
Abstract
Dipeptidyl peptidase 4 (DPP4) inhibitors are a class of antidiabetic medications that cause glucose-dependent increase in incretins in diabetic patients. One of the two incretins, glucagon-like peptide-1 (GLP-1), beside its insulinotropic activity, has been studied for extra pancreatic effects. Most of DPP4 inhibitors (DPP4i) have been investigated in in vivo and in vitro models of diabetic and nondiabetic cardiovascular diseases including heart failure, hypertension, myocardial ischemia or infarction, atherosclerosis, and stroke. Results of preclinical studies proved prominent therapeutic potential of DPP4i in cardiovascular diseases, regardless the presence of diabetes. This review aims to present an updated summary of the cardiovascular protective and therapeutic effects of DPP4 inhibitors through the past 5 years focusing on the molecular mechanisms beneath these effects. Additionally, based on the results summary presented here, future studies may be conducted to elucidate or illustrate some of these findings which can add clinical benefits towards management of diabetic cardiovascular complications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes mellitus (DM) is associated with various cardiovascular (CV) complications including hypertension, ischemic heart disease, heart failure, and atherosclerosis. Diabetic patients tend to develop cardiovascular disease at a younger age and at higher incidence than non-diabetic patients (Chen et al. 2016; Leon and Maddox 2015). In addition to the deleterious effects of hyperglycemia, studies proved that diabetic patients exhibit an impaired response to intestinal hormones called incretins compared to individuals with normal blood glucose level. This sheds lights on a new family of antidiabetic medications, the dipeptidyl peptidase 4 inhibitors (DPP4i) or gliptins, which target incretin hormones. Several DPP4i are commercially available including alogliptin, linagliptin, saxagliptin, sitagliptin, anagliptin, vildagliptin, teneligliptin, and omarigliptin, in addition to several others which are under development.
Incretins are hormones released by the intestinal mucosa to stimulate insulin secretion following oral nutrient intake. Humans express two types of incretins: glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP). GLP-1 is secreted by the neuroendocrine L cells in the ileum, while GIP is secreted by K cells in the duodenum and jejunum. Both hormones promote insulin release from pancreatic β-cells by activation of intracellular cAMP. They also stimulate β-cell proliferation and inhibit apoptosis, which increases β-cell mass (Drucker 2006). In addition, GLP-1, but not GIP, inhibits gastric emptying, food intake, and pancreatic release of glucagon, the hormone which stimulates hepatic glucose production.
GLP-1 has extra pancreatic effects and its receptor is expressed on many sites including the brain, intestine, lung, adipose tissue, myocardium, and vascular smooth muscle cells (Bullock et al. 1996). On the other hand, GIP has no major role in the cardiovascular system (Nauck and Meier 2018). GLP-1(7–36) is the active form that is degraded to an inactive form, GLP-1 (9–36), within 1 to 2 min by the enzyme DPP4 (Mafong and Henry 2009). DPP-4 is a widely expressed enzyme that exists in 2 forms, an extracellular protein that, when activated, initiates intracellular signal transduction pathways independent of its enzymatic activity, as well as a circulating soluble form which is enzymatically active (Drucker 2006).
Current developments in incretin research have demonstrated a significant role of incretins in cardiovascular pathology. This comprises 2 approaches. Firstly, the role of GLP-1 which is mediated through receptor-dependent and receptor-independent mechanisms. GLP-1 signaling seems to play a role in cardiac development, because mice with a targeted gene deletion of the GLP-1 receptors showed enlarged hearts (Gros et al. 2003). Diebold et al. (2018) demonstrated a significant role of GLP-1 secretion for left ventricular contractility during myocardial infarction. Mechanistically, it was shown that DPP-4 inhibition increased AMP-activated protein kinase (AMPK) activity and stimulated the mitochondrial respiratory capacity of non-infarcted myocardial tissues.
Secondly, it is now known that DPP-4 also cleaves several other peptides, some of which have direct actions on the cardiac and vascular cells. Hence, DPP4 inhibition provides favorable cardiovascular outcomes independent of GLP-1. DPP-4 is responsible for degradation of B-type natriuretic peptide (BNP, Brandt et al. 2006), stromal cell-derived factor 1α (SDF-1α, Zaruba et al. 2009), and substance P (Wang et al. 1991). These findings suggest DPP4i as potential cardioprotective agents in diabetic and non-diabetic patients specially when considering that DPP4i have no risk of hyperglycemia or weight gain.
This review presents an updated overview on the cardiovascular protective effects of DPP4i in experimental preclinical studies during the past 5 years. In addition, this review describes and evaluates the underlying molecular mechanisms for these effects.
Methodology
Literature search
To gather the most recent experimental studies on DPP4i in animal models of cardiovascular disease, a literature search was performed in “PubMed” and “Google Scholar” search engines up to 20 February 2022. The keywords “DPP4 inhibitors or gliptins or linagliptin or vildagliptin or saxagliptin or sitagliptin or alogliptin or anagliptin or teneligliptin or omarigliptin,” were matched with “cardiovascular or myocardial ischemia reperfusion or myocardial infarction or heart failure or atherosclerosis or stroke,” and with “experimental animals.” We included studies published in trustful scientific journals starting from 2017 and thereafter. To ensure a comprehensive search, we also included some related articles from references. Articles were carefully read, and data related to disease models, DPP4i dose, treatment duration, and main findings as well as the underlying molecular mechanisms were summarized in a tabular form.
Figure creation
Figure 2 was created using BioRender software tool.
Results
Results of our search of DPP4i cardiovascular effects are summarized in a tabular form. Results are classified based on the CV disease then the type of gliptin (Table 1).
Discussion
This review covers the recent research of the cardiovascular (CV) protective potential of DPP4 inhibitors (DPP4i) during the past 5 years. It is believed that the protective effects of DPP4i are maintained through 2 approaches, (I) Glucagon-like peptide-1 (GLP-1)–mediated mechanisms and (II) conservation of some peptides that are physiologically degraded by the DPP4 enzyme. Researchers reported that DPP4 is responsible for degradation of B-type natriuretic peptide (BNP), stromal cell–derived factor 1α (SDF-1α), and substance P. Gliptins effectively mitigate the deleterious effects of postprandial hyperglycemia on oxidative stress, inflammation, and CV remodeling. However, in models of CV disease in non-diabetic animals, gliptins show protective effects through various pathways that will be discussed here.
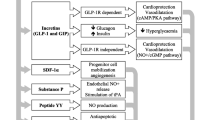
GLP-1 deficiency is a consequence rather than a cause of diabetes as GLP-1 secretion is decreased in hyperglycemia. Also, DPP4 activity is increased in diabetes, both type 1 and 2, and is negatively correlated with adiponectin levels (Vollmer et al. 2009). The effect of DPP4 antagonism was studied in models of myocardial ischemia/reperfusion (I/R) and infarction in normoglycemic and hyperglycemic animal models of CV disease by genetic deletion of DPP4 or by using DPP4i. Both approaches resulted in improved cardiac function and hemodynamics (Sauvé et al. 2010). These promising finding may be attributed to (I) increased GLP-1 level which exerts its effects through receptor-dependent as well as receptor-independent mechanisms. (II) DPP4 inhibition preserves some peptides that have advantages on CV function in various disease conditions (see Fig. 1).

Biological consequences of DPP4 inhibition
I-DPP4 inhibition increases GLP-1
It is well known that hyperglycemia underlines diabetic cardiovascular complications through various pathways. Glucotoxicity increases reactive oxygen species (ROS) production through many mechanisms including polyol and hexosamine pathways, increased advanced glycation end products (AGEs) production, activation of protein kinase C (PKC) and poly(ADP-ribose) polymerase-1 (PARP-1) enzyme (Mapanga and Essop 2016). Increased ROS production triggers a closed cycle of myocardial and vascular inflammation and oxidative stress leading to apoptosis, endothelial dysfunction, and cardiomyopathies. In healthy individuals, GLP-1 is secreted by the neuroendocrine L cells in the ileum in response to food ingestion and stimulates pancreatic beta cells to release insulin. Postprandial insulin augmentation helps regulate blood glucose with minimal hypoglycemic effect. As GLP-1 is impaired in diabetes, inhibiting its degradation helps decreasing blood glucose in diabetic patients and consequently accentuate the rush through these oxidative and nonoxidative glucotoxicity pathways. In addition, enhanced insulin secretion improves cardiac function and increase inotropy, regulate blood flow to myocardial tissue, and exerts anti-inflammatory, antioxidant, and antiapoptotic effects (Ng et al. 2012).
Beside the glycemic effect of GLP-1, it exerts direct cardiovascular protection. Since the discovery of GLP-1 receptor expression in the cardiac muscle, cardioprotective role of GLP-1 was extensively studied and was proved. Infusions of GLP-1 in animal models and human subjects with heart failure have demonstrated significant improvement in cardiac parameters (Ban et al., 2008). In patients with type 2 diabetes (T2DM), GLP-1 infusion significantly improved endothelial function, irrespective of changes in insulin sensitivity (Bullock et al. 1996). Moreover, infusion of GLP-1 in patients with T2DM and established coronary artery disease significantly improved endothelial dysfunction as measured by flow-mediated vasodilation (Nyström et al. 2004). Observations suggest that part of the cardioprotective and vasodilatory effects of GLP-1 on myocardial metabolism is direct, insulin-independent, and GLP-1-receptor–independent (Ban et al. 2008). A study by Gros et al. (2003) on GLP-1 receptor knock out mice (GLP-1R−/−) showed that lack of GLP-1R is associated with decreased resting heart rate and increased left ventricular end diastolic (LVED) pressure and LV thickness compared with CD-1 wild-type controls. In addition, GLP-1R deletion resulted in impaired LV contractility and diastolic function after insulin or epinephrine administration. In another way, a study on a mouse model of I/R found that GLP-1(9–36), the primary metabolite of GLP-1, has nearly identical results, suggesting the presence of an alternative signaling mechanism for GLP-1 and its metabolite independent of its known receptor.
II-DPP4 inhibition preserves physiologically active cardioprotective peptides
Beside GLP-1, other peptides are substrates of DPP4 enzyme including atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), stromal cell–derived factor-1α (SDF-1α), and substance P. ANP is synthesized in the atria while BNP is produced by heart ventricles. They act locally and systemically to exert several biological functions including diuresis, natriuresis, and vasodilatation as well as inhibition of renin and aldosterone secretion (Nishikimi et al. 2006). Physiological levels of ANP and BNP are low but they increase as a compensatory mechanism in heart failure. The active form, BNP(1–32), is degraded by DPP4 by removing the two N-terminal amino acids (serine and proline) to produce BNP(3–32), which has reduced biological activity. Elevated levels of NPs were proved in hyperglycemia and decrease by improved glycemic control (Dal et al. 2014). However, in cardiac pathology, it seems that DPP4 is implicated in high levels of NPs as it was found that genetic deletion of DPP4 improved the elevated levels of ANP and BNP in rats subjected to myocardial ischemia/reperfusion (Ku et al. 2011). However, a recent meta-analysis of clinical studies on diabetic patients treated with DPP4i reported no significant effect of DPP4 on NP levels (Mu et al. 2022) and this finding needs further explanation.
SDF-1α is a chemokine that promotes cardiac homing of endothelial progenitor cells, to stimulate angiogenesis, which consequently improves myocardial perfusion. SDF-1α is a substrate of DPP4 enzyme and DPP4 inhibition preserves SDF-1α actions and promotes cardiac recovery after I/R (Pala and Rotella 2013), acute myocardial infarction (Li et al. 2019), or stroke (Chiazza et al. 2018). Another DPP4 substrate is substance P which has role in regulating heart rate and blood pressure. Substance P showed protective effects in some animal models of heart disease through inhibiting apoptosis, myocardial cell injury (Chen et al. 2022), and fibrosis (Widiapradja et al. 2021). On the contrary, in vitro studies reported that DPP4 inactivates fibrin by cleavage of fibrin a-chain leading to inhibition of fibrin polymerization and clot formation (Mentlein and Heymann 1982). This points to possible thrombolytic effect of DPP4 enzyme.
Molecular pathways behind DPP4i effects on CV diseases
Diabetes is linked to a variety of cardiovascular diseases that lower the life quality of diabetic patients. Diabetic patients have many fold increase in the risk of atherosclerosis, myocardial ischemia, myocardial infarction, and heart failure. A common theme shared among these pathologies is massive ROS production that affects glucose metabolism and increase fatty acid oxidation. Additionally, ROS activates proinflammatory mediators, NRLP3 inflammasomes, and proatherogenic transcription factors. They also reduce mediators of tissue repair such as Nrf-2, sirtuin, and AMPK. Moreover, ROS stimulates mitochondrial fission leading to reduced efficiency of the mitochondrial electron transport chain and ATP synthesis, hence, myocardial ischemia and endothelial dysfunction. DPP4 mediates ROS production through several mechanisms of which glucotoxicity is major (see Fig. 1).
DPP4i inhibit oxidative stress via controlling glucotoxicity and lipotoxicity
Studies showed that many gliptins significantly decreased ROS, RNS, DNA fragmentation, AGEs, and Nox4 and increased antioxidants in most of animal models of CV disease. In addition, a recent study by Wang et al. (2021) on liver inflammation in diabetic mice found direct ROS scavenging activity of sitagliptin (Wang et al. 2021). DPP4i work through several mechanisms that can improve myocardial perfusion. DPP4i preserve endothelial function by increasing eNOS phosphorylation and decreasing Ang II-mediated Nox-4 production. They decrease ischemia-induced damage by minimizing oxidative stress. They also increase the level of intracellular cAMP and activate cAMP-dependent protein kinase (PKA) and SDF-1α. Besides, they enhance eNOS activity with subsequent augmentation of endothelial-dependent vasodilatation and myocardial perfusion. Incretins might target postprandial lipid metabolism and thereby favorably influence several endothelial and cardiovascular functions. DPP4 release strongly correlates with adipocyte size and is considered risk factor for obesity (Pala and Rotella 2013). Several studies covered by this review found decreased serum TG and total cholesterol and LDL in models of obesity or insulin resistance. DPP4i improve insulin sensitivity which is mediated partially by Sirt-1 and Sirt-6 beside other mechanisms that collectively decrease oxLDL and saturated fatty acids.
DPP4i improve CV inflammation
Preclinical studies show that DPP4i reduce myocardial inflammation via inhibition of cytokine release, monocyte activation, and chemotaxis. It is known that DPP4 significantly activate MAPK and NF-κB signaling pathway leading to vascular aging and dysfunction. Recent studies summarized in the “Results” section show that DPP4i control the release of many proinflammatory mediators such as NFkB, TNF-α, ILs, COX, MAPK, TLR4, CCl2, MCP-1, and MMPs. Inhibition of MMP activity maintains cellular architecture and prevents remodeling and fibrosis. DPP4 at least indirectly is implicated in endothelial and vascular smooth muscle cells structural remodeling and aging through inflammation and oxidative stress. In addition, ROS increases mitochondrial stress leading to energy shortage and subsequent CV senescence. The recent studies on animal models of myocardial infarction, I/R, and diabetes or obesity-induced cardiac remodeling reported that DPP4i significantly reduced hypertrophy, left ventricular interstitial, and periarterial fibrosis as evidenced by decreased expression of TGF-β, collagen, and components of cAMP/PKA/RhoA/ROCK2 pathway. Decreased fibrotic lesions in myocardial tissue will improve heart conductance and contractility.
DPP4i inhibit cell death
DPP4i decrease overall cell death as evidenced by decreasing infarct size, serum cardiac enzymes (CK, LDH), and troponin T. In addition, they decrease proapoptotic markers MMP-2, HSP-70, and caspase-3 but increased antiapoptotic marker Bcl-2. In another way, DPP4i promote tissue repair mechanisms such as Sirt1,6 (vildagliptin, saxagliptin and sitagliptin) and Nrf-2 (saxagliptin). Also, DPP4 inhibition by sitagliptin or vildagliptin modulated the disturbed autophagy responses in CV disease (LC3II and P65 levels) and vildagliptin enhanced mitochondrial fusion (increased mfn-2 level). These findings are partly resulted from decreased oxidative stress and inflammation due to improved hyperglycemia but may also be attributed to direct action of GLP-1 on its receptor in the CVS.
DPP4i improve CV hemodynamics
Diabetic cardiomyopathies affect almost all parameters of CV hemodynamics with negative impact on both conductance and contractility. Diabetes in animal and human increases peripheral resistance due to endothelial dysfunction and atherosclerosis. Long-term increase in blood pressure attenuates cardiac output specially in the presence of other dependent or independent by CV pathologies. Results of our search in models of diabetes-, obesity-, or drug-induced cardiomyopathy showed that treatment with gliptins resulted in increased cardiac output and left ventricular ejection fraction, but reduced systolic, diastolic, mean arterial, and left ventricular end diastolic pressures. These changes help preserve cardiac function. The mechanism of gliptin-mediated decrease in BP may be attributed in part to reduced plasma renin concentration and cardiac angiotensin II (Ang II) contents as well as increasing ACE2 and Angiotensin 1–7 (see Fig. 2). It can be also due to less atherosclerotic lesions in large arteries due to controlled lipotoxicity and CV lipid accumulation which is evident in many studies. Taken together, the studies presented in this review clearly found that gliptins improved systolic and diastolic left ventricular dysfunction in many CV disease models and so improved overall cardiac performance.

A summary of molecular mechanisms that underly cardiovascular protective effects of gliptins
Conclusions and prospective
Uncontrolled diabetes is associated with CV complications and predispose the patient to end-organ damage such as stroke and heart failure. Since their introduction into drug market, DPP4i have gained attention due to their effective glucose regulation and due to their beneficial CV effects. Studies on DPP4i proved that these effects are partly due to control of hyperglycemia and are also due to direct effect on the CV system via receptor-dependent and possibly receptor-independent effects too. DPP4i modulate not only the level of GLP-1, but also the concentration of other peptides that might exert vasoactive, and CV protective effects such as BNP, SDF-1α, and substance P. In this review, we summarized the result of most recent preclinical studies on CV protective effects of gliptins during the past 5 years. DPP4i control hyperglycemia, decreasing oxidative stress and inflammation, leading to less mitochondrial stress and cell death. They also enhance tissue repair and preserve endothelial function leading to improved myocardial perfusion. Moreover, DPP4i can significantly decrease cardiac Ang II but increase Ang1-7 which can also improve cardiac perfusion. These consistent findings in various CV diseases suggest promising cardioprotective potential of DPP4i, especially when considering their ability to improve glucose control without affecting body weight or causing hypoglycemia. However, further investigations on their mechanism and long-term safety data are required before recommending gliptins as CV-protecting agents in diabetes.
References
Abbas SS, Mahmoud HM, Schaalan MF, El-Abhar HS (2018) Involvement of brain natriuretic peptide signaling pathway in the cardioprotective action of sitagliptin. Pharmacological Reports : PR 70(4):720–729. https://doi.org/10.1016/j.pharep.2018.02.010
Al Zoubi S, Chen J, Murphy C et al (2018) Linagliptin attenuates the cardiac dysfunction associated with experimental sepsis in mice with pre-existing type 2 diabetes by inhibiting NF-κB. Front Immunol 9:2996. https://doi.org/10.3389/fimmu.2018.02996
Al-Awar A, Almási N, Szabó R, et al. (2018) Novel potentials of the DPP-4 inhibitor sitagliptin against ischemia-reperfusion (I/R) injury in rat ex-vivo heart model. International journal of molecular sciences 19(10) https://doi.org/10.3390/ijms19103226
Alves BEO, de Alencar AKN, Gamba LER et al (2019) Reduction of cardiac and renal dysfunction by new inhibitor of DPP4 in diabetic rats. Pharmacological Reports : PR 71(6):1190–1200. https://doi.org/10.1016/j.pharep.2019.07.005
Arinno A, Apaijai N, Kaewtep P et al (2019) Combined low-dose testosterone and vildagliptin confers cardioprotection in castrated obese rats. J Endocrinol. https://doi.org/10.1530/joe-18-0673
Aroor AR, Habibi J, Kandikattu HK et al (2017) Dipeptidyl peptidase-4 (DPP-4) inhibition with linagliptin reduces western diet-induced myocardial TRAF3IP2 expression, inflammation and fibrosis in female mice. Cardiovasc Diabetol 16(1):61. https://doi.org/10.1186/s12933-017-0544-4
Augestad IL, Pintana H, Larsson M et al (2020) Regulation of glycemia in the recovery phase after stroke counteracts the detrimental effect of obesity-induced type 2 diabetes on neurological recovery. Diabetes 69(9):1961–1973. https://doi.org/10.2337/db20-0095
Aziz TA (2021) Cardioprotective effect of quercetin and sitagliptin in doxorubicin-induced cardiac toxicity in rats. Cancer Management and Research 13:2349–2357. https://doi.org/10.2147/cmar.s300495
Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M (2008) Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 117(18):2340–2350. https://doi.org/10.1161/circulationaha.107.739938
Batchu SN, Yerra VG, Liu Y, Advani SL, Klein T, Advani A (2020) The dipeptidyl peptidase-4 inhibitor linagliptin directly enhances the contractile recovery of mouse hearts at a concentration equivalent to that achieved with standard dosing in humans. International journal of molecular sciences 21(16) https://doi.org/10.3390/ijms21165756
Bayrami G, Alihemmati A, Karimi P et al (2018) Combination of vildagliptin and ischemic postconditioning in diabetic hearts as a working strategy to reduce myocardial reperfusion injury by restoring mitochondrial function and autophagic activity. Advanced pharmaceutical bulletin 8(2):319–329. https://doi.org/10.15171/apb.2018.037
Bayrami G, Karimi P, Agha-Hosseini F, Feyzizadeh S, Badalzadeh R (2018b) Effect of ischemic postconditioning on myocardial function and infarct size following reperfusion injury in diabetic rats pretreated with vildagliptin. J Cardiovasc Pharmacol Ther 23(2):174–183. https://doi.org/10.1177/1074248417729881
Beraldo JI, Benetti A, Borges-Júnior FA, et al. (2019) Cardioprotection conferred by sitagliptin is associated with reduced cardiac angiotensin ii/angiotensin-(1–7) balance in experimental chronic kidney disease. International journal of molecular sciences 20(8) https://doi.org/10.3390/ijms20081940
Birnbaum Y, Tran D, Bajaj M, Ye Y (2019) DPP-4 inhibition by linagliptin prevents cardiac dysfunction and inflammation by targeting the Nlrp3/ASC inflammasome. Basic Res Cardiol 114(5):35. https://doi.org/10.1007/s00395-019-0743-0
Bradic J, Milosavljevic I, Bolevich S et al (2021) Dipeptidyl peptidase 4 inhibitors attenuate cardiac ischaemia-reperfusion injury in rats with diabetes mellitus type 2. Clin Exp Pharmacol Physiol 48(4):575–584. https://doi.org/10.1111/1440-1681.13450
Brandt I, Lambeir AM, Ketelslegers JM, Vanderheyden M, Scharpé S, De Meester I (2006) Dipeptidyl-peptidase IV converts intact B-type natriuretic peptide into its des-SerPro form. Clin Chem 52(1):82–87. https://doi.org/10.1373/clinchem.2005.057638
Brown SM, Smith CE, Meuth AI et al (2017) Dipeptidyl peptidase-4 inhibition with saxagliptin ameliorates angiotensin II-induced cardiac diastolic dysfunction in male mice. Endocrinology 158(10):3592–3604. https://doi.org/10.1210/en.2017-00416
Bullock BP, Heller RS, Habener JF (1996) Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology 137(7):2968–2978. https://doi.org/10.1210/endo.137.7.8770921
Chen R, Ovbiagele B, Feng W (2016) Diabetes and stroke: epidemiology, pathophysiology, pharmaceuticals and outcomes. Am J Med Sci 351(4):380–386. https://doi.org/10.1016/j.amjms.2016.01.011
Chen Z, Yu J, Fu M et al (2020) Dipeptidyl peptidase-4 inhibition improves endothelial senescence by activating AMPK/SIRT1/Nrf2 signaling pathway. Biochem Pharmacol 177:113951. https://doi.org/10.1016/j.bcp.2020.113951
Chen FX, Wan Q, Li QL, Fang J, Peng L, Hu J (2022) Substance P prevents doxorubicin‑induced cardiomyocyte injury by regulating apoptosis and autophagy: in vitro and in vivo evidence. Molecular medicine reports 25(2) https://doi.org/10.3892/mmr.2021.12566
Chiazza F, Tammen H, Pintana H et al (2018) The effect of DPP-4 inhibition to improve functional outcome after stroke is mediated by the SDF-1α/CXCR4 pathway. Cardiovasc Diabetol 17(1):60. https://doi.org/10.1186/s12933-018-0702-3
Cuijpers I, Papageorgiou AP, Carai P et al (2021) Linagliptin prevents left ventricular stiffening by reducing titin cleavage and hypophosphorylation. J Cell Mol Med 25(2):729–741. https://doi.org/10.1111/jcmm.16122
Dal K, Ata N, Yavuz B et al (2014) The relationship between glycemic control and BNP levels in diabetic patients. Cardiol J 21(3):252–256. https://doi.org/10.5603/CJ.a2013.0109
Diebold S, Moellmann J, Kahles F et al (2018) Myocardial infarction is sufficient to increase GLP-1 secretion, leading to improved left ventricular contractility and mitochondrial respiratory capacity. Diabetes Obes Metab 20(12):2911–2918. https://doi.org/10.1111/dom.13472
Drucker DJ (2006) The biology of incretin hormones. Cell Metab 3(3):153–165. https://doi.org/10.1016/j.cmet.2006.01.004
El-Marasy SA, Abdel-Rahman RF, Abd-Elsalam RM (2018) Neuroprotective effect of vildagliptin against cerebral ischemia in rats. Naunyn Schmiedebergs Arch Pharmacol 391(10):1133–1145. https://doi.org/10.1007/s00210-018-1537-x
Esposito G, Cappetta D, Russo R et al (2017) Sitagliptin reduces inflammation, fibrosis and preserves diastolic function in a rat model of heart failure with preserved ejection fraction. Br J Pharmacol 174(22):4070–4086. https://doi.org/10.1111/bph.13686
Fan S, Xiong Q, Zhang X, Zhang L, Shi Y (2020a) Glucagon-like peptide 1 reverses myocardial hypertrophy through cAMP/PKA/RhoA/ROCK2 signaling. Acta Biochim Biophys Sin 52(6):612–619. https://doi.org/10.1093/abbs/gmaa038
Fan SH, Xiong QF, Wang L, Zhang LH, Shi YW (2020b) Glucagon-like peptide 1 treatment reverses vascular remodelling by downregulating matrix metalloproteinase 1 expression through inhibition of the ERK1/2/NF-κB signalling pathway. Mol Cell Endocrinol 518:111005. https://doi.org/10.1016/j.mce.2020.111005
Fan X, Yang Y, Qi L (2020c) Vildagliptin protects hypoxia/reoxygenation-induced injury of cardiac microvascular endothelial cells. Minerva medica https://doi.org/10.23736/s0026-4806.20.06682-3
Fleenor BS, Ouyang A, Olver TD et al (2018) Saxagliptin prevents increased coronary vascular stiffness in aortic-banded mini swine. Hypertension (Dallas, Tex : 1979) 72(2):466–475. https://doi.org/10.1161/hypertensionaha.118.10993
Furukawa N, Koitabashi N, Matsui H et al (2021) DPP-4 inhibitor induces FGF21 expression via sirtuin 1 signaling and improves myocardial energy metabolism. Heart Vessels 36(1):136–146. https://doi.org/10.1007/s00380-020-01711-z
Gao P, Li L, Wei X et al (2020) Activation of transient receptor potential channel vanilloid 4 by DPP-4 (dipeptidyl peptidase-4) inhibitor vildagliptin protects against diabetic endothelial dysfunction. Hypertension (Dallas, Tex : 1979) 75(1):150–162. https://doi.org/10.1161/hypertensionaha.119.13778
Gros R, You X, Baggio LL et al (2003) Cardiac function in mice lacking the glucagon-like peptide-1 receptor. Endocrinology 144(6):2242–2252. https://doi.org/10.1210/en.2003-0007
Gu Y, Ma CT, Gu HL, Shi L, Tian XT, Xu WQ (2018) Sitagliptin improves cardiac function after myocardial infarction through activation of autophagy in streptozotocin-induced diabetic mice. Eur Rev Med Pharmacol Sci 22(24):8973–8983. https://doi.org/10.26355/eurrev_201812_16668
Hardigan T, Yasir A, Abdelsaid M et al (2016) Linagliptin treatment improves cerebrovascular function and remodeling and restores reduced cerebral perfusion in type 2 diabetes. Am J Physiol Regul Integr Comp Physiol 311(3):R466–R477. https://doi.org/10.1152/ajpregu.00057.2016
He Y, Yang G, Yao F et al (2019) Sitagliptin inhibits vascular inflammation via the SIRT6-dependent signaling pathway. Int Immunopharmacol 75:105805. https://doi.org/10.1016/j.intimp.2019.105805
Ideishi A, Suematsu Y, Tashiro K et al (2021) Combination of linagliptin and empagliflozin preserves cardiac systolic function in an ischemia-reperfusion injury mice with diabetes mellitus. Cardiology research 12(2):91–97. https://doi.org/10.14740/cr1194
Ji Y, Ge Y, Xu X et al (2019) Vildagliptin reduces stenosis of injured carotid artery in diabetic mouse through inhibiting vascular smooth muscle cell proliferation via ER stress/NF-κB pathway. Front Pharmacol 10:142. https://doi.org/10.3389/fphar.2019.00142
Khodeer DM, Bilasy SE, Farag NE, Mehana AE, Elbaz AA (2019) Sitagliptin protects diabetic rats with acute myocardial infarction through induction of angiogenesis: role of IGF-1 and VEGF. Can J Physiol Pharmacol 97(11):1053–1063. https://doi.org/10.1139/cjpp-2018-0670
Ku HC, Chen WP, Su MJ (2011) DPP4 deficiency preserves cardiac function via GLP-1 signaling in rats subjected to myocardial ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol 384(2):197–207. https://doi.org/10.1007/s00210-011-0665-3
Lei Y, Yang G, Hu L et al (2017) Increased dipeptidyl peptidase-4 accelerates diet-related vascular aging and atherosclerosis in ApoE-deficient mice under chronic stress. Int J Cardiol 243:413–420. https://doi.org/10.1016/j.ijcard.2017.05.062
Leon BM, Maddox TM (2015) Diabetes and cardiovascular disease: epidemiology, biological mechanisms, treatment recommendations and future research. World J Diabetes 6(13):1246–1258. https://doi.org/10.4239/wjd.v6.i13.1246
Li M, Wang Z, Xia H, Yu L, Hu Z (2019) Vildagliptin and G-CSF improved angiogenesis and survival after acute myocardial infarction. Arch Med Res 50(3):133–141. https://doi.org/10.1016/j.arcmed.2019.07.004
Li X, Meng C, Han F et al (2021) Vildagliptin attenuates myocardial dysfunction and restores autophagy via miR-21/SPRY1/ERK in diabetic mice heart. Front Pharmacol 12:634365. https://doi.org/10.3389/fphar.2021.634365
Lin FY, Shih CM, Huang CY et al (2021) Dipeptidyl peptidase-4 inhibitor decreases allograft vasculopathy via regulating the functions of endothelial progenitor cells in normoglycemic rats. Cardiovasc Drugs Ther 35(6):1111–1127. https://doi.org/10.1007/s10557-020-07013-w
Mafong DD, Henry RR (2009) The role of incretins in cardiovascular control. Curr Hypertens Rep 11(1):18–22. https://doi.org/10.1007/s11906-009-0005-x
Mansour SM, Aly S, Hassan SHM, Zaki HF (2021) Protective effect of sitagliptin and whole-body γ-irradiation in diabetes-induced cardiac injury. Can J Physiol Pharmacol 99(6):676–684. https://doi.org/10.1139/cjpp-2020-0454
Mapanga RF, Essop MF (2016) Damaging effects of hyperglycemia on cardiovascular function: spotlight on glucose metabolic pathways. Am J Physiol Heart Circ Physiol 310(2):H153–H173. https://doi.org/10.1152/ajpheart.00206.2015
Mentlein R, Heymann E (1982) Dipeptidyl peptidase IV inhibits the polymerization of fibrin monomers. Arch Biochem Biophys 217(2):748–750. https://doi.org/10.1016/0003-9861(82)90556-2
Mišúth S, Uhrinová M, Klimas J, Vavrincová-Yaghi D, Vavrinec P (2021) Vildagliptin improves vascular smooth muscle relaxation and decreases cellular senescence in the aorta of doxorubicin-treated rats. Vascul Pharmacol 138:106855. https://doi.org/10.1016/j.vph.2021.106855
Mu L, Wang Z, Ren J, Xiong X, Jin Z, Liu X (2022) Impact of DPP-4 inhibitors on plasma levels of BNP and NT-pro-BNP in type 2 diabetes mellitus. Diabetol Metab Syndr 14(1):30. https://doi.org/10.1186/s13098-022-00797-x
Nakajima Y, Ito S, Asakura M et al (2019) A dipeptidyl peptidase-IV inhibitor improves diastolic dysfunction in Dahl salt-sensitive rats. J Mol Cell Cardiol 129:257–265. https://doi.org/10.1016/j.yjmcc.2019.03.009
Nauck MA, Meier JJ (2018) Incretin hormones: their role in health and disease. Diabetes Obes Metab 20(Suppl 1):5–21. https://doi.org/10.1111/dom.13129
Ng KW, Allen ML, Desai A, Macrae D, Pathan N (2012) Cardioprotective effects of insulin: how intensive insulin therapy may benefit cardiac surgery patients. Circulation 125(5):721–728. https://doi.org/10.1161/circulationaha.111.063784
Nishikimi T, Maeda N, Matsuoka H (2006) The role of natriuretic peptides in cardioprotection. Cardiovasc Res 69(2):318–328. https://doi.org/10.1016/j.cardiores.2005.10.001
Nyström T, Gutniak MK, Zhang Q et al (2004) Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am J Physiol Endocrinol Metab 287(6):E1209–E1215. https://doi.org/10.1152/ajpendo.00237.2004
Okabe K, Matsushima S, Ikeda S et al (2020) DPP (dipeptidyl peptidase)-4 inhibitor attenuates Ang II (angiotensin II)-induced cardiac hypertrophy via GLP (glucagon-like peptide)-1-dependent suppression of Nox (nicotinamide adenine dinucleotide phosphate oxidase) 4-HDAC (histone deacetylase) 4 pathway. Hypertension (Dallas, Tex: 1979) 75(4):991–1001. https://doi.org/10.1161/hypertensionaha.119.14400
Pala L, Rotella CM (2013) The role of DPP4 activity in cardiovascular districts: in vivo and in vitro evidence. J Diabetes Res 2013:590456. https://doi.org/10.1155/2013/590456
Pirzeh L, Babapour V, Badalzadeh R, Panahi N (2019) Pretreatment with vildagliptin boosts ischemic-postconditioning effects on cardioprotection and expression profile of genes regulating autophagy and mitochondrial fission/fusion in diabetic heart with reperfusion injury. Naunyn Schmiedebergs Arch Pharmacol 392(11):1371–1382. https://doi.org/10.1007/s00210-019-01660-z
Qiao S, Mao G, Li H et al (2018) DPP-4 inhibitor sitagliptin improves cardiac function and glucose homeostasis and ameliorates β-cell dysfunction together with reducing S6K1 activation and IRS-1 and IRS-2 degradation in obesity female mice. J Diabetes Res 2018:3641516. https://doi.org/10.1155/2018/3641516
Salim HM, Fukuda D, Higashikuni Y et al (2017) Teneligliptin, a dipeptidyl peptidase-4 inhibitor, attenuated pro-inflammatory phenotype of perivascular adipose tissue and inhibited atherogenesis in normoglycemic apolipoprotein-E-deficient mice. Vascul Pharmacol 96–98:19–25. https://doi.org/10.1016/j.vph.2017.03.003
Sato A, Suzuki S, Watanabe S et al (2017) DPP4 inhibition ameliorates cardiac function by blocking the cleavage of HMGB1 in diabetic mice after myocardial infarction. Int Heart J 58(5):778–786. https://doi.org/10.1536/ihj.16-547
Sauvé M, Ban K, Momen MA et al (2010) Genetic deletion or pharmacological inhibition of dipeptidyl peptidase-4 improves cardiovascular outcomes after myocardial infarction in mice. Diabetes 59(4):1063–1073. https://doi.org/10.2337/db09-0955
Sivasinprasasn S, Tanajak P, Pongkan W, Pratchayasakul W, Chattipakorn SC, Chattipakorn N (2017) DPP-4 inhibitor and estrogen share similar efficacy against cardiac ischemic-reperfusion injury in obese-insulin resistant and estrogen-deprived female rats. Sci Rep 7:44306. https://doi.org/10.1038/srep44306
Suda M, Shimizu I, Yoshida Y et al (2017) Inhibition of dipeptidyl peptidase-4 ameliorates cardiac ischemia and systolic dysfunction by up-regulating the FGF-2/EGR-1 pathway. PLoS ONE 12(8):e0182422. https://doi.org/10.1371/journal.pone.0182422
Tanajak P, Sa-Nguanmoo P, Sivasinprasasn S et al (2018) Cardioprotection of dapagliflozin and vildagliptin in rats with cardiac ischemia-reperfusion injury. J Endocrinol 236(2):69–84. https://doi.org/10.1530/joe-17-0457
Tanajak P, Sa-Nguanmoo P, Apaijai N, et al. (2017) Comparisons of cardioprotective efficacy between fibroblast growth factor 21 and dipeptidyl peptidase-4 inhibitor in prediabetic rats. Cardiovascular therapeutics 35(4) https://doi.org/10.1111/1755-5922.12263
Vollmer K, Gardiwal H, Menge BA et al (2009) Hyperglycemia acutely lowers the postprandial excursions of glucagon-like peptide-1 and gastric inhibitory polypeptide in humans. J Clin Endocrinol Metab 94(4):1379–1385. https://doi.org/10.1210/jc.2008-2197
Wang LH, Ahmad S, Benter IF, Chow A, Mizutani S, Ward PE (1991) Differential processing of substance P and neurokinin A by plasma dipeptidyl(amino)peptidase IV, aminopeptidase M and angiotensin converting enzyme. Peptides 12(6):1357–1364. https://doi.org/10.1016/0196-9781(91)90220-j
Wang X, Ke J, Zhu YJ et al (2021) Dipeptidyl peptidase-4 (DPP4) inhibitor sitagliptin alleviates liver inflammation of diabetic mice by acting as a ROS scavenger and inhibiting the NFκB pathway. Cell Death Discovery 7(1):236. https://doi.org/10.1038/s41420-021-00625-7
Widiapradja A, Kasparian AO, McCaffrey SL, et al. (2021) Replacement of lost substance P reduces fibrosis in the diabetic heart by preventing adverse fibroblast and macrophage phenotype changes. Cells 10(10) https://doi.org/10.3390/cells10102659
Wu L, Wang K, Wang W et al (2018) Glucagon-like peptide-1 ameliorates cardiac lipotoxicity in diabetic cardiomyopathy via the PPARα pathway. Aging Cell 17(4):e12763. https://doi.org/10.1111/acel.12763
Xu J, Wang J, He M et al (2018) Dipeptidyl peptidase IV (DPP-4) inhibition alleviates pulmonary arterial remodeling in experimental pulmonary hypertension. Laboratory investigation; a journal of technical methods and pathology 98(10):1333–1346. https://doi.org/10.1038/s41374-018-0080-1
Yamaguchi T, Watanabe A, Tanaka M et al (2019) A dipeptidyl peptidase-4 (DPP-4) inhibitor, linagliptin, attenuates cardiac dysfunction after myocardial infarction independently of DPP-4. J Pharmacol Sci 139(2):112–119. https://doi.org/10.1016/j.jphs.2018.12.004
Yamamoto M, Ishizu T, Seo Y et al (2018) Teneligliptin prevents cardiomyocyte hypertrophy, fibrosis, and development of hypertensive heart failure in Dahl salt-sensitive rats. J Cardiac Fail 24(1):53–60. https://doi.org/10.1016/j.cardfail.2017.09.001
Zaruba MM, Theiss HD, Vallaster M et al (2009) Synergy between CD26/DPP-IV inhibition and G-CSF improves cardiac function after acute myocardial infarction. Cell Stem Cell 4(4):313–323. https://doi.org/10.1016/j.stem.2009.02.013
Zhang G, Kim S, Gu X, Yu SP, Wei L (2020) DPP-4 inhibitor linagliptin is neuroprotective in hyperglycemic mice with stroke via the AKT/mTOR pathway and anti-apoptotic effects. Neurosci Bull 36(4):407–418. https://doi.org/10.1007/s12264-019-00446-w
Zhang Q, Xiao X, Zheng J et al (2021) Vildagliptin, a dipeptidyl peptidase-4 inhibitor, attenuated endothelial dysfunction through miRNAs in diabetic rats. Archives of Medical Science : AMS 17(5):1378–1387. https://doi.org/10.5114/aoms.2019.86609
Zhou Y, Wang H, Man F et al (2018) Sitagliptin protects cardiac function by reducing nitroxidative stress and promoting autophagy in Zucker diabetic fatty (ZDF) rats. Cardiovasc Drugs Ther 32(6):541–552. https://doi.org/10.1007/s10557-018-6831-9
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
Esraa M. Zakaria raised the idea and contributed to all steps of manuscript construction and revision. Walaa M. Tawfeek participated in constructing the manuscript. Mohammed Y. Hassaballah contributed to data collection, extraction, and manuscript revision. Mohammed Hassan created the figure. All authors approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zakaria, E.M., Tawfeek, W.M., Hassanin, M.H. et al. Cardiovascular protection by DPP-4 inhibitors in preclinical studies: an updated review of molecular mechanisms. Naunyn-Schmiedeberg's Arch Pharmacol 395, 1357–1372 (2022). https://doi.org/10.1007/s00210-022-02279-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-022-02279-3