Goicolea MA, Barrio RJ. Multi-residue analysis of pharmaceutical compounds in wastewaters by dual solid-phase microextraction coupled to liquid chromatography electrospray ionization ion trap mass spectrometry. J Chromatogr A. 2010;1217:3392–9.
Rao RN, Raju SS, Vali RM. Ionic-liquid based dispersive liquid-liquid microextraction followed by high performance liquid chromatographic determination of anti-hypertensives in rat serum. J Chromatogr B Anal Technol Biomed Life Sci. 2013;931:174–80.
Code availability
Not applicable
Funding
The support of one of us (MAA) through a Graduate Scholarship for Professionals to the USA by the Egyptian Cultural and Educational Bureau and the Central Department for Missions is gratefully appreciated.
Author information
Authors and Affiliations
Contributions
The research idea originated with MA Adelaziz and FR Mansour who both also wrote the original manuscript draft. ND Danielson advised MA Abdelaziz (who did most of the experimental work at Miami University) during the research project and revised the manuscript draft into its present form.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 687 kb)
Rights and permissions
About this article

Cite this article
Abdelaziz, M.A., Mansour, F.R. & Danielson, N.D. A gadolinium-based magnetic ionic liquid for dispersive liquid–liquid microextraction. Anal Bioanal Chem 413, 205–214 (2021). https://doi.org/10.1007/s00216-020-02992-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-020-02992-z