
Abstract
The mechanism of the Diels–Alder reactions between perfluorobicyclo[2.2.0]hex-1(4)-ene (1a) and bicyclo[2.2.0]hex-1(4)-ene (1b) with benzene (2a) and naphthalene (2b) has been studied within the density functional theory at the MPWB1K/6-311G(d,p) level. The bonding pattern in these reactions is analyzed in the topology of the electron localization function within the bonding evolution theory perspective. The bonding electron density changes along the reaction paths reveal that the C–C bond formation takes place through a synchronous and non-concerted one-step mechanism and proceeds with a moderate activation energy. The reactivity order with 1a is 2a–2b. The reactions begin by the rupture of the double bond in the strained 1a-b molecules, and then two pseudoradical centers at the 1a-b fragments are created. Finally, at the same time, two new single bonds are formed in the cycloaddition products. The TSs proceed with high global electron density transfer providing a polar character at these reactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The extremely low, potential reactivity of the parent benzene in the addition processes is generally known as the logical consequence of the tendency for retention of ideal-aromatic nature of their six-membered ring. However, the arene systems with some disturbances in their aromatic nature show some tendency to participate in cycloaddition reactions with suitably strong reactive partners. This tendency correlates with the disturbance of the aromatic nature of the molecule.
So, many examples of Diels–Alder (DA) reactions involving furan as 1,3-diene analogue have been described [1,2,3,4,5,6]. In the case of DA cycloadditions of furan with electrophilic activated ethenes, these type reactions may proceed under relatively mild conditions. For example, DA reaction of furan with (E)-3,3,3-trichloro-1-nitroprop-1-ene [2] (global electrophilicity is equal to 3.27 eV) [7] proceeds easily even at the room temperature (Scheme 1).

DA reaction of furan with (E)-3,3,3-trichloro-1-nitroprop-1-ene [2]
Carbocyclic aromatic systems are less reactive. For example, anthracene reacts with the mentioned above (E)-3,3,3-trichloro-1-nitroprop-1-ene at 180 °C in 19 h (Scheme 2) [8].

DA reaction between anthracene and (E)-3,3,3-trichloro-1-nitroprop-1-ene [8]
Less, than in the case of anthracene disordered aromatic system, observed in the case of naphthalene, require more dramatic conditions. For example, naphthalene reacts with the N-phenyl-maleimide only under high-pressure conditions. Despite high pressure as well as higher temperature, the conversion of addends is realized in 80 h (Scheme 3) [9].

DA reaction naphthalene with N-phenyl-maleimide [9]
For the contrast, with dienophiles mentioned above under conditions described, the benzene does not react via DA cycloaddition scheme. Some examples DA reactions of benzene are known, but under photochemical conditions [10, 11]. On the other hand, some strong electrophilic and extremely active alkenes such as 2-nitroprop-1-ene can react with benzene. There are, however, not DA cycloadditions, but SEAr reaction. Thermal allowed DA reactions involving benzene ring are extremely difficult and with satisfactory yield are known regarding for only one example. This interesting process is based on the perfluorobicyclo[2.2.0]hex-1(4)-ene and is determined by presence of fluorine atoms with highly strained of >C=C< moiety (Scheme 4) [12].

DA reaction of perfluorobicyclo[2.2.0]hex-1(4)-ene with benzene [12]
On the other hand, Houk and coworkers considered on the basis of quantum chemical calculations the stepwise, biradical mechanism of the hypothetical cycloaddition of benzene with allene. In this case, the biradical exists on the potential energy hypersurface (PES) as the common intermediate for [2 + 2] and [4 + 2] processes. Unfortunately, this study was not supported by experimental study due to tendency of allene systems for the rapid dimerization [13]. Recently, Bickelhaupt and coworkers studied the effect of alkali cations catalyzing aromatic DA reactions [14]. The catalytic effect in the DA reaction barrier decreases in the order M+ = none > Cs > Rb > K > Li. The authors conclude that augmented reactivity of the metal-catalyzed DA reactions is due to reduction of Pauli repulsions between benzene cation and acetylenes within the context of the activation strain model (ASM) [15]. More recently, it was reported, within the so-called molecular electron density theory (MEDT) scheme, a theoretical study of the lithium cation-catalyzed benzene DA reactions, which showed that the coordination of Li+ to the complex of benzene with crown ether improved the electrophilic character of benzene and the feasibility of the DA reaction [16]. It is important to mention that the polar Diels–Alder (P-DA), as will see later, takes relevance when the electrophilic/nucleophilic interactions occur at the TS controlling the feasibility of the DA reactions [17]. Recently, other theoretical study using MEDT [18] scheme of a series of DA reactions of ethylene with aza aromatic compounds [19] finds an enhancement of the reactivity of the latter species mainly due to the loss of their aromatic character that destabilizes the reagents.
The aim of this work is to enhance the understand and the description of bonding breaking/forming involved on the mechanism of the DA reactions, in particular in cycloadditions involving very stable reagents as aromatic species; hence, we present a complete study of the DA reaction given by a strong electrophile as perfluorobicyclo[2.2.0]hex-1(4)-ene 1a with benzene 2a and naphthalene 2b and, on the other hand, the corresponding DA reaction mechanism given by the marginal electrophile as bicyclo[2.2.0]hex-1(4)-ene 1b with benzene 2a and naphthalene 2b. To achieve that will be used known theoretical tools defined within the context of the density functional theory (DFT) [20, 21]. A qualitative bonding evolution theory (BET) [22] analysis along these DA reactions is performed to characterize these bonding changes.
2 Computational details
All calculations associated with the P-DA reactions were performed using the GAUSSIAN 16 package [23] in the Prometheus computer cluster of the CYFRONET regional computer center in Cracow. The geometries of all reactants, transition state structures (TSs) and products of the reactions were fully optimized using the MPWB1K [24] functional together with the 6-311G(d,p) basis set. Stationary points were characterized by frequency calculations. All reactants and products had positive Hessian eigenvalues. All TSs had only one negative eigenvalue in their diagonalized Hessian matrices, and their associated eigenvectors were confirmed to correspond to the motion along the reaction coordinate under consideration. TSs were located using the (QST2) algorithm. Intrinsic reaction coordinate (IRC) calculations [25] were performed in all cases to verify that the located TSs are connected to the corresponding minimum stationary points associated with reactants, products and intermediates. The solvent effects of benzene were simulated using a relatively simple self-consistent reaction field (SCRF) [26,27,28] based on the polarizable continuum model (PCM) of Tomasi’s group [29, 30]. The stability of the wave function for all reagents, products and TSs, and the points of the IRC for the most favorable pathway has been checked. The values of energies, enthalpies, entropies and Gibbs free energies were calculated for temperature 393 K.
Global reactivity indices at the ground states (GS) of the reagents and products, within of the conceptual density functional theory (CDFT) [31, 32], were calculated at the B3LYP/6-31G(d) level. The global electron density transfer (GEDT) [33] values were calculated as the sum of the natural atomic charges (q), obtained by a natural population analysis (NPA) [34, 35], by the equation: GEDT (f) = \(\sum_{\mathrm{q}\in \mathrm{f}}\mathrm{q}\), for all atoms belonging to each f fragment (i.e., electrophile and nucleophile) of the TSs at the same level of theory.
Global electronic properties of the reactants were estimated according to the equations recommended in references [31, 32]. In particular, the electronic chemical potentials (μ) and chemical hardness (η) were evaluated in terms of one-electron energies of FMO (EHOMO and ELUMO) using the following equations: μ≈(EHOMO + ELUMO)/2, η ≈ ELUMO − EHOMO. Next, the values of μ and η were used for the calculation of global electrophilicity (ω) according to the formula: ω = μ2/2η [36]. Subsequently, global nucleophilicity (N) [37] can be expressed in terms of the equation: N = EHOMO − EHOMO(tetracyanoethylene), where tetracyanoethylene is used as reference. Local reactivity indices, electrophilic \(P_{k}^{ + }\) and nucleophilic \(P_{k}^{ - }\) Parr functions [38] were obtained from the changes of atomic spin density (ASD) of the reagents to identify electrophilic and nucleophilic centers in the molecules.
Electron localization function (ELF) [39] studies were performed with the TopMod package [40] considering the standard cubical grid of step size of 0.1 Bohr. The bonding changes along corresponding reactions were analyzed, according to the bonding evolution theory (BET) [22], by performing the topological analysis of the ELF for 198 nuclear configurations along the IRC path. The ELF molecular geometries and basin attractor positions were visualized using the GaussView program [41]. ELF localization domains were represented by using the ParaView software at an isovalue of 0.75 a.u [42, 43]. A similar approach has been successfully used to explain the mechanism of different types of reactions [44,45,46,47,48,49,50].
3 Results and discussion
The present study is organized as follows: In Sect. 3.1, an analysis of the electronic structure and ELF topological analysis of reagents is performed. Section 3.2 contains an analysis of the CDFT reactivity indices of the reagents involved in the DA reactions of perfluorobicyclo[2.2.0]hex-1(4)-ene 1a, bicyclo[2.2.0]hex-1(4)-ene 1b and 2a-b. In Sect. 3.3, the reaction profiles associated with the DA reactions of 1a-b with 2a-b are analyzed. Finally, in Sect. 3.4 a BET study of the DA reactions between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and 2a-b is performed.
3.1 ELF topological analysis of reagents
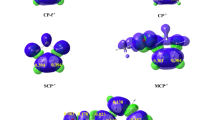
Quantum chemical analysis of Becke and Edgecombe’s ELF [38] allows the electronic structure to be determined by a direct combination of electronic density distribution and chemical structure. Therefore, in order to characterize the electronic structures of perfluorobicyclo[2.2.0]hex-1(4)-ene 1a, bicyclo[2.2.0]hex-1(4)-ene 1b, benzene 2a and naphthalene 2b, a topological analysis of the ELF of reagents was first performed. Lewis-like structures, ELF localization domains and basin attractor positions, together with the most significant valence basin populations, are shown in Fig. 1 and Fig. 2.

MPWB1K/6-311G(d,p) localization domains (isovalue = 0.75) and basin attractor positions together with the most significant valence basin populations and proposed Lewis-like structures of perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and bicyclo[2.2.0]hex-1(4)-ene 1b

MPWB1K/6-311G(d,p) localization domains (isovalue = 0.75) and basin attractor positions together with the most significant valence basin populations and proposed Lewis-like structures of benzene 2a and naphthalene 2b
ELF topological analysis of perfluorobicyclo[2.2.0]hex-1(4)-ene 1a, in the most important region, shows the presence of two V(C1,C2) and V’(C1,C2) disynaptic basins integrating 1.64 e and 1.55 e, respectively. In the bicyclo[2.2.0]hex-1(4)-ene 1b, we also find, in the most important region, V(C1,C2) and V’(C1,C2) disynaptic basins integrating a total population of 3.27 e. These disynaptic basins are associated with C1-C2 double bonds in 1a and 1b molecules.
On the other hand, an ELF topological analysis of benzene 2a shows the presence of six V(C3,C4), V(C4,C5), V(C5,C6), V(C6,C7), V(C7,C8) and V(C8,C3) disynaptic basins integrating in all cases 2.75–2.79 e (Fig. 2). These disynaptic basins are associated with partial double bonds in benzene 2a. In turn, in the case of naphthalene 2b, we find three V(C4,C5), V(C6,C7) and V(C8,C3) disynaptic basins integrating 3.03 e, 3.03 and 2.63 e, respectively. These disynaptic basins are related to partial double bond, while the other three V(C3,C4), V(C5,C6) and V(C7,C8) disynaptic basins are associated with single bonds.
3.2 Analysis of the global and local CDFT Reactivity Indices at the GS of the Reagents
In order to understand the participation of these reagents in the DA reactions, an analysis of the global indices at the ground states (GS) of these species was performed, defined within the context of the CDFT [31, 32] and calculated at the B3LYP/6-31G(d) level of theory. In Table 1 are collected electronic chemical potential μ, chemical hardness η, global electrophilicity ω, and global nucleophilicity N values of the GS of the reagents. The order is given in decreasing electrophilicity values.
The electronic chemical potential [21] μ of 2a is − 3.30 and that of 2b is − 3.37 eV, which are higher than that of 1a, μ = − 5.38 eV, indicating that GEDT [33] in these cycloaddition reactions will proceed from benzene, naphthalene and butadiene toward 1a. Note that 1b presents the highest value of μ (− 2.46 eV).
It can be seen that the electrophilicity [36] ω and nucleophilicity [37] N indices of 1a are 2.08 and 0.26 eV, respectively, classifying it as a strong electrophile and marginal nucleophile within the electrophilicity [50] and nucleophilicity [51] scales. This behavior predicts a P-DA reaction with strong and moderate nucleophiles. Instead, 1b presents ω = 0.41 eV and N = 2.95 eV, being a marginal electrophile and in the borderline of moderate nucleophile. Clearly, the presence of four fluorine centers in 1a makes it more electrophilic than 1b.
On the other hand, the electrophilicity ω and nucleophilicity N indices of benzene 2a are 0.80 eV and 2.42 eV, being classified in the borderline of moderate electrophile and moderate nucleophile. The reactivity indices of two fused benzene rings to give naphthalene 2b, strongly increases both the electrophilicity and nucleophilicity, ω = 1.28 eV and * = 3.33 eV, predicting feasible participation in P-DA reactions as a nucleophilic species. Additionally, note that 1a and 1b are strained species, while 2a and 2b are recognized aromatic species.
It is well known that in polar processes, the nucleophilic species transfer electron density toward electrophilic ones. The electron density changes on the electrophile and nucleophile can be sensed by the nucleophilic \(P_{k}^{ - }\) and electrophilic \(P_{k}^{ + }\) Parr functions [38], which are good predictors for local reactivity in polar processes. The electrophilic \(P_{k}^{ + }\) and the nucleophilic \(P_{k}^{ - }\) and Parr functions for the GS of the reagents are gathered in Table 2 and Fig. 3.

3D representations of the ASD of the radical anion 1a·− and 1b·− and the radical cation 2a·+ and 2b·+, together with the electrophilic \({P}_{k}^{+}\) Parr functions of 1a-b and the nucleophilic \({P}_{k}^{-}\) Parr functions of 2a-b
An analysis of the electrophilic \(P_{k}^{ + }\) Parr functions of 1a indicates that C1 and C2 are the most electrophilic center in the molecule, \(P_{k}^{ + }\) = 0.49, while all CF centers show negligible electrophilic and nucleophilic character. The electrophilic \(P_{k}^{ + }\) and nucleophilic \(P_{k}^{ - }\) Parr functions of 1b, at C1 and C2, \(P_{k}^{ + }\) = 0.49 and \(P_{k}^{ - }\) = 0.44, show electrophilic and nucleophilic behavior predicting that this reagent will react with a strong nucleophiles or electrophiles via a polar reaction. The local electrophilic and nucleophilic character of CH is insignificant.
The nucleophilic \(P_{k}^{ - }\) Parr functions of 2a at C3 and C6, \(P_{k}^{ - }\) = 0.48, show the strong nucleophilic character at these centers. Note that electrophilic \(P_{k}^{ + }\) values at the same centers are very similar; however, the nucleophilic character of 2a, shown in Table 1 (N = 2.42 eV), predicts that its reactive centers will have nucleophilic character in polar reactions. It is worth to mention that centers C4, C5, C7 and C8 (see Table 2) develop unimportant nucleophilic/electrophilic character.
For 2b, nucleophilic \(P_{k}^{ - }\) Parr functions at C4 and C7, \(P_{k}^{ - }\) = 0.26, presenting the nucleophilic character in agreement with the high value of nucleophilicity, N = 3.33 eV. Very similar electrophilic \(P_{k}^{ + }\) values at C4 and C7, \(P_{k}^{ + }\) = 0.25, were obtained. Their local electrophilic character is lower than 2a due to the presence of more centers in the two fused benzene rings. The electrophilic/nucleophilic Parr functions at the other centers of 2b are negligible.
In summary, along polar reactions, the most favorable process will be associated with the two centers interaction between the most electrophilic center of 1a and the most nucleophilic centers of 2a–2b. Note that 1a and 1b are strained reagents; therefore, it is expected that they react more favorably releasing that strain. On the other hand, 2a and 2b are aromatic species, so it is expected they react with their partners minimizing that loss.
3.3 Study of the DA reaction between 1a-b and 2a-b
The P-DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and 2a-b takes place through a one-step mechanism (see Scheme 5). The MPWB1K(PCM)/6-311G(d,p) relative enthalpies, Gibbs free energy and entropies of the stationary points in the DA reactions of perfluorobicyclo[2.2.0]hex-1(4)-ene 1a with 2a-b are given in Table 3 and in Supplementary Information (Tables S2 and S4). In turn, total electronic energies in benzene and gas phase for DA reactions 1a-b with 2a-b are given in Supplementary Information (Tables S1 and S3).

DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and 2a-b. MPWB1K/6-31G(d,p) relative energies to the separated reagents, in benzene, in orange, and in gas phase, in blue, are given in kcal mol−1
The reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and benzene 2a begins with formation of molecular complex MC1, which is slightly stabilized by only 1.6 kcal mol−1 with respect to the separated reagents. From MC1, the activation energy associated with TS1 is 18.0 kcal mol−1, the reaction being exothermic by 25.8 kcal mol−1. In turn, the relative energy of the TS3 with respect to separated reagents is 16.6 kcal mol−1 for DA reaction 1a with naphthalene 2b. It is worth to mention that the main factor responsible for higher activation energies of TS1 and TS3 and less favorable values for the formation of 3a and 4a is associated with the loss of aromatic character along the path evading DA reactions with aromatic species. DA reactions of 1a with 2a-b in the gas phase proceed also according to the one-step mechanism. The activation energy for TS1 (18.7 kcal mol−1) is slightly higher compared to the reaction in benzene environment. In turn, in the case of reactions 1a with 2b, we observe lower activation energy, 16.3 kcal mol−1 (Scheme 5).
Values of relative enthalpies, Gibbs free energy and entropies of stationary points involved in the DA reaction of 1a with 2a-b are summarized in Table 3. Thus, the activation Gibbs free energy associated with the DA reaction 1a with 2a, via TS1, is 32.3 kcal mol−1, the formation of 3a being exergonic by 9.1 kcal mol−1. In the case of DA reaction 1a with naphthalene 2b, the Gibbs free energy is 2.5 kcal mol−1 less than the reaction 1a with 2a. Note that relative energies (Scheme 5) present the same trend that relative enthalpy and Gibbs free energy values (Table 3).
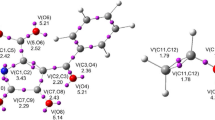
Optimized TSs involved in the DA reactions between 1 and 2a–b, including some selected distances, are given in Fig. 4. At TS1, the distance between the C1 and C3, and the C2 and C6 interacting atoms are 2.206 Å and 2.203 Å, respectively. These parameters suggest a synchronous bond formation process in which the C1-C3 and C2-C6 bonds are created at the same time. The same dependence is observed for the formation of C1-C4 and C2-C7 bonds in the transition state TS3 in DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and 2b (Fig. 4). Formation of C1-C4 and C2-C7 bonds in TS3, beginning at the same value for both bonds, and are 2.287 Å. The latter shows insignificant differences between the two C–C bonding formation process. Hence, it is worth to mention that all these distances, higher than 2.0 Å, indicate the C–C bond formation have not begun at these TSs [33].

MPWB1K(PCM)/6-311G(d,p) optimized geometries for the TSs associated with the DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and 2a-b. Distances are given in angstroms Å
The DA reactions of 1a and 2a–b were analyzed by computing the GEDT [33] (Fig. 4). Thus, the GEDT, for the transition state of reaction 1a with benzene 2a–b, is for TS1 0.37e and for TS3 0.34e, respectively. GEDT values indicate that these reactions show strong polar character of cycloaddition between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and 2a–b. The DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and benzene 2a shows the most polar character. The reaction paths associated with the DA reactions present high values of GEDT; there is not a great effect in the activation energy sue to the aromatic character of the dienes. However, while GEDT is higher, more energetic is the corresponding TS.
The MPWB1K(PCM)/6-311G(d,p) calculations for DA reaction between bicyclo[2.2.0]hex-1(4)-ene 1b and 2a-b indicate that these reactions take place according to one-step mechanism (Scheme 6). The relative enthalpies, Gibbs free energy and entropies of the stationary points in the DA reactions of 1b with 2a-b are given in Table 4.

DA reaction between bicyclo[2.2.0]hex-1(4)-ene 1b and 2a-b. MPWB1K/6-31G(d,p) relative energies to the separated reagents, in benzene, in orange, and in gas phase, in blue, are given in kcal mol−1
The activation energy associated with the DA reaction of bicyclo[2.2.0]hex-1(4)-ene 1b and benzene 2a, begins with the creation of molecular complex MC2. Consequently, the creation of a MC2 entails the drop of energy by 0.5 kcal mol−1. Thereafter, the molecular complex is recast to transition state (TS2), which is associated with an increase in the energy of activation over 23.2 kcal mol−1 (Scheme 6). The formation of the final cycloadduct 3b is exothermic by 29.0 kcal mol−1. In the case of reaction between 1 and 2b, we also observed the similar course of reactions, but we notice a slightly lower energy of activation and for TS4 is 17.7 kcal mol−1. In the case of DA reaction between 1b and 2a-b, we observe lower energy of activation, for TS2 23.0 kcal mol−1 and for TS4 17.3 kcal mol−1. Similar behavior is found in benzene.
The relative enthalpies, entropies and Gibbs free energies of the stationary points involved in the DA reaction bicyclo[2.2.0]hex-1(4)-ene 1b with 2a-b are displayed in Table 4. Thus, the Gibbs free energy associated with the bicyclo[2.2.0]hex-1(4)-ene 1b and benzene 2a, via TS2 is 37.0 kcal mol−1, the formation of the cycloadduct 3b being exergonic by 13.7 kcal mol−1. In the case of reaction 1b with 2b, we observe a lower Gibbs free energy, for TS4 31.5 kcal mol−1 (Table 4). Again, it is important to note that relative energies (Scheme 6) present the same trend that relative enthalpy and Gibbs free energy values (Table 4).
The geometries of TS2 and TS4 are given in Fig. 5. At TS2, the lengths of the two forming bonds are 2.245 Å for C1–C3 and C2–C6. These parameters suggest a synchronous bond formation process in which the C1–C3 and C2–C6 bonds are created at the same time. In the case of transition state TS4, we also observed synchronous bond formation.

MPWB1K(PCM)/6-311G(d,p) optimized geometries for the TSs associated with the DA reaction between bicyclo[2.2.0]hex-1(4)-ene 1b and 2a-b. Distances are given in angstroms Å
Finally, in order to evaluate the polar nature of the DA reaction bicyclo[2.2.0]hex-1(4)-ene 1b with 2a-c, the GEDT [33] at the TSs was analyzed (Fig. 5). The GEDT values computed at the TSs are: 0.05e at TS2 and 0.01e at TS4. These very low values emphasize the nonpolar character of this DA reactions. The nonpolar character of these processes is a result of the marginal electrophilic character of 1b.
3.4 BET analysis of the DA reactions
3.4.1 BET study of the P-DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and benzene 2a
In order to understand the bonding changes along these polar DA reactions, a BET study of the DA reaction perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and benzene 2a was carried out. The molecular mechanism represented by Lewis-like structures result from the ELF topology is shown in Scheme 7. Populations of the most significant valence basins of selected structures of the IRC are collected in Table 5, together with other important parameters.

Simplified representation of the molecular mechanism of the P-DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and benzene 2a by Lewis-like structures arising from the topological analysis of the ELF along the reaction path
The bonding changes along this P-DA reaction are characterized by six phases. The topological features of the ELF of the corresponding molecular complex, MC1, which is the first point of the IRC, are very similar to those of the isolated reagents. Along Phase I, a slight depopulation of the C1–C2 bonding region of the 1a fragment by 0.04 e [V(C1,C2)] is observed, while the populations around the C3–C4, C5–C6, C6–C7, C7–C8 and C8–C3 bonding regions at the benzene moiety remain almost without changes (see Table 5). Phase II begins at the structure S1F. Along this phase, the two V(C1,C2) and V’(C1,C2) disynaptic basins present in Phase I have merged into one V(C1,C2) disynaptic basins, integrating 3.21 e. Phase III starts at the structure S2F. This phase begins with the creation of two C1 and C2 pseudoradical centers [52] at the perfluorobicyclo[2.2.0]hex-1(4)-ene 1a moiety, integrating 0.34 e each one (see V(C1) and V(C2) in Table 5, Scheme 7 and Fig. 6) at a length C–C of 2.26 Å. The formation of these pseudoradical centers is mainly prompted by the depopulation of the C1–C2 bonding region of the 1a fragment by 0.50 e and probably by depopulation of some bonding regions of benzene (see V(C1,C2) and V(C3,C4) and V(C5,C6) in Table 5). At this phase, the TS (TS1) of the reaction is found. The two V(C1) and V(C2) monosynaptic basins have reached populations by ca 0.57 e. Note that the GEDT at TS is the highest, 0.37 e, along this reaction. Along Phase IV, which begins at the structure S3F, two new C3 and C6 pseudoradical centers at the benzene 2a moiety integrating a population of 0.09 e, each one, are created (see V(C3) and V(C6) in Table 5, Scheme 7 and Fig. 6). The electron density of these pseudoradical centers is a consequence to the depopulation of the C3–C4 and C5–C6 bonding regions by ca 0.08 e [V(C3,C4) and V(C5,C6)]. Along this phase, it may be seen that the population associated with the C1 and C2 pseudoradical centers [52] increase to 0.62 e [see V(C1) and V(C2) in Table 5). Phase V starts at S4F. The most relevant topological change is here evidenced: the two pairs of C1, C2 and C3, C6 pseudoradical centers merged into two new C1–C3 and C2–C6 bonding regions [33] with an initial population of 1.10 e each one (see V(C1,C3) and V(C2,C6) in Table 5, Scheme 7 and Fig. 6). These electron density changes indicate that the formation of the two new C–C single bonds begins at a distance of 2.08 Å by sharing the non-bonding electron densities of the two pairs of C1 and C2 toward C3 and C6 centers. Along this phase while the population associated with C3–C4, C5–C6, C6–C7 and C8–C3 bonding regions slightly decreases, those associated with the C4–C5 and C7–C8 bonding regions slightly increase by ca 0.07 e. Phase VI, begins at S5F and ends at the cycloadduct 3a. At the beginning of this phase, while the V(C1,C3) and V(C2,C6) disynaptic basins have reached a population by ca 1.20 e, the V(C4,C5) and V(C7,C8) disynaptic basins present at S4F have been split into two new pairs of disynaptic basins, V(C4,C5) and V’(C4,C5), and V(C7,C8) and V’(C7,C8), respectively, integrating a total population of 3.18 and 3.21 e. Finally, at bicyclic compound 3a, the electron population is relaxed: the C1–C3 and C2–C6 bonding regions integrate 1.90 e and 2.09 e. The C4–C5 and C7–C8 bonding regions reach incremented and symmetric populations of 3.45 e, acquiring the expected population for double bonds, while that associated with the C1––C2, C3–C4(C8), C6–C5(C7) bonding regions reaches values around 2.0 e, characterizing the expected single bonds at 3a.

Attractor positions of the ELF valence basins of the IRC structures S2F, S3F and S4F involved in the C–C single bond formation along the DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and benzene 2a. The electron populations, in average number of electrons, is given in e
Interesting conclusions can be obtained from BET analysis of the P-DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and benzene 2a. (i) This P-DA reaction takes place along six phases. The maximum of GEDT proceeds along Phase III (0.37 e). This very high GEDT is a consequence of the strong electrophilic character perfluorobicyclo[2.2.0]hex-1(4)-ene 1a; (ii) TS1 shows the highest energy of the path, 19.6 kcal·mol−1. The activation energy of this reaction is mainly associated with the continuous depopulations of the C1–C2, C3– C4[C8], C6– C5[C7] bonding regions, demanded, at the first time, for the creation of the two C1 and C2 pseudoradical centers at the perfluorobicyclo[2.2.0]hex-1(4)-ene moiety 1a. (iii) The formation of the two C1–C3 and C2–C6 single bonds takes place simultaneously at a C–C distance of 2.08 Å, by sharing the non-bonding electron densities of the two pairs of C1 and C2 toward C3 and C6 pseudoradical centers in a 81:19 ratio. (iv) The C–C bonding formation process is the P-DA reaction that is completely synchronic. Finally, (v) the P-DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and benzene 2a takes place through a non-concerted one-step mechanism.
3.4.2 BET study of the P-DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and naphthalene 2b
A BET analysis of the DA reaction perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and naphthalene 2b was performed. Lewis-like structures resulting from the ELF topology representing the mechanism are shown in Scheme 8. Populations of the most significant valence basins of selected structures of the IRC and other important parameters are given in Table 6.

Simplified representation of the molecular mechanism of the P-DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and naphthalene 2b by Lewis-like structures arising from the topological analysis of the ELF along the reaction path
The bonding changes along this P-DA reaction are characterized by six phases. The topological features of the ELF of the corresponding molecular complex, MC3, being the first point of the IRC, are very similar to those of the separated reagents 1a and 2b. Along Phase I, very slight depopulation of the C1–C2 bonding region of the 1a fragment by 0.06 e [V(C1,C2)] is observed, while the populations around the C3–C4, C4–C5, C5–C6, C6–C7, C7–C8 and C8–C3 bonding regions at the naphthalene 2b fragment remain without observable changes (see Table 6). Phase II begins at the structure S1F2b. Along this phase, the two V(C1,C2) and V’(C1,C2) disynaptic basins present in the above Phase merge into one V(C1,C2) disynaptic basins, achieving a population of 3.21 e. Along Phase III, which starts at the structure S2F2b, it can be seen the creation of two C1 and C2 pseudoradical center [52] at the perfluorobicyclo[2.2.0]hex-1(4)-ene 1a moiety, integrating 0.40 e, each one at a C–C length of 2.31 Å (see V(C1) and V(C2) in Table 6, Scheme 8 and Fig. 7). The formation of these pseudoradical centers is mainly encouraged by the depopulation of the C1–C2 bonding region of the 1a fragment by ca. 0.54 e and by depopulation of C3–C4 and C7–C8 bonding regions by ca. 0.12 e and 0.14 e, respectively (see V(C1,C2) and V(C3,C4) and V(C7,C8) in Table 6). At this phase, the TS3 of the reaction is found. The two V(C1) and V(C2) monosynaptic basins have reached populations by ca. 0.48 e. Note that the GEDT at TS is the highest, 0.34 e, along this reaction. Along Phase IV, which begins at the structure S3F2b, two new C4 and C7 pseudoradical [52] centers are created at the naphthalene 2b moiety integrating a population of 0.15 e, each one (see V(C4) and V(C7) in Table 6, Scheme 8 and Fig. 7). The population attained by these pseudoradical centers mainly is a result of the depopulation of the C4–C5 and C6–C7 bonding regions by ca. 0.20 e (V(C4,C5) and V(C6,C7)). Along this phase, it may be seen that the population associated with the C1 and C2 pseudoradical centers increase to 0.77 e (see V(C1) and V(C2) in Table 6). Along Phase V, which starts at S4F2b, evidencing an important topological change, the two pairs of C1, C2 and C4, C7 pseudoradical centers have merged into two new C1–C4 and C2–C7 bonding regions [33] with an initial population of 1.10 e, each one (see V(C1,C4) and V(C2,C7) in Table 6, Scheme 8 and Fig. 7). The changes of the electron density indicate that the formation of the two new C–C single bonds begins at a distance of 2.13 Å by sharing the non-bonding electron densities of the two pairs of C1 and C2 toward C4 and C7 centers. Along this phase, the population associated with C1–C2, C3–C4, C4–C5, C6–C7 and C7–C8 bonding regions slightly decreases, while those associated with the C5––C6 and C8–C3 bonding regions very slightly increase. Phase VI, begins at S5F2b and finishes at the cycloadduct 4a. At the begin of this phase, while the V(C1,C4) and V(C2,C7) disynaptic basins have reached a population by ca. 1.30 e, the V(C5,C6) disynaptic basin is split into two new pairs of disynaptic basins, V(C5,C6) and V’(C5,C6) reaching a total population of 3.19 e. Lastly, at compound 4a, the electron population is relaxed: the C1–C4 and C2–C7 bonding regions integrate 1.89 e, each one. The C3–C4, C4–C5, C6–C7 and C7–C8 bonding regions reach populations by ca. 2.0 e, in agreement to the expected single bonds, C5–C6 bonding region attains population of 3.48 e, an expected value for double bonds, while C8–C3 bonding region integrates a population of 2.85 e, in consistency with delocalized double bonds at the benzene ring of 4a.

Attractor positions of the ELF valence basins of the IRC structures S2F2b, S3F2b and S4F2b involved in the C–C single bond formation along the DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and naphthalene 2b. The electron populations, in average number of electrons, are given in e
From the BET analysis of the P-DA reaction between perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and naphthalene 2b, it may be concluded follows: (i) this is a P-DA reaction, similar to that between 1a and benzene 2a, which also is developed through six different phases. The maximum of GEDT proceeds along Phase III (0.34 e), behavior mainly given by the strong electrophilic character perfluorobicyclo[2.2.0]hex-1(4)-ene 1a, ω = 2.08 eV. (ii) The highest energy of the path is found at the TS3, 18.1 kcal mol−1. The activation energy of this reaction is mainly associated with the continuous depopulation of the C1–C2 bonding region, required for the creation of the two C1 and C2 pseudoradical centers at the perfluorobicyclo[2.2.0]hex-1(4)-ene 1a moiety. (iii) The formation of the two C1–C4 and C2–C7 single bonds takes place simultaneously at a C–C distance of 2.13 Å, by sharing the non-bonding electron densities of the two pairs of C1 and C2 toward C4 and C7 pseudoradical centers in a 81:19 ratio. (iv) The current BET analysis shows many similarities to that observed between 1a and benzene 2a. Note that the formation of the two C1 and C2 pseudoradicals in 1a is slightly more advanced in the reaction with benzene 2a than with naphthalene 2b, C–C length 2.27 Å and 2.31 Å, respectively. (vi) At last, this P-DA takes place through a synchronic non-concerted one-step mechanism.
A detailed BET study of the nonpolar DA reaction of bicyclo[2.2.0]hex-1(4)-ene 1b with benzene 2a and naphthalene 2b is given in Supporting Information (see pages S15-S22). It is worth to mention here the following concluding remarks for all three DA reactions.
From the BET analysis of the nonpolar DA (N-DA) reaction between bicyclo[2.2.0]hex-1(4)-ene 1b and benzene 2a, it can be concluded: (i) this N-DA reaction proceeds along six different phases, in a similar mode than that between 1 and 2a. The nonpolar character is given by the negligible GEDT observed along the entire process. Note that this result is a fallout of the low electrophilic character of 1b (ω = 0.41 eV). (ii) TS2 shows the highest energy of this path, 23.2 kcal mol−1. The activation energy of this reaction is mainly associated with the continuous depopulations of the C1–C2 bonding region, required, at the first time, for the creation of the two C1 and C2 pseudoradical centers [52] at the bicyclo[2.2.0]hex-1(4)-ene 1b. (iii) Note that even though the activation energies of TS1 and TS2 are in a narrow range (3.6 kcal mol−1); the populations of the V(C1) and V(C2) monosynaptic basins are different, 0.57 e and 0.40 e, mainly due to the strong electrophilic character of 1a (ω = 2.08 eV). (iv) The first two monosynaptic basins appear in compounds 1a and 1b at the same C–C length of 2.26 Å, with similar population, 0.34 e. However, these changes, in the reaction of 1a with 2a, have an energy cost of almost 1.1 kcal·mol−1 lesser than that for 1b with 2a (see Supporting Information, pages S15-S18). (v) The formation of the two C1–C3 and C2–C6 single bonds takes place simultaneously at a C–C distance of 2.05 Å, by sharing the non-bonding electron densities of the two pairs of C1 and C2 toward C3 and C6 pseudoradical centers in a 65:35 ratio. At the end, (vi) the C–C bonding formation process is also non-concerted and fully synchronic.
From the BET study of the DA reaction between bicyclo[2.2.0]hex-1(4)-ene 1b and naphthalene 2b, the concluding remarks are: (i) this is a N-DA reaction, similar to that between 1b and benzene 2a. The difference is the current reaction is developed only in five phases. (ii) The highest energy of the path is found at the TS4, 18.7 kcal·mol−1. The activation energy of this reaction is mainly associated with the continuous depopulation of the C1–C2 bonding region required for the creation, in the following phase, the two C1 and C2 pseudoradical centers [52] at the bicyclo[2.2.0]hex-1(4)-ene 1b fragment. (iii) The associated energy cost is very similar to that found for the reaction between 1a and 2b. Note that TS4 does not present formation of pseudoradical centers, in comparison with TS3, probably due to the insignificant GEDT and the marginal electrophilic character of 1b (ω = 0.41 eV). (iv) Note that the creation of the pseudoradical centers occurs at a C–C length of 2.27 Å, which is more advanced than that for the reaction of 1a with 2b, 2.31 Å. (v) The formation of the two C1–C4 and C2–C7 single bonds takes place simultaneously at a C–C distance of 2.05 Å, by sharing the non-bonding electron densities of the two pairs of C1 and C2 toward C4 and C7 pseudoradical centers in a 62:38 ratio. At last, (vi) this N-DA also is completely synchronic in the C–C bond forming process (for details see Supporting Information, pages S19-S22).
4 Conclusions
The molecular mechanism of the DA reactions of perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and bicyclo[2.2.0]hex-1(4)-ene 1b with benzene 2a and naphthalene 2b has been studied using DFT calculations at the MPWB1K/6-311G(d,p) computational level.
Analysis of the CDFT reactivity indices indicates that 1a can be classified as a strong electrophile, ω = 2.08 eV, while 1b presents ω = 0.41 eV and N = 2.95 eV, being classified a marginal electrophile and a moderate nucleophile. Clearly, the presence of four fluorine centers in 1a makes it more electrophilic than 1b. The most favorable process will be associated with the two centers interaction between the most electrophilic center of 1a and the most nucleophilic centers of 2a-b. Note that 1a and 1b are strained reagents; therefore, it is expected that they react more favorably releasing that strain. On the other hand, 2a and 2b are aromatic species, so it is expected they react with their partners minimizing that loss. On the other hand, ELF topological analysis of perfluorobicyclo[2.2.0]hex-1(4)-ene 1a and bicyclo[2.2.0]hex-1(4)-ene 1b, in the most important region, shows the presence of two V(C1,C2) and V’(C1,C2) disynaptic basins, which are related to C1-C2 double bonds in 1a and 1b molecules.
Analysis of the stationary points involved in this DA reaction indicates that it takes place through a one-step mechanism which begins with formation of molecular complex. From MC1, the activation energy associated with TS1 is 18.0 kcal mol−1, the reaction being exothermic by 25.8 kcal mol−1. In turn, the relative energies of the TSs with respect to separated reagents are: 16.6 kcal mol−1 for DA reaction 1a with naphthalene 2b (TS3). The activation energy associated with the DA reaction of bicyclo[2.2.0]hex-1(4)-ene 1b and benzene 2a, begins also with the creation of molecular complex MC2. Thereafter, the molecular complex is recast to transition state (TS2), which is associated with an increase in the energy of activation over 23.2 kcal mol−1. The formation of the final cycloadduct 3b is exothermic by 29.0 kcal mol−1. In the case of reaction between 1 and 2b, we also observed the similar course of reactions, but we notice a slightly lower energy of activation and for TS4 is 17.7 kcal mol−1. It is worth mentioning that the reactivity order with 1a is 2b–2a. The last two behaviors may be related to the loss of the aromatic character of these compounds. On the other hand, the reactivity order with 1b is 2b > 2a, even though the activation energies are higher than those observed with 1a, the process is more exothermic. In summary, fluorinated bicyclic 1a is more reactive than non-fluorinated bicyclic 1b compound. Note that the high energy barriers of perfluorobicyclo[2.2.0]hex-1(4)-ene with benzene and naphthalene are in agreement with the drastic experimental conditions; however, the interest of the current study is to get insight into the reaction mechanism of strained alkene with aromatic species like benzene and naphthalene.
Analysis of the TSs geometries in the case of DA reactions of 1a with 2a-b indicates that in all reactions, the formation of two bonds are created at the same time, which confirmed the synchronicity one-step process. Also, in the case of DA reactions between 1b and 2a-b, the same trend is observed.
BET/ELF analysis of the molecular mechanism associated with the DA reaction of perfluorobicyclo[2.2.0]hex-1(4)-ene 1a with benzene 2a indicates that it takes place through a synchronic non-concerted one-step mechanism, which is initialized by rupture of the C1-C2 double bond and next we observed the formation of two C1 and C2 pseudoradical centers at the perfluorobicyclo[2.2.0]hex-1(4)-ene 1a moiety. The formation of the two C1–C4 and C2–C7 single bonds takes place simultaneously at a C–C distance of 2.13 Å, by sharing the non-bonding electron densities of the two pairs of C1 and C2 toward C4 and C7 pseudoradical centers. Similar relationships are observed in the case of DA reaction 1a with 2b. In the case of the DA reaction of 1b with 2a-b, a similar course of reactions was observed. The bonding pattern found here is similar to that found in other DA reactions [53,54,55]. The knowledge of the molecular mechanism will fully contribute to the design of further syntheses involving benzene.
References
Bur S, Padwa A (2014) [4+2] Cycloaddition chemistry of substituted Furans. In: Nishiwaki N (ed) Methods and applications of cycloaddition reactions in organic syntheses. Wiley, pp 355–406. https://doi.org/10.1002/9781118778173
Balthazor TA, Gaede B, Korte DE, Shieh H-S (1984) Reaction of 1, 1, 1-trichloro-3-nitro-2-propene with furans: a reexamination. J Org Chem 49:4547–4549
Anderson WK, Milowsky AS (1985) A retro Diels–Alder synthesis of 3-pyrrolines. J Org Chem 50:5423–5424
Jarvest RL, Readshaw SA (1992) Synthesis of some 2, 5-substituted 7-oxabicyclo [2.2. 1] heptanes: stereochemistry of Diels–Alder adducts of a 3-alkylfuran. Synthesis 10:962–965
Batt DG, Jones DG, La Greca S (1991) Regioselectivity in the acid-catalyzed isomerization of 2-substituted 1, 4-dihydro-1, 4-epoxynaphthalenes. J Org Chem 56:6704–6708
Kozikowski AP, Floyd WC, Kuniak MP (1977) 1,3-Diethoxycarbonylallene: an active dienophile and ethoxycarbonylketen equivalent in the synthesis of antibiotic C-nucleosides. J Chem Soc Chem Commun. https://doi.org/10.1039/c39770000582
Szczepanek A, Jasińska E, Kącka A, Jasiński R (2015) An experimental and quantumchemical study of [2+3] cycloaddition between (Z)-C-(m, m, p-trimethoxyphenyl)-N-(p-methyphenyl)-nitrone and (E)-3,3,3-trichloro-1-nitroprop-1-ene: mechanistic aspects. Curr Chem Lett 4:33–44
Atherton JCC, Jones S (2003) Diels-Alder reactions of anthracene, 9-substituted anthracenes and 9,10-disubstituted anthracenes. Tetrahedron 59:9039–9057
Dinulescu IG, Avram M, Nenitzescu CD (1960) Dien-Synthesen des 2.3-Dihydro-naphthalins und des Naphthalins mit N-Phenyl-maleinimid. Chem Ber 93:1795–1799
Lange GL, Neidert E (1973) Photochemistry of 2, 4-Cyclooctadienone I. In Benzene and Toluene. Can J Chem 51:2207–2214
Streit U, Bochet ChG (2011) The arene–alkene photocycloaddition. Beilstein J Org Chem 7:525–542
He Y, Junk ChP, Lemal DM (2003) Diels–Alder reactions of Perfluorobicyclo [2.2. 0] hex-1 (4)-ene with aromatics. Org Lett 5:2135–2136
Houk KN, Nendel M, Wiest O, Storer JW (1997) The vinylcyclopropane–cyclopentene rearrangement: a prototype thermal rearrangement involving competing diradical concerted and stepwise mechanisms. J Am Chem Soc 119:10545–10546
Vermeeren P, Brinkhuis F, Hamlin TA, Bickelhaupt MF (2020) How alkali cations catalyze aromatic Diels–Alder reactions. Chem Asian J 15:1167–1174
Bickelhaupt FM (1999) Understanding reactivity with Kohn-Sham molecular orbital theory: E2–SN2 mechanistic spectrum and other concepts. J Comput Chem 20:114–128
Domingo LR, Pérez P (2020) Lithium Cation-catalyzed benzene Diels−Alder reaction: insights on the molecular mechanism within the molecular electron density theory. J Org Chem 85:13121–13132
Domingo LR, Sáez J (2009) Understanding the mechanism of polar Diels–Alder reactions. Org Biomol Chem 7:3576–3583
Domingo LR (2016) Molecular electron density theory: a modern view of reactivity in organic chemistry. Molecules 21:1319
Domingo LR, Ríos-Gutiérrez M, Pérez P (2020) A molecular electron density theory study of the participation of tetrazines in aza-Diels–Alder reactions. RSC Adv 10:15394–15405
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford University Press, New York
Krokidis X, Noury S, Silvi B (1997) Characterization of elementary chemical processes by catastrophe theory. J Phys Chem A 101:7277–7283
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven TJ, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima Y, Honda O, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas MC, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2016) Gaussian 16 rev A.1 Gaussian Inc, Wallingford CT
Zhao Y, Truhlar GD (2004) Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: the MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and Van Der Waals interactions. J Phys Chem A 108:6908–6918
Fukui K (1970) Formulation of the reaction coordinate. J Phys Chem 74:4161–4163
Tapia O (1992) Solvent effect theories: Quantum and classical formalisms and their applications in chemistry and biochemistry. J Math Chem 10:139–181
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev 94:2027–2094
Simkin Y, Sheikhet I (1995) Quantum chemical and statistical theory of solutions: a computational approach. Ellis Horwood, Chichester
Cances MT, Mennunci V, Tomasi J (1997) Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: theoretical bases, computational implementation, and numerical applications. J Chem Phys 107:3032–3041
Cossi M, Barone V, Cammi R, Tomasi J (1996) Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem Phys Lett 255:327–335
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103:1793–1874
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21:748
Domingo LR (2014) new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4:32415–32428
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926
Parr RG, Szentpaly LV, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924
Domingo LR, Chamorro E, Perez P (2008) Understanding the reactivity of captodative ethylenes in polar Cycloaddition reactions. A Theoretical Study J Org Chem 73:4615–4624
Domingo LR, Pérez P, Sáez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486–1494
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular-systems. J Chem Phys 92:5397–5403
Noury S, Krokidis X, Fuster F, Silvi B (1999) Computational tools for the electron localization function topological analysis. Comput Chem 23:597–604
Dennington R, Keith TA, Millam JM (2016) Semichem Inc., Shawnee Mission, KS, GaussView, Version 6.1, Semichem Inc., Shawnee Mission, KS
Ahrens J, Law GBC (2005) ParaView: an end-user tool for large data visualization. Elsevier, Visualization Handbook
Ayachit U (2015) The ParaView Guide: a parallel visualization application, Kitware.
Kącka-Zych A (2020) Push-pull nitronates in the [3+2] cycloaddition with nitroethylene: molecular electron density theory study. J Mol Graph Model 97:107549
Kącka-Zych A (2019) Understanding the molecular mechanism of the rearrangement of internal nitronic ester into nitronorbornene in light of the MEDT study. Molecules 24:462
Kącka-Zych A, Jasiński R (2019) Unexpected molecular mechanism of trimethylsilyl bromide elimination from 2-(trimethylsilyloxy)-3-bromo-3-methyl-isoxazolidines. Theor Chem Acc 138:81–86
Kącka-Zych A, Ríos-Gutiérrez M, Domingo LR (2019) A molecular electron density theory study of the Lewis acid–catalyzed decomposition reaction of nitroethyl benzoate using aluminum derivatives. J Phys Org Chem 32:e3938
Domingo LR, Ríos-Gutiérrez M, Pérez P (2020) Unveiling the Lewis acid catalyzed Diels–Alder reactions through the molecular electron density theory. Molecules 25:2535
Domingo LR, Ríos-Gutiérrez M, Pérez P (2020) A molecular electron density theory study of the enhanced reactivity of aza aromatic compounds participating in Diels–Alder reactions. Org Biomol Chem 18:292–304
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 58:4417–4423
Jaramillo P, Domingo LR, Chamorro E, Pérez P (2008) A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J Mol Struct THEOCHEM 865:68–72
Domingo LR, Chamorro E, Pérez P (2010) Understanding the high reactivity of the Azomethine Ylides in [3 + 2] cycloaddition reactions. Lett Org Chem 7:432–439
Domingo LR, Pérez P, Sáez J (2012) Origin of the synchronicity in bon-formation in polar Diels–Alder reactions. An ELF analysis of the reaction between cyclopentadiene and tetracyanoethylene. Org Biomol Chem 10:3841–3851
Domingo LR, Aurell MJ, Pérez P (2014) The mechanism of ionic Diels–Alder reactions. A DFT study of the oxa-Povarov reaction. RSC Adv 4:16567–16577
Yepes D, Valenzuela J, Martinez-Araya J, Pérez P, Jaque P (2019) Effect of exchange-correlation functional on the synchronicity/nonsynchronicity in bond formation in Diels–Alder reactions: a reaction force constant analysis. Phys Chem Chem Phys 21:7412–7428
Acknowledgements
Partial support of this research by PL-Grid Infrastructure is gratefully acknowledged. The authors acknowledge the support of the PROM programme no. PPI/PRO/2019/1/00018, which is co-financed by the European Social Fund under the Knowledge Education Development Operational Programme. P.P. also acknowledges continuous support provided by FONDECYT—Chile—through Project No. 1180348. The authors thank Professor R. Jasiński and Professor L. R. Domingo for their valuable comments and suggestions.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kącka-Zych, A., Pérez, P. Perfluorobicyclo[2.2.0]hex-1(4)-ene as unique partner for Diels–Alder reactions with benzene: a density functional theory study. Theor Chem Acc 140, 17 (2021). https://doi.org/10.1007/s00214-020-02709-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-020-02709-6