
Abstract
Dengue virus, particularly serotype 2 (DENV-2), poses a significant global health threat, and understanding the molecular basis of its interactions with host cell proteins is imperative for developing targeted therapeutic strategies. This study elucidated the interactions between proline-enriched motifs and Src homology 3 (SH3) domain. The SH3 domain is pivotal in mediating protein–protein interactions, particularly by recognizing and binding to proline-rich regions in partner proteins. Through a computational pipeline, we analyzed the interactions and binding modes of proline-enriched motifs with SH3 domains, identified new potential DENV-2 interactions with the SH3 domain, and revealed potential hot spot residues, underscoring their significance in the viral life cycle. This comprehensive analysis provides crucial insights into the molecular basis of DENV-2 infection, highlighting conserved and serotype-specific interactions. The identified hot spot residues offer potential targets for therapeutic intervention, laying the foundation for developing antiviral strategies against Dengue virus infection. These findings contribute to the broader understanding of viral–host interactions and provide a roadmap for future research on Dengue virus pathogenesis and treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dengue Virus (DENV) has emerged as a significant global health threat, demanding an in-depth understanding of its intricate pathogenesis (Idrees and Ashfaq 2012; Chala and Hamde 2021; WHO 2021). As a prominent arthropod-transmitted viral disease causing substantial human morbidity and mortality, numerous studies have probed into Dengue’s pathogenesis (Bhatt et al. 2021). However, despite extensive research, the precise molecular mechanisms governing Dengue disease remain elusive, necessitating further exploration (Srikiatkhachorn 2009; Bhatt et al. 2013; Idrees and Ashfaq 2013; Kraemer et al. 2015; Guo et al. 2017). A hypothesis rooted in molecular mimicry suggests that certain Dengue-induced antibodies may cross-react with host proteins, and a study validated a direct association between the severity of secondary Dengue disease in humans and pre-existing anti-DENV antibody levels (Chuang et al. 2014). The interactions between Dengue and host proteins can be via Short Linear Motifs (SLiMs), that help viruses in hijacking the endosomal sorting complexes required for transport (ESCRT) (Idrees and Paudel 2023a, b; Idrees et al. 2023). Upon viral invasion, ESCRTs are recruited to the viral entry site by viral proteins with SLiMs (Ren and Hurley 2011; Venkatakrishnan et al. 2020; Idrees et al. 2023).
Previous studies have shown the significance of SH3 interactions in viral replication and pathogenicity. Notable instances include alphaviruses relying on NsP3-SH3 binding for RNA replication, HIV/SIV Nef proteins using an “R-clamp” strategy for kinase activation, HCV NS5A’s high-affinity interaction with the Fyn SH3 domain, and HIV/SIV Nef PxxP motifs promoting viral growth via SH3 binding (Saksela et al. 1995; Shelton and Harris 2008; Neuvonen et al. 2011; Ren and Hurley 2011; Zhao et al. 2021). Moreover, a study on mature DENV-2 virus has previously elucidated interactions within the E glycoprotein chains, partly mediated by the PXXP motif (Thomas and Endy 2011; Torres-Flores et al. 2022; Palanichamy Kala et al. 2023). As seen in related viruses and considering the therapeutic implications of similar motifs, further exploration of SH3 interactions in DENV-2 can provide insights into host–virus interactions and can aid in developing potential targets for antiviral strategies (Kaneko et al. 2008; Gould et al. 2009; Gadkari and Srinivasan 2010).
Therefore, in this study, we performed a network and structural approach to predict proline mediated interactions between DENV-2 proteins and the SH3 domain by investigating virus–host protein–protein interactions (PPIs) to understand DENV-2 molecular mechanisms and have identified hot spot residues involved in dengue infection which can be used as therapeutic targets.
Methods
Data retrieval and processing
Dengue virus (DENV) protein–protein interactions (PPIs) [n = 213] (Table S1) were downloaded from the Pathogen Host Interaction Search Tool (PHISTO) database (Durmus Tekir et al. 2013). These PPIs were used to predict new SLiM instances and domain–motif interactions (DMIs). UniProt IDs were mapped to their respective gene symbols, and viral protein sequence data for DENV-2 was retrieved from the UniProt database (Apweiler et al. 2004). Known structural domain data was downloaded from the 3did database (Mosca et al. 2014) that included SH3 resolved PDB structure (PDB ID: 5FW9).
SLiM and DMI prediction
To predict new DMIs involving DENV-2, SLiM instances of known Eukaryotic Linear Motifs (ELM) were predicted using SLiMProb v2.5.1 (Edwards and Palopoli 2015). As SLiMs mostly reside in intrinsically disordered regions (IDRs) we used the disordered masking feature (IUPred score ≥ 0.2) to restrict the analysis to IDRs and refraining from any evolutionary filtering of results (Hagai et al. 2011). The predicted viral SLiMs were then used to predict DMIs using SLiMEnrich v1.5.1 (Idrees et al. 2018) through ELMc-Domain strategy (predicted viral SLiMs mapped to Pfam-domain-containing human partner proteins) available in SLiMEnrich (Idrees et al. 2018). SLiMEnrich works based on all ELM classes (e.g., LIG, DOC, DEG, MOD, CLV) by default. We restricted our analysis to proline-enriched motifs by downloading only LIG_SH3 motifs from ELM database and feeding it to SLiMEnrich, along with our PPI data. As the ELMc-Domain stringency is associated with a higher false discovery rate, we employed a computational structural approach to infer the likelihood of the identified interactions being real.
Molecular docking analysis
3D structure of the SH3 domain belonging to SPTAN1 protein was downloaded from the 3DID database [PDB ID: 5FW9 (1.55 Å)]. Optimization of the structure was done by first removing H2O molecules and then doing 3D protonation to change the state to ionization level. This was done using “Dock Prep” function in UCSF Chimera software (Pettersen et al. 2004). Moreover, energy minimization was done using parameters such as force field: MMFF94X + Solvation, gradient 0.05, and chiral constraint: current geometry using UCSF Chimera software (Pettersen et al. 2004). This minimized structure was then used for docking through CABS-dock server. CABS-dock, based on the Computational Alanine Scanning (CABS) model, emphasizes the flexibility and dynamics of ligand–receptor interactions during docking, providing a more realistic portrayal of molecular dynamics. Particularly adept with unbound or partially flexible structures, CABS-dock accommodates significant conformational changes in binding partners, making it suitable for predicting binding modes in flexible proteins. Its computational efficiency, driven by an efficient sampling algorithm, enables the exploration of extensive conformational spaces within reasonable timeframes, proving advantageous for high-throughput or large-scale docking studies (Kurcinski et al. 2019). Following protein docking, the docked complexes underwent hydrogen bonding and buried surface interaction area analysis using PDBePISA (protein interfaces, surfaces, and assemblies service) server (Krissinel and Henrick 2007). Subsequently, FoldX (Schymkowitz et al. 2005) was employed to determine the binding energies within the peptide–protein complexes. FoldX offers a rapid and quantitative assessment of interaction significance in maintaining protein and protein complex stability. In addition, alanine scanning was performed using FoldX, a widely utilized method (Stein and Aloy 2008; London et al. 2010), particularly for predicting hot spot residues. Initially, the “RepairPDB” command rectified residues with unfavorable torsion angles, van der Waals clashes, or total energy issues. Subsequently, the “complex_alascan” command was employed to assess the contribution of each residue on the peptide’s interaction interface to the protein and peptide binding. This was achieved by estimating the binding free energy differences (ΔΔG) following the mutation of the residue to Ala as shown.
where ΔGmutate and ΔGwild are the binding free energies of the mutant complex and the wild-type complex, respectively.
Conservation analysis
Polyprotein sequences of different serotypes of the Dengue virus were downloaded from the UniProt database (Apweiler et al. 2004). The UniProt Ids of sequences were serotype 1: P17763, serotype 2: P29991, serotype 3: Q6YMS4 and serotype 4: Q2YHF0. Sequences were aligned using the CLC workbench, and conservation analysis was done assessing the degree of similarity and preservation of specific sequences or structural motifs across different serotypes of Dengue virus. Conservation analysis helps researchers understand the evolutionary relationships, identify key functional elements, and inform the development of broad-spectrum treatments or vaccines.
Results
SLiM and DMI prediction
SLiMs were predicted using SLiMProb providing DENV-2 virus protein sequences involved in PPIs. SLiMs are mostly found in IDRs; therefore, the disordered masking feature was applied during SLiMProb run to predict new instances of known motifs that were in the IDRs. The predicted SLiMs along with PPIs were fed to SLiMEnrich v.1.5.1 to predict DMIs. SLiMEnrich works by mapping SLiMs to their interacting domain partners in the host proteins, giving the set of all possible DMIs. PPI data are then mapped to these DMIs to predict actual/real DMIs. A total of 23 DMIs were predicted involving 13 unique motifs instances from 3 ELM (i.e., LIG_SH3_1, LIG_SH3_2, LIG_SH3_3) in the DENV-2 polyprotein sequence (E, NS1, NS3 and NS5) interacting with SH3 domain of three unique human protein partners (Table 1).
Docking and interaction analysis
The identified DMIs were then docked using the CABS-dock server (Kurcinski et al. 2019), where motif instances (peptides) identified in different DENV-2 viral proteins were docked with the native SH3 domain structure (Receptor, PDB ID: 5FW9) and looked at close contact possible interactors, i.e., pairs of peptide/receptor residues closer than 4.0 Å in the selected complex. To see, if there were actual interactions between any of these residues, we conducted hydrogen bond analysis using PDBePISA (Krissinel and Henrick 2007) server by uploading our newly docked domain–motif complexes and identified hydrogen bond between only different residues of SH3 domain and DENV-2 proteins, suggesting a possible interaction between both residues. In simpler terms, we found specific connections (like hydrogen bonds and salt bridges) between different DENV-2 proteins, e.g., E, NS1, NS3, NS5, and SH3 receptor providing important details about how these proteins work and remain stable during a DENV-2 infection (Table 2).
Energy calculations and alanine scanning
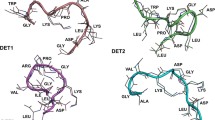
The FoldX package was utilized to compute the binding energy (ΔG) of the peptide–domain complex. In addition, alanine scanning mutagenesis, a widely adopted technique involving the systematic replacement of selected residues with alanine, was conducted using FoldX to identify crucial peptide residues for binding. This method systematically replaces selected residues in a target protein with alanine through site-directed mutagenesis. Alanine scanning facilitates the identification of specific amino acid residues crucial for peptide binding, as alanine substitutions eliminate side-chain interactions without affecting the main-chain conformation or introducing steric or electrostatic effects, preserving the native protein structure. Six hot spot residues were identified, including TYR 8, ASP 9, ASN 30, GLN 11, ARG 16, and TYR 10. A hot spot is defined as a residue exhibiting a ΔΔG ≥ 1.0 kcal/mol upon mutation to alanine, following the criteria established by Kortemme et al. (2004) and Grosdidier and Fernandez-Recio (2008). These residues exhibited various interaction types, with identified DENV-2 peptides forming strong hydrogen bond interactions. In addition, ARG 16 engaged in both hydrogen bond interactions and salt bridges. The reduction in peptide binding upon substituting an essential amino acid serves as a relative measure of its importance in the context of the substitution (Table 3, Fig. 1).

Hot spot residues of SH3 domain identified to be interacting with DENV-2 proteins
Conservation analysis of SH3 motifs
Polyprotein sequences representing diverse serotypes of the Dengue virus were sourced from the UniProt database (Apweiler et al. 2004). The UniProt identifiers for these sequences are as follows: P17763 for serotype 1, P29991 for serotype 2, Q6YMS4 for serotype 3, and Q2YHF0 for serotype 4. Employing the CLC Workbench, we aligned these sequences, keeping DENV-2 as reference and conducted a conservation analysis to evaluate the degree of similarity and preservation of specific sequences or structural motifs across the distinct Dengue serotypes. The envelope protein revealed residues, such as GLN at position 411 and THR at position 418, with conserved interactions, emphasizing their pivotal role in binding. Conversely, ILE at position 419 displayed non-conservation, highlighting the context-specific nature of certain interactions. In addition, VAL at position 420 exhibited specificity for serotype 4, adding a layer of complexity to the binding environment. In the NS1 protein, residues like GLN at position 810 and GLU at position 812 displayed strong conserved interactions, underscoring their significance in the viral life cycle. Notably, the identified hot spot residues in NS1, including SER at position 813 and PRO at position 814, further contribute to our understanding of the intricate molecular mechanisms governing DENV-2 infection. Within the NS3 protein, the conserved interactions of TRP at position 1480, SER at position 1484, and PRO at position 1485, contrasted with the non-conservation of GLU at position 1895, showcase the diverse landscape of interactions within the viral polyprotein. Meanwhile, ARG at position 1896 underscores the strategic conservation of certain residues critical for binding. The NS5 protein revealed a network of conserved interactions, with residues such as VAL at positions 2697, 3068, 3069, 3071, and others, indicating their essential roles in viral–host interactions. Notably, ARG at position 3070 exhibited serotype specificity, adding a layer of selectivity to the binding interactions. The diverse array of interactions identified in this study contributes to our understanding of the molecular basis of DENV-2 infection and provides a foundation for the design of targeted therapeutic interventions. These findings pave the way for further exploration into the specific roles of these residues in viral entry, replication, and immune evasion. In addition, the conservation patterns highlighted in this study may have implications for the development of broad-spectrum antiviral strategies targeting shared elements across different serotypes (Table 4, Fig. 2).

Conservation analysis of identified proline-enriched peptides in different Dengue serotypes, A envelope protein peptides, B NS1 protein peptides, C NS3 protein peptides, D NS5 protein peptides. Conservation is shown at the bottom in green. Dot (.) shows identical/conserved residues. Alanine (A) residues represented as grey, arginine (R) and lysine (K) as light blue, asparagine (D) and glutamate (E) as red, cysteine (C) and methionine (M) as yellow, glycine (G) as white, histidine (H) as purple, isoleucine (I), leucine (L), and valine (V) as green. Phenylalanine (F), tyrosine (T) as dark blue, proline (P) as flesh, serine (S) and threonine (T) as orange, tryptophan (W) as orchid (color figure online)
Discussion
Dengue infection has become a global threat to human health. Dengue virus has four serotypes, which hinders development of a successful vaccine (Idrees and Ashfaq 2012, 2013). The comprehensive analysis of Dengue Virus Serotype 2 (DENV-2) molecular interactions presented in this study sheds light on the viral–host protein-protein interactions (vhPPIs), providing valuable insights into potential therapeutic targets. Our exploration encompassed docking studies, interaction analyses, energy calculations, and alanine scanning mutagenesis, identifying specific residues critical for the stability and functionality of viral proteins during infection. The SH3 domain interactions with proline-rich motifs have been extensively studied in various viruses. SH3 domains are modular protein–protein interaction domains that specifically recognize and bind to proline-rich motifs, often found in viral and cellular proteins (Kaneko et al. 2008; Neuvonen et al. 2011; Zhao et al. 2021; Tossavainen et al. 2022). The presence of proline-rich motifs in DENV-2 proteins, such as those identified in this study, suggests a potential mechanism for viral proteins to exploit host cell machinery through SH3 domain interactions.
In this study, the application of SLiMProb, specifically considering IDRs through the disordered masking feature, yielded a comprehensive prediction of SLiMs within the DENV-2 polyprotein. SLiMs are often located in IDRs, and their prediction within these regions provides insights into potential interaction interfaces crucial for the virus life cycle. In this study, we focused on three proline-enriched motifs (LIG_SH3_1, LIG_SH3_2, LIG_SH3_3) within IDRs. The subsequent integration of predicted SLiMs into SLiMEnrich facilitated the prediction of DMIs, mapping SLiMs to their interacting domain partners in host proteins. This step is pivotal for understanding the broader context of vhPPIs, as it moves beyond individual motifs to predict the specific domains within host proteins that may be targeted by the virus. The focus on SH3 domains, known for their involvement in various cellular processes including signal transduction, emphasizes the strategic selection of host cellular components by the virus. The results showcased a total of 23 predicted DMIs involving 13 unique motif instances positioned in DENV-2 polyproteins, including E, NS1, NS3, and NS5, engaging with two unique domains in three distinct human protein partners (TP53BP2, SPTAN1, FYB). The variety of motifs and host protein partners underscore the intricacies of DENV-2 interactions, demonstrating the virus’s ability to target multiple cellular pathways. Molecular docking analyses then further assessed the predicted DMIs, providing a detailed view of specific peptide–residue interactions within the DENV-2 polyprotein and host SH3 domains. The strength and likeliness of interactions were further confirmed using hydrogen bond analysis. As we know, hydrogen bonds between specific amino acid residues in the protein and functional groups on the ligand contribute to the specificity of these interactions. This is crucial for processes such as enzyme–substrate binding or receptor–ligand interactions (Morrow and Zhang 2012). The incorporation of hot spots in subsequent analyses enhances our understanding of the contribution of specific residues to the overall binding free energy. The computation of binding energy (ΔΔG) using the FoldX package identified six hot spot residues crucial for binding. These residues exhibited various interaction types, such as hydrogen bonds and salt bridges, emphasizing their significance in maintaining the stability of vhPPIs. The concept of hot spots is instrumental in identifying key residues contributing significantly to the binding free energy. The distribution of energy across PPIs is not uniform, and a subset of residues, termed “hot spots,” significantly contributes to binding free energy (Clackson and Wells 1995; Keskin et al. 2005). Clackson and Wells’ pioneering study established the concept of hot spots, highlighting residues whose mutation to alanine results in a significant decrease in binding free energy (ΔΔG) (Clackson and Wells 1995). Hot spots occupy a fraction of the larger interface area and exhibit structural conservation (Bogan and Thorn 1998). Extensive research on protein–protein interfaces has identified specific hot spots. These regions, comprising only a small fraction of interfacial residues, play a crucial role in determining binding affinity and specificity (DeLano 2002). Structural conservation of hot spots is crucial in understanding the cooperative nature of these residues. Hot spots have been considered in drug design, serving as attractive targets for small molecule inhibitors to disrupt unwanted PPIs. The conservation of hot spots and their correlating binding affinity make them promising targets for drug development, offering avenues for small molecule inhibition of specific interactions. The consideration of hot spots in the design of small molecule inhibitors presents two key avenues for drug development. First, the presence of hot spots aids in predicting the binding site, guiding the docking, and screening of potential ligands. Second, the relatively less flexible nature of hot spots can be exploited in rigid docking approaches, improving the accuracy of protein docking by considering dominant conformational states obtained from molecular dynamics simulations (Gonzalez-Ruiz and Gohlke 2006; Morrow and Zhang 2012). The binding energy (ΔΔG) of the peptide–domain complexes was computed using the FoldX package. Furthermore, a conservation analysis was conducted across different serotypes of Dengue virus polyproteins (DENV 1-4), revealing the preservation of specific residues involved in interactions. The conservation patterns highlighted the significance of certain residues in the context of viral–host interactions, offering potential targets for therapeutic interventions. In general, the incorporation of hot spots in the analysis of DENV-2 molecular interactions enhances our understanding of the energetics and structural aspects of vhPPIs. This knowledge holds promise for the design of targeted therapeutic interventions aimed at disrupting key PPIs essential for DENV infection.
Conclusion
This study investigated interactions between the proline-enriched motif and SH3 domains in Dengue virus serotype 2 (DENV-2). Through a comprehensive analysis, we have uncovered significant insights into the role of proline-enriched motifs in mediating crucial interactions with SH3 domains, shedding light on DENV-2 interactions. Our findings suggest that the proline-enriched motifs play a pivotal role in facilitating interactions with SH3 domains, contributing to the network of protein–protein interactions that govern viral replication and host cell manipulation. Moreover, the identification and characterization of specific proline-enriched motifs involved in SH3 interactions offer potential targets for antiviral drug development. By disrupting these critical interactions, we may be able to impede the progression of DENV-2 infection and mitigate its impact on human health.
Data availability
The PPI data used in this study are available as supplementary file.
References
Apweiler R et al (2004) UniProt: the universal protein knowledgebase. Nucl Acids Res 32:D115-119. https://doi.org/10.1093/nar/gkh131
Bhatt S et al (2013) The global distribution and burden of dengue. Nature 496:504–507. https://doi.org/10.1038/nature12060
Bhatt P, Sabeena SP, Varma M, Arunkumar G (2021) Current understanding of the pathogenesis of dengue virus infection. Curr Microbiol 78:17–32. https://doi.org/10.1007/s00284-020-02284-w
Bogan AA, Thorn KS (1998) Anatomy of hot spots in protein interfaces. J Mol Biol 280:1–9. https://doi.org/10.1006/jmbi.1998.1843
Chala B, Hamde F (2021) Emerging and re-emerging vector-borne infectious diseases and the challenges for control: a review. Front Public Health 9:715759. https://doi.org/10.3389/fpubh.2021.715759
Chuang YC, Lin YS, Liu HS, Yeh TM (2014) Molecular mimicry between dengue virus and coagulation factors induces antibodies to inhibit thrombin activity and enhance fibrinolysis. J Virol 88:13759–13768. https://doi.org/10.1128/JVI.02166-14
Clackson T, Wells JA (1995) A hot spot of binding energy in a hormone–receptor interface. Science 267:383–386. https://doi.org/10.1126/science.7529940
DeLano WL (2002) Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol 12:14–20. https://doi.org/10.1016/s0959-440x(02)00283-x
Durmus Tekir S et al (2013) PHISTO: pathogen–host interaction search tool. Bioinformatics 29:1357–1358. https://doi.org/10.1093/bioinformatics/btt137
Edwards RJ, Palopoli N (2015) Computational prediction of short linear motifs from protein sequences. Methods Mol Biol 1268:89–141. https://doi.org/10.1007/978-1-4939-2285-7_6
Gadkari RA, Srinivasan N (2010) Prediction of protein–protein interactions in dengue virus coat proteins guided by low resolution cryoEM structures. BMC Struct Biol 10:17. https://doi.org/10.1186/1472-6807-10-17
Gonzalez-Ruiz D, Gohlke H (2006) Targeting protein–protein interactions with small molecules: challenges and perspectives for computational binding epitope detection and ligand finding. Curr Med Chem 13:2607–2625. https://doi.org/10.2174/092986706778201530
Gould CM, Kannan N, Taylor SS, Newton AC (2009) The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J Biol Chem 284:4921–4935. https://doi.org/10.1074/jbc.M808436200
Grosdidier S, Fernandez-Recio J (2008) Identification of hot-spot residues in protein–protein interactions by computational docking. BMC Bioinform 9:447. https://doi.org/10.1186/1471-2105-9-447
Guo C et al (2017) Global epidemiology of dengue outbreaks in 1990–2015: a systematic review and meta-analysis. Front Cell Infect Microbiol 7:317. https://doi.org/10.3389/fcimb.2017.00317
Hagai T, Azia A, Toth-Petroczy A, Levy Y (2011) Intrinsic disorder in ubiquitination substrates. J Mol Biol 412:319–324. https://doi.org/10.1016/j.jmb.2011.07.024
Idrees S, Ashfaq UA (2012) A brief review on dengue molecular virology, diagnosis, treatment and prevalence in Pakistan. Genet Vaccines Ther 10:6. https://doi.org/10.1186/1479-0556-10-6
Idrees S, Ashfaq UA (2013) RNAi: antiviral therapy against dengue virus. Asian Pac J Trop Biomed 3:232–236. https://doi.org/10.1016/S2221-1691(13)60057-X
Idrees S, Paudel KR (2023a) Bioinformatics prediction and screening of viral mimicry candidates through integrating known and predicted DMI data. Arch Microbiol 206:30. https://doi.org/10.1007/s00203-023-03764-w
Idrees S, Paudel KR (2023b) Proteome-wide assessment of human interactome as a source of capturing domain–motif and domain–domain interactions. J Cell Commun Signal. https://doi.org/10.1002/ccs3.12014
Idrees S, Perez-Bercoff A, Edwards RJ (2018) SLiMEnrich: computational assessment of protein–protein interaction data as a source of domain–motif interactions. PeerJ 6:e5858. https://doi.org/10.7717/peerj.5858
Idrees S, Paudel KR, Sadaf T, Hansbro PM (2023) How different viruses perturb host cellular machinery via short linear motifs. EXCLI 22:1113–1128
Kaneko T, Li L, Li SS (2008) The SH3 domain—a family of versatile peptide- and protein-recognition module. Front Biosci 13:4938–4952
Keskin O, Ma B, Nussinov R (2005) Hot regions in protein–protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol 345:1281–1294. https://doi.org/10.1016/j.jmb.2004.10.077
Kortemme T, Kim DE, Baker D (2004) Computational alanine scanning of protein–protein interfaces. Sci STKE 2004:pl2. https://doi.org/10.1126/stke.2192004pl2
Kraemer MU et al (2015) The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. Elife 4:e08347. https://doi.org/10.7554/eLife.08347
Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. https://doi.org/10.1016/j.jmb.2007.05.022
Kurcinski M et al (2019) CABS-dock standalone: a toolbox for flexible protein–peptide docking. Bioinformatics 35:4170–4172. https://doi.org/10.1093/bioinformatics/btz185
London N, Movshovitz-Attias D, Schueler-Furman O (2010) The structural basis of peptide–protein binding strategies. Structure 18:188–199. https://doi.org/10.1016/j.str.2009.11.012
Morrow JK, Zhang S (2012) Computational prediction of protein hot spot residues. Curr Pharm Des 18:1255–1265. https://doi.org/10.2174/138161212799436412
Mosca R, Ceol A, Stein A, Olivella R, Aloy P (2014) 3did: a catalog of domain-based interactions of known three-dimensional structure. Nucl Acids Res 42:D374-379. https://doi.org/10.1093/nar/gkt887
Neuvonen M, Kazlauskas A, Martikainen M, Hinkkanen A, Ahola T, Saksela K (2011) SH3 domain-mediated recruitment of host cell amphiphysins by alphavirus nsP3 promotes viral RNA replication. PLoS Pathog 7:e1002383. https://doi.org/10.1371/journal.ppat.1002383
Palanichamy Kala M, St John AL, Rathore APS (2023) Dengue: update on clinically relevant therapeutic strategies and vaccines. Curr Treat Options Infect Dis 15:27–52. https://doi.org/10.1007/s40506-023-00263-w
Pettersen EF et al (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
QIAGEN CLC Genomics Workbench. https://digitalinsights.qiagen.com/
Ren X, Hurley JH (2011) Proline-rich regions and motifs in trafficking: from ESCRT interaction to viral exploitation. Traffic 12:1282–1290. https://doi.org/10.1111/j.1600-0854.2011.01208.x
Saksela K, Cheng G, Baltimore D (1995) Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J 14:484–491. https://doi.org/10.1002/j.1460-2075.1995.tb07024.x
Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L (2005) The FoldX web server: an online force field. Nucl Acids Res 33:W382-388. https://doi.org/10.1093/nar/gki387
Shelton H, Harris M (2008) Hepatitis C virus NS5A protein binds the SH3 domain of the Fyn tyrosine kinase with high affinity: mutagenic analysis of residues within the SH3 domain that contribute to the interaction. Virol J 5:24. https://doi.org/10.1186/1743-422X-5-24
Srikiatkhachorn A (2009) Plasma leakage in dengue haemorrhagic fever. Thromb Haemost 102:1042–1049. https://doi.org/10.1160/TH09-03-0208
Stein A, Aloy P (2008) Contextual specificity in peptide-mediated protein interactions. PLoS ONE 3:e2524. https://doi.org/10.1371/journal.pone.0002524
Thomas SJ, Endy TP (2011) Vaccines for the prevention of dengue: development update. Hum Vaccin 7:674–684. https://doi.org/10.4161/hv.7.6.14985
Torres-Flores JM, Reyes-Sandoval A, Salazar MI (2022) Dengue vaccines: an update. BioDrugs 36:325–336. https://doi.org/10.1007/s40259-022-00531-z
Tossavainen H et al (2022) Structure of SNX9 SH3 in complex with a viral ligand reveals the molecular basis of its unique specificity for alanine-containing class I SH3 motifs. Structure 30:828-839 e826. https://doi.org/10.1016/j.str.2022.03.006
Venkatakrishnan AJ, Kayal N, Anand P, Badley AD, Church GM, Soundararajan V (2020) Benchmarking evolutionary tinkering underlying human-viral molecular mimicry shows multiple host pulmonary-arterial peptides mimicked by SARS-CoV-2. Cell Death Discov 6:96. https://doi.org/10.1038/s41420-020-00321-y
WHO (2021) Dengue and severe dengue. https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue
Zhao Z et al (2021) Evolutionary plasticity of SH3 domain binding by Nef proteins of the HIV-1/SIVcpz lentiviral lineage. PLoS Pathog 17:e1009728. https://doi.org/10.1371/journal.ppat.1009728
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. The authors received no funding for this study.
Author information
Authors and Affiliations
Contributions
Conceptualization and design: SI; performed analysis: MB, SI; writing: SI, MB, KRP, RM; review and editing: SI, KRP. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Additional information
Communicated by Yusuf Akhter.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Banik, M., Paudel, K.R., Majumder, R. et al. Prediction of virus–host interactions and identification of hot spot residues of DENV-2 and SH3 domain interactions. Arch Microbiol 206, 162 (2024). https://doi.org/10.1007/s00203-024-03892-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-024-03892-x