
Abstract
Summary
Children with sickle cell disease (SCD) have the potential for extensive and early-onset bone morbidity. This study reports on the diversity of bone morbidity seen in children with SCD followed at three tertiary centers. IV bisphosphonates were effective for bone pain analgesia and did not trigger sickle cell complications.
Introduction
To evaluate bone morbidity and the response to intravenous (IV) bisphosphonate therapy in children with SCD.
Methods
We conducted a retrospective review of patient records from 2003 to 2019 at three Canadian pediatric tertiary care centers. Radiographs, magnetic resonance images, and computed tomography scans were reviewed for the presence of avascular necrosis (AVN), bone infarcts, and myositis. IV bisphosphonates were offered for bone pain management. Bone mineral density was assessed by dual-energy X-ray absorptiometry (DXA).
Results
Forty-six children (20 girls, 43%) had bone morbidity at a mean age of 11.8 years (SD 3.9) including AVN of the femoral (17/46, 37%) and humeral (8/46, 17%) heads, H-shaped vertebral body deformities due to endplate infarcts (35/46, 76%), and non-vertebral body skeletal infarcts (15/46, 32%). Five children (5/26, 19%) had myositis overlying areas of AVN or bone infarcts visualized on magnetic resonance imaging. Twenty-three children (8/23 girls) received IV bisphosphonate therapy. They all reported significant or complete resolution of bone pain. There were no reports of sickle cell hemolytic crises, pain crises, or stroke attributed to IV bisphosphonate therapy.
Conclusion
Children with SCD have the potential for extensive and early-onset bone morbidity. In this series, IV bisphosphonates were effective for bone pain analgesia and did not trigger sickle cell complications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sickle cell disease (SCD) is the most common inherited hemoglobinopathy whereby sickling of hemoglobin S (HbS) causes red blood cells to polymerize, occlude vasculature, hemolyze, and induce oxidative stress [1]. At the skeletal level, chronic anemia promotes marrow hyperplasia and local hypoxia can induce bone necrosis. This creates an environment of high bone turnover and weakened architecture with further susceptibility to vaso-occlusive episodes and bone tissue death [2, 3]. As such, SCD has significant effects on bone structure including widened marrow cavity, thinning of the cortex, and low bone mineral density (BMD) [2]. Chronic bone morbidity is one of the most common complications of SCD including osteoporosis, vertebral body and non-vertebral body skeletal infarcts, and avascular necrosis (AVN) [2, 4].
AVN is a severe chronic bone complication in SCD that progresses to joint collapse and functional impairment. It is most common in the femoral head followed by the humeral head. AVN often presents in childhood, with approximately 28% of children with SCD experiencing femoral head AVN and the consequent impact on quality of life, physical capacity, and school functioning [5]. Femoral head AVN is present in half of adults with SCD with 77% progressing to joint collapse [6, 7]. Nearly a quarter of adults will require a total hip arthroplasty by their third decade of life [6, 8]. International guidelines for monitoring SCD recommend evaluating all children and adults for hip pain and if present, to consider imaging for AVN [9].
Bone infarcts are areas of localized necrosis that often present during vaso-occlusive episodes as acute, severe pain, and localized erythema, but they can also be found incidentally on imaging [10]. Long bones and vertebral endplates are common locations for bone infarcts; however, any skeletal bone can be affected [11]. Vertebral endplate infarcts cause H-shaped vertebral deformities due to sickling and ischemia in the vulnerable long arterial blood vessels that feed the central vertebral body [12, 13]. Extramedullary hematopoiesis leads to protrusion of the disk into the infarcted central vertebral body, creating the characteristic biconcave deformity seen in H-shaped vertebral bodies [11]. Myositis is thought to be a rare and often overlooked complication of SCD that occurs due to infection or due to underlying bone marrow changes and bone infarct [14, 15]. It is attributed to vaso-occlusive episodes in the muscle and can have a fulminant presentation with acute fasciitis, necrotizing myositis, and compartment syndrome [16]. Vitamin D deficiency is present in a third of individuals with SCD which may further impair their bone health [17].
Despite the frequency of bone morbidity in SCD, there is currently no standard of care for its prevention or treatment. Bisphosphonates are anti-resorptive medications used to treat osteoporosis, and act as an analgesic for bone pain in a variety of settings, including fibrous dysplasia, cancer-related AVN, and bone metastases [18,19,20,21]. Bisphosphonates may therefore be useful in patients with SCD to treat bone morbidity and for pain control. Zoledronic acid was administered to nine adults in a study evaluating vertebral involvement in sickle cell bone disease, but the indication, response to treatment, and adverse effects of bisphosphonates were not reported [22]. Another group demonstrated improved BMD T-scores at 6 and 12 months after treatment of alendronate in adults with SCD and osteoporosis [23]. Major clinical questions remain unanswered such as the indication for bisphosphonate therapy in SCD, efficacy of reducing or halting bone morbidity, and safety of bisphosphonate use. Given the limited information about skeletal morbidity and its treatment in SCD, we aim to describe the skeletal features of pediatric SCD bone morbidity and the response to intravenous (IV) bisphosphonate therapy including pain management, side effects, impact on BMD, and bone histomorphometry.
Materials and methods
Study design and setting
This was a retrospective study of children < 18 years of age with SCD and bone morbidity followed at three Canadian tertiary pediatric centers as part of routine clinical care. Affected children and adolescents with SCD from 2015 to 2020 were identified at the Stollery Children’s Hospital, University of Alberta, Edmonton, Alberta, and their bone imaging reports were reviewed. Data from 2013 to 2020 were also collected retrospectively from children with SCD who were referred to the Pediatric Bone and Metabolism Clinic at the Centre Hospitalier Universitaire Sainte-Justine (CHU St. Justine), Montreal, Québec. In addition, data from 2001 to 2020 were collected retrospectively from children with SCD who were referred to the Genetic and Metabolic Bone Disease Clinic at the Children’s Hospital of Eastern Ontario (CHEO), Ottawa, Ontario. Years over which data were collected differed from center to center due to access to electronic medical records and the establishment of bone and metabolism clinics. Currently, these centers follow approximately 750 children with SCD. The study was approved by the Research Ethics Boards (REBs) in each of the participating institutions (REB approval numbers: Stollery Children’s Hospital—Pro 0090451, CHEO—20190342, and CHU St. Justine—2021–3219).
Clinical data
Study data were collected and managed using the REDCap electronic data capture tool hosted at the University of Alberta. Clinical data were extracted using a structured data form by clinicians familiar with the electronic health system. Clinical data included sex, age, height, and weight Z-scores based on WHO growth curves, hemoglobinopathy genotype, whether hydroxyurea was prescribed at the time of bone morbidity diagnosis, age at initial radiographic diagnosis of bone morbidity, and age upon presentation to a bone health clinic. The diagnosis of SCD was confirmed by electrophoresis and genetic testing for HbS variants. Biochemical markers of bone metabolism were collected at time of identification of bone morbidity, at time of referral to a bone health clinic, and pre-/post-IV bisphosphonate therapy. This included serum calcium, phosphate, parathyroid hormone, 25-hydroxyvitamin D, and alkaline phosphatase. Biochemical tests were completed as per local laboratory protocols. Serum 25-hydroxyvitamin D levels were defined as sufficiency > 50 nmol/L, insufficiency 30–50 nmol/L, and deficiency < 30 nmol/L [24].
Identification of bone morbidity and myositis
Bone morbidity was identified by reports from pediatric radiologists at each tertiary care center including radiographs, computed tomography (CT), and magnetic resonance imaging (MRI). AVN and bone infarcts were diagnosed by MRI and/or radiographs. MRI and radiographs were ordered in the context of severe pain during acute vaso-occlusive episodes, persistent pain, or decreased joint range of motion. The presence of myositis was also extracted from MRI reports. CT was used to identify cranial bone infarcts and localized hematomas in patients presenting with acute swelling and proptosis. All imaging was carried out at the discretion of the treating physicians (CG, MER, NA, AB, YP, ES, and LMW), targeting areas that were linked to loss of functional mobility and/or bone pain.
Treatment of bone morbidity with intravenous bisphosphonate therapy
IV bisphosphonate therapy was offered at CHEO and CHU St. Justine as a part of local standard of care for chronic bone pain causing functional limitation and data were collected from these two centers. IV bisphosphonates were not routinely offered at the Stollery Children’s Hospital; one patient at the Stollery Children’s Hospital received IV bisphosphonate after consultation with a bone and metabolism expert at CHEO. Data on IV bisphosphonate therapy included the formulation of bisphosphonate, indication, duration of therapy, analgesic response to treatment, and reported side effects. IV bisphosphonate therapy was administered based on each center’s routine bone pain/osteoporosis protocol. Patients were treated with one of three regimens: (1) IV pamidronate (annual dose, 4.5 to 9 mg/kg body weight/year given as 1 mg/kg/day for each of 3 days [9 mg/kg/year], or 1.5 mg/kg on a single day [4.5 mg/kg/year], both every 4 months), (2) IV zoledronic acid (annual dose 0.05–0.1 mg/kg body weight, divided into two doses every 6 months), or (3) an initial dose of pamidronate followed by zoledronic acid to minimize first infusion side effects, given studies that suggest zoledronic acid may have more side effects than pamidronate [25, 26]. In some patients, zoledronic acid was given more frequently than every 6 months for pain management, but in all cases, the maximum annual dose was not exceeded. 25-Hydroxyvitamin D levels were optimized through vitamin D supplementation to a minimum of 50 nmol/L prior to IV bisphosphonate therapy, and it was a pre-requisite that a serum calcium level was normal prior to IV bisphosphonate therapy. Patients were followed by phone or clinic visits afterwards.
Bisphosphonate infusion side effects were assessed by phone visits 24–72 h after the infusion. Pain was assessed by phone and at follow-up clinic visits. This included objective assessments of pain scales from 0 to 10, and/or subjective assessments of significant improvement or complete resolution of pain. Complete resolution was defined as pain that had fully resolved while significant improvement was defined as baseline pain that had resolved but would still occur with intense physical activity. Charts were reviewed for the duration of bisphosphonate treatment to assess for possible complications related to infusions. Clinic notes and hospital admissions up to two weeks post-bisphosphonate infusion were reviewed for painful crises, clinical or radiographic signs of stroke, concerns of hemolytic crises, or other clinical concerns that could be provoked SCD complications. Osteonecrosis of the jaw (ONJ) is a rare but potentially severe side effect of bisphosphonates. For the duration of bisphosphonate infusions, clinic notes and dental radiograph reports were reviewed for observations of jaw pain or dental problems.
Evaluation of the response to treatment — quantitative skeletal imaging and bone histomorphometry
BMD was collected pre- and post-IV bisphosphonate therapy. Lumbar spine (vertebrae L1–L4) and total body less head BMD were measured by dual-energy X-ray absorptiometry (DXA) in the anterior–posterior direction (Lunar Prodigy and Lunar iDxa; General Electronic; Madison, WI, USA). Lunar raw results were cross-calibrated to Hologic units using calibration factors from a previous multi-center study [27]. Lumbar spine (LS) and total body less head (TBLH) areal BMD (aBMD) raw results were then transformed to age-, sex-, and race-specific Z-scores using previously published normative data which included black children, the ethnicities relevant to this report [28]. LS and TBLH aBMD Z-scores were adjusted for height-for-age Z-score (HAZ) using adjustment equations [29].
Trans-iliac bone biopsies were performed on one patient prior to IV bisphosphonate therapy and approximately 2 years after therapy initiation. Biopsies were taken at a site 2 cm posterior to the superior anterior iliac spine 4 or 5 days after dual labeling with demeclocycline (15–20 mg/kg per day taken orally for 2 days and repeated for 2 more days after a 10-day free interval). Biopsy preparation and histomorphometric analyses were performed as described previously [30]. Measurements were carried out using a digitizing table with OsteoMeasure software (Osteometrics, Ing. Atlanta, GA, USA). Results were compared to the reference data of healthy age- and sex-matched controls and expressed as percentages of the average value [30].
Statistical analyses
Results of clinical, biochemical, and radiological data were analyzed descriptively. Categorical values were summarized using frequency and percentage. Normality was assessed using the Shapiro–Wilk test. Normally distributed continuous variables were summarized using mean and standard deviation (SD). Non-normally distributed continuous variables were summarized using median and interquartile range (IQR). Statistical analyses were carried out using STATA SE software (version 17.0, TX, USA).
Results
Clinical characteristics
Forty-six children (20 girls and 26 boys) with SCD and bone morbidity were included in the study (Table 1). Seventeen children were identified at CHEO, 16 children at CHU St. Justine, and 13 at the Stollery Children’s Hospital. The mean age of initial bone morbidity diagnosis was 11.8 years (SD 3.9) with the youngest diagnosed at 2.4 years of age. Thirty-seven of these 46 children (80%) were referred for a specialized bone health assessment at a mean age of 13.3 years (SD 3.3); the remainder were identified at the Stollery Children’s Hospital before a bone and metabolism clinic was available. The majority of children had HbSS genotype (40/46, 87%) and the remainder had HbS/beta-0 thalassemia (3/46, 7%), HbSC (2/46, 4%), or HbSS/Arab-Indian haplotype (1/46, 2%). Thirty-five of 46 children (76%) were prescribed hydroxyurea prior to diagnosis of bone morbidity. Baseline 25-hydroxyvitamin D levels were assessed upon referral. Vitamin D insufficiency was prevalent with a median 25-hydroxyvitamin D level of 43.0 nmol/L (n = 46, IQR 27.0, 72.8). Vitamin D status was as follows: 29% of children (13/46) had vitamin D deficiency (25-hydroxyvitamin D < 30 nmol/L) and 30% of children (14/46) had vitamin D insufficiency (25-hydroxyvitamin D 30–50 nmol/L), consistent with previous reports [17].
Bone infarcts and joint avascular necrosis
Bone morbidity observed in children with SCD included femoral and humeral head AVN, vertebral body infarcts resulting in H-shaped vertebral deformities, and non-vertebral body skeletal infarcts (Fig. 1, Table 1). Twenty-eight of 46 children (61%) had two or more types of bone morbidity. There were two children who sustained non-vertebral fractures, both pre-bisphosphonate therapy. One child sustained a humerus fracture at 5 years of age when he fell down the stairs and a humerus fracture on the contralateral side when he fell off a bike at eight years of age. The second child sustained a proximal radial metaphyseal fracture while playing at one year of age (also pre-bisphosphonate therapy).

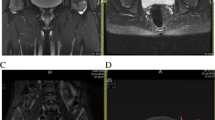
a Lumbar H-shaped vertebral body deformities. b Vertebral bone infarcts visualized on T2-weighted MRI. Note the patchy increased signal intensity throughout the thoracic, lumbar, and sacral vertebral bodies. c Patchy and serpiginous increased T2 signal intensity on MRI reflecting bone infarcts in the bilateral acetabuli and proximal femoral diaphysis, and early femoral head AVN. Note also the right hip effusion. d Osteosclerotic lesions in the right femur, bilateral tibia, and right fibula, reflecting bone infarcts. e Chronic humeral head AVN visualized on T1-weighted MRI as increased epiphyseal marrow T1 signal with serpiginous margins. f CT demonstrating a left orbital infarct with subgaleal hematoma. Note the proptosis of the left eye and tissue swelling around the left orbit. g Bilateral femoral head AVN on STIR MRI. Note the patchy increased STIR signal in the femoral heads, additional focus of increased signal in right femoral necks, and small right hip effusion. Abbreviations: MRI, magnetic resonance imaging; CT, computed tomography; SCD, sickle cell disease; AVN, avascular necrosis
Bone infarcts of the vertebral body were the most common bone morbidity, seen in 76% of children with bone morbidity (35/46). Twenty-four of 46 children (69%) with vertebral body infarcts also had additional bone morbidity; vertebral infarcts were present in 59% of children with femoral head AVN, 75% of children with humeral head AVN, and 73% of children with non-vertebral body skeletal infarcts. Non-vertebral body skeletal infarcts were seen in 15 of 46 children (33%), with locations including the femoral, tibial, fibular, ulnar and radial diaphysis, pelvis, vertebral posterior elements, orbital bone, and mandible. Thirteen of 16 children (81%) with non-vertebral body skeletal infarcts had multiple sites of infarct (as shown in Fig. 1 c and d). Femoral head AVN was present in 17 of 46 children (37%), with 53% (9/17) of these having bilateral involvement. Humeral head AVN was present in eight of 46 children (17%) with 38% (3/8) of these children having bilateral involvement.
Pain assessments were reported in 29 children and pain was present in 27 children (93%), localized to the back, hip, shoulder, arm, or knee. Twelve children with H-shaped vertebral bodies had pain assessments and ten of these children reported back pain (83%). Hip pain was present in ten of 11 children (91%) with femoral head AVN. All three children (100%) with humeral head AVN had shoulder pain. Eight of 29 children (28%) who underwent pain assessments had also reported functional impairment including missed school days and exercise limitation.
Myositis
Twenty-six children had MRI for bone morbidity and of these, five (19%) had myositis overlying areas of AVN or bone infarct. Locations of myositis included soft tissue overlying the femoral, humeral and ulnar diaphysis, and the sacrum. One child had extensive soft tissue edema of his proximal thigh, pelvis, and lower abdomen in the setting of widespread infarcts of the proximal femoral diaphysis, pelvis, and sacrum. Creatinine kinase (CK) levels (N 40–220 U/L) were evaluated at the time of imaging in two children. One child had a normal CK (34 U/L) while the other had a significantly elevated CK (666 U/L). We note that one child had a follow-up MRI a year later and the myositis had resolved.
Response to intravenous bisphosphonates
Treatment with IV bisphosphonates varied by center, and the indication largely depended on the degree of painful bone morbidity. As CHEO and CHU St. Justine gained experience with IV bisphosphonates, IV bisphosphonate administration became standard local practice for pain and 23 of 46 children (50%) were treated with IV bisphosphonates. Indications included bone pain and AVN. Fifteen of 23 children (65%) received zoledronic acid, seven of 23 children (30%) received pamidronate, and one of 23 children (4%) received an initial dose of pamidronate followed by zoledronic acid. The mean duration of treatment was 1.5 years (SD 0.8) in the 15 of 23 children who have completed their IV bisphosphonate therapy. IV bisphosphonate therapy was ongoing in eight of 23 children at the time of the preparation of this manuscript.
Children who received IV bisphosphonate therapy had a mean 25-hydroxyvitamin D level of 58 nmol/L (n = 23, SD 34.9). Ten of these 23 children (43%) had vitamin D insufficiency/deficiency (25-hydroxyvitamin D ≤ 50 nmol/L) with a mean 25-hydroxyvitamin D level of 27.3 nmol/L (SD 12.1). 25-Hydroxyvitamin D levels were optimized through vitamin D supplementation to a minimum of 50 nmol/L prior to IV bisphosphonate therapy.
IV bisphosphonates were well-tolerated and some of the adverse effects included acute phase reaction (n = 4/23, 17%), and one case of symptomatic hypocalcemia that was treated for five days with oral calcium and calcitriol. There were no episodes of painful or hemolytic crises, stroke, or other sickle cell complications associated with IV bisphosphonate therapy. There were no reports of osteonecrosis of the jaw for the duration of bisphosphonate infusions. Pain improvement was noted within 1–6 months of IV bisphosphonate therapy and persisted throughout the duration of bisphosphonate therapy (mean 1.5 years, SD 0.8), after which children were discharged from clinic or referred to adult endocrinology. No child was re-referred for recurrence of pain.
Eleven children had pain assessments following initiation of bisphosphonates. Ten of the 11 children (91%) reported significant or complete resolution of pain six months after initiation of IV bisphosphonate therapy. Six of 11 children (55%) reported complete resolution of pain after two years of IV bisphosphonate therapy. Four children of 11 children (36%) had significant improvement on their pain which was defined as resolution of their baseline pain, but still intermittent sickle cell painful episodes or their baseline pain were reduced from 9/10 to 1–2/10. The one child who did not have resolution or improvement of pain only had follow-up for five months after receiving one infusion. He also had near total collapse of his femoral head and was anticipated to undergo a hip arthroplasty. The sample size was not sufficient to determine if there was a measurable difference in the effect of IV pamidronate vs zoledronic acid on bone pain. See Supplemental Data for several cases that demonstrate the severity of SCD bone morbidity and response to IV bisphosphonate therapy.
Potential for femoral head and vertebral body reshaping in SCD
We observed reshaping of femoral head AVN and vertebral bodies in three skeletally immature children (Fig. 2). Interestingly, two children demonstrated reshaping of the femoral head after extensive collapse due to AVN. One child had femoral head collapse at 11.4 years of age, and he did not receive IV bisphosphonate therapy as per his center’s practice (Stollery Children’s Hospital). He had spontaneous and complete reshaping by 16.5 years of age (Fig. 2 a and b). One child had femoral head collapse at 8.3 years of age. In this patient, pamidronate was initiated when femoral head AVN was diagnosed, and infusions were ongoing when partial femoral head AVN reshaping was observed at 10.4 years of age (Fig. 2 c and d). He remains on bisphosphonate infusions and reshaping has been ongoing. One adolescent with delayed puberty had multiple H-shaped vertebral bodies at 14.9 years of age, before his pubertal growth spurt, and by 18.8 years of age, he had reshaping of the T5 vertebra with stabilization and partial reshaping of T7/T8/T12 (Fig. 2 e and f). He received three doses of zoledronic acid between 15 and 17 years of age. He has since transitioned to adult care, and it is unknown if his reshaping continued after cessation of bisphosphonate infusions. Other children received IV bisphosphonate therapy; however, follow-up lateral spine radiographs were not ordered routinely as a part of post-IV bisphosphonate monitoring.

Femoral head and vertebral body reshaping in SCD. a, b Left femoral head AVN and acetabular sclerosis in an 11.4-year-old boy (a). He had spontaneous and complete reshaping of the femoral head by 16.5 years of age (b). c, d Right femoral head AVN in an 8.3-year-old (c). He received pamidronate for two years and at 10.4 years of age, he had partial reshaping of the femoral head with progressive resorption of the necrotic fragment (d). e, f A 14.9-year-old boy had multiple H-shaped vertebral bodies (e). He received three doses of zoledronic acid and at 18.8 years of age, reshaping of T5 and stabilization with partial reshaping of T7/T8/T12 are evident (f)
Bone mineral density and trans-iliac bone histomorphometry
Fourteen children had pre- and post-IV bisphosphonate DXA BMD measurements of the LS and TBLH (Table 2), and the average age at completion of IV bisphosphonate therapy was 15.9 (SD 3.28). Post-IV bisphosphonate DXA did not show statistically significant gains in height-adjusted LS or TBLH aBMD Z-scores (LS HAZ aBMD Z-score 0.4, SD 0.5, TBLH HAZ BMD Z-score 0.4, SD 0.6). Trans-iliac bone histomorphometry was available for one patient, an adolescent male before and after two years of zoledronic acid therapy, at 16.5 and 18.5 years of age (Fig. 3). He was approaching adult height during the interval between paired biopsies, having grown 3.5 cm over the 2.5 years. Cortical width was 46% of the healthy mean pre-zoledronic acid and increased to 62% of the healthy average post-IV bisphosphonate. His bone formation rate/bone surface was elevated at 265% of the healthy mean pre-IV bisphosphonate and fell to 27% of the healthy average post-zoledronic acid. 25-Hydroxyvitamin D levels pre- and post-IV bisphosphonate therapy were above the lower limit of acceptability at 56 and 53 nmol/L, respectively.

Trans-iliac bone samples pre- and post-bisphosphonate. Trans-iliac bone biopsies in an adolescent boy at 16.5 years of age pre-bisphosphonate (a) and 18.5 years of age post-bisphosphonate (b). Trans-iliac bone biopsy post-bisphosphonate demonstrated a 16% increase in cortical width, with raw values indicated below each of the external (left) and internal (right) cortices
Discussion
Our findings demonstrate that a wide range of SCD bone morbidity can present in childhood, and we report successful pain management with IV bisphosphonate therapy. Vertebral deformities due to endplate infarction were the most common type of bone morbidity, followed by femoral head AVN, non-vertebral body skeletal infarcts, and humeral head AVN. We illustrate the diversity of non-vertebral body skeletal infarct locations in SCD including long bone diaphyses, vertebral posterior elements, pelvis, and cranial bones, including cases of a mandibular bone infarct that resulted in facial nerve palsy and an orbital infarct that led to a subgaleal hematoma (see Supplemental Data). Our findings of AVN in children with SCD are consistent with other reports demonstrating that AVN may present during childhood, including frequent bilateral joint involvement [5, 31,32,33]. In contrast to other reports, we found a greater proportion of children with vertebral deformities in our cohort.
We were also surprised to observe a large proportion of children with myositis as 19% of children with MRIs for AVN or bone infarct had local myositis over the area of bone disease. Myositis has been previously reported in SCD; however to our knowledge, our study is the first to describe myositis in children who underwent MRI for bone morbidity. The frequency of myositis seen on MRI demonstrates that such a finding may not be a rare complication, despite the sparsity of literature on this observation [14, 15]. CK is an enzyme released during muscle inflammation and elevated CK has been seen in SCD myositis [15, 16]. While CK was not frequently measured during vaso-occlusive episodes or during assessment of bone morbidity in our cohort, we describe one child with significantly elevated CK in the setting of multiple bone infarcts and overlying myositis. Further studies are needed to explore the frequency of myositis in vaso-occlusive episodes, the etiology of the myositis, the utility of CK as a marker of myositis in this setting, and whether myositis is associated with more severe bone morbidity.
At our institutions, IV bisphosphonate administration has become more widely used in SCD. We observed that IV bisphosphonates consistently improved or completely resolved pain in our cohort and were well tolerated. Importantly, our experience suggests that IV bisphosphonate therapy in SCD is safe as none of our patients experienced pain crises, hemolytic crises, stroke, or other sickle cell complications that might have been attributed to bisphosphonate administration. Although osteonecrosis of the jaw in children receiving IV bisphosphonate therapy has not been described, the possibility of spontaneous vaso-occlusive events of the jaw in sickle cell disease [34] merits prudence with respect to this theoretical complication. Dental extractions should be carried out prior to starting bisphosphonate therapy wherever possible, good oral hygiene and regular dental evaluations should be maintained, and monitoring for ONJ complications should be routine [35]. Bone histomorphometry in an adolescent before and after IV zoledronic acid therapy demonstrated a high rate of bone turnover that declined on IV bisphosphonate therapy, as expected given the drug’s mechanism of action. Cortical width increased by 16% in this older adolescent, in keeping with the known anti-resorptive effect of IV bisphosphonate therapy on the endocortical surface [36]. Children who had post-bisphosphonate BMDs did not demonstrate a significant aBMD increase. This may be expected as this group was nearing final adult height at completion of IV bisphosphonate therapy, and they still have SCD as an ongoing risk factor for osteoporosis.
Beyond the reduction of pain following IV bisphosphonate administration, we also observed spontaneous (that is, medication-unassisted) femoral head reshaping following AVN. Reshaping of both the femoral heads and vertebral bodies also occurred in two other children, both of whom had been treated with IV bisphosphonate therapy. IV bisphosphonate therapy promotes vertebral body reshaping in pediatric osteoporosis populations such as osteogenesis imperfecta and Duchenne muscular dystrophy (the latter only occurs if glucocorticoid therapy is withdrawn and normal growth resumes) [37,38,39]. Partial femoral head reshaping following AVN has been seen in a previous report of five children with SCD AVN, without medical or surgical intervention [40]. To our knowledge, our experience is the first report of vertebral body reshaping in SCD. These findings demonstrate the potential for structural improvement in SCD bone morbidity although it is unclear why some cases recover while others progress to further bone destruction. Overall, a larger prospective study is required to better understand the potential for recovery from structural collapse in pediatric SCD.
From a clinical perspective, the most important observation arising from our study is that IV bisphosphonates appeared to significantly reduce the severity of chronic bone pain in our cohort. Over 50% of adults with SCD will experience chronic pain with significant consequences including depression, anxiety, impaired sleep, and decreased quality of life [41]. Bone pain is a complex pathway involving increased osteoclast differentiation and activation, release of cytokines, recruitment of immune cells, local hydrogen ion production, and nociceptor sensitization [42,43,44,45]. As pyrophosphate analogues, bisphosphonates are taken up by osteoclasts causing osteoclast apoptosis, thereby suppressing pain arising from bone resorption while also decreasing osteoclast release of hydrogen ions and microenvironment acidification, an important sensitizer of nociceptor. In addition, bisphosphonates have immune-modulating actions including inhibition of macrophage activation interfering with pro-inflammatory mediators [42, 46]. The latter is presumed to be the primary mechanism of bisphosphonate-mediated bone analgesia.
Whether IV bisphosphonates can positively impact the evolution of the SCD itself remains to be explored. Vaso-occlusive crises, the clinical signature of SCD, are caused by Hb sickling in the microvasculature which leads to localized ischemia–reperfusion injury [2, 14, 16]. Skeletal vaso-occlusive episodes lead to inflammation and cytokine release, increased receptor-activator of nuclear factor kappa β ligand (RANKL) signalling, osteoclast recruitment, and osteoclast activation [47,48,49]. A mouse model of SCD showed increased osteoclast activity and recruitment as well as interleukin-6 production in response to hypoxia-reperfusion injury and osteoclast activity, and recruitment was markedly reduced by zoledronic acid [48]. Thus, vaso-occlusive episodes activate pathways, similar to those which trigger bone pain, providing rationale for future studies to explore the impact of IV bisphosphonates on the development of such sickle cell–related vaso-occlusive crises in murine and human models, and their impact on muscle and bone inflammatory and osteoclastogenesis pathways.
The limitations of this study include its retrospective design and therefore data collection challenges inherent to retrospective studies, as follows. First, referral practices for pain and bone morbidity assessments and consideration for IV bisphosphonate therapy were pursued more pro-actively at CHEO and CHU St. Justine compared with the Stollery Children’s Hospital. This reflected the fact that there were no specific referral guidelines in place at the Stollery Children’s Hospital until after this study was completed. These differences in referral/practice patterns introduced heterogeneity into the number of patients who were treated with IV bisphosphonates across the three centers. In addition, reports of pain were not assessed in a standardized fashion across the three centers and there was a lack of consistent objective pain score data. As a result, our study provides a description of the potential bone morbidities that can arise from pediatric SCD, their frequencies, and their response to IV bisphosphonate therapy as evaluated pragmatically during routine clinical care. Ultimately, a randomized controlled trial would be necessary to definitively determine whether IV bisphosphonates have measurable benefit to skeletal health compared with disease-targeted treatment alone.
Another consideration in the interpretation of our results is that the chart review encompassed 20 years of clinical care, and practice has changed significantly over that period. Hydroxyurea has more widely become a standard of care and most children with recurrent pain crises receive this therapy. Since 2014, it has further been recommended to introduce hydroxyurea by 9–12 months of age even in the absence of symptoms [9]. It remains uncertain, however, whether hydroxyurea alters the bone morbidity trajectory in SCD. In particular, there are conflicting reports on whether hydroxyurea may be associated with reduced or increased risk of AVN [8, 50, 51]. In addition, new therapies for SCD are on the forefront, including gene therapy (clinicaltrials.gov NCT03282656), and some patients undergo bone marrow transplant. The impact of these targeted approaches on bone morbidity also requires further study. Hematopoietic stem cell transplantation is an evolving curative therapy for SCD, and gene therapy trials are investigating the infusion of modified autologous stem cells that express anti-sickling beta globulins. It remains to be seen if these curative therapies could modify the course of bone morbidity in SCD [52].
The observations in this study have important implications for clinical care of children with SCD, as they suggest the need for early and ongoing monitoring for bone morbidity. The possibility of spontaneous femoral head and vertebral body reshaping in this population warrants further understanding around which children have the potential for structural recovery, and the safety of IV bisphosphonate therapy in children with SCD at published doses is supported by our report. Moreover, these data suggest IV bisphosphonates have a positive effect on the treatment of painful bone morbidity, an observation that requires further testing in a randomized, controlled setting.
References
Piel FB, Steinberg MH, Rees DC (2017) Sickle cell disease. N Engl J Med 376:1561–1573. https://doi.org/10.1056/NEJMra1510865
Osunkwo I (2013) An update on the recent literature on sickle cell bone disease. Curr Opin Endocrinol Diabetes Obes 20:539–546. https://doi.org/10.1097/01.med.0000436192.25846.0b
Green M, Akinsami I, Lin A et al (2015) Microarchitectural and mechanical characterization of the sickle bone. J Mech Behav Biomed Mater 48:220–228. https://doi.org/10.1016/j.jmbbm.2015.04.019
Almeida A, Roberts I (2005) Bone involvement in sickle cell disease. Br J Haematol 129:482–490. https://doi.org/10.1111/j.1365-2141.2005.05476.x
Adekile AD, Gupta R, Al-Khayat A, et al (2018) Risk of avascular necrosis of the femoral head in children with sickle cell disease on hydroxyurea: MRI evaluation. Pediatr Blood Cancer 1–5https://doi.org/10.1002/pbc.27503
Milner PF, Kraus AP, Sebes JI et al (1991) Sickle cell disease as a cause of osteonecrosis of the femoral head. N Engl J Med 325:1476–1481. https://doi.org/10.1056/NEJM199111213252104
Hernigou PH, Habibi A, Bachir D, Galacteros F (2006) The natural history of asymptomatic osteonecrosis of the femoral head in adults with sickle cell disease. J bone Jt Surg 88:2565
Adesina O, Brunson A, Keegan THM, Wun T (2017) Osteonecrosis of the femoral head in sickle cell disease: prevalence, comorbidities, and surgical outcomes in California. Blood Adv 1:1287–1295. https://doi.org/10.1182/bloodadvances.2017005256
Yawn BP, Buchanan GR, Afenyi-Annan AN et al (2014) Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA - J Am Med Assoc 312:1033–1048
Ware HE, Brooks AP, Toye R, Berney SI (1991) Sickle cell disease and silent avascular necrosis of the hip. J Bone Joint Surg Br 73:947–949
Ejindu VC, Hine MAL, Mohammad F et al (2007) Musculoskeletal manifestations of sickle cell disease. Radiographics 27:1005–1022. https://doi.org/10.1007/BF00357859
Jaremko JL, Siminoski K, Firth GB et al (2015) Common normal variants of pediatric vertebral development that mimic fractures: a pictorial review from a national longitudinal bone health study. Pediatr Radiol 45:593–605
Aguilar C, Vichinsky E, Neumayr L (2005) Bone and joint disease in sickle cell disease. Hematol Oncol Clin North Am 19:929–941. https://doi.org/10.1016/j.hoc.2005.07.001
Turaga LP, Boddu P, Kipferl S et al (2017) Myonecrosis in sickle cell anemia case study. Am J Case Rep 18:100–103. https://doi.org/10.12659/AJCR.900538
Tageja N, Racovan M, Valent J, Zonder J (2010) Myonecrosis in sickle cell anemia—overlooked and underdiagnosed. Case Rep Med 2010:1–3. https://doi.org/10.1155/2010/659031
Mani S, Duffy TP (1993) Sickle myonecrosis revisited. Am J Med 95:525–530. https://doi.org/10.1016/0002-9343(93)90336-n
Grégoire-Pelchat P, Alos N, Ribault V et al (2018) Vitamin D intake and status of children with sickle cell disease in Montreal, Canada. J Pediatr Hematol Oncol 40:531–536. https://doi.org/10.1097/MPH.0000000000001306
Wong RKS, Wiffen PJ (2009) Bisphosphonates for the relief of pain secondary to bone metastases. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD002068
August KJ, Dalton A, Katzenstein HM et al (2011) The use of zoledronic acid in pediatric cancer patients. Pediatr Blood Cancer. https://doi.org/10.1002/pbc.22681
Javaid MK, Boyce A, Appelman-Dijkstra N et al (2019) Best practice management guidelines for fibrous dysplasia/McCune-Albright syndrome: a consensus statement from the FD/MAS international consortium. Orphanet J Rare Dis 14:1–17. https://doi.org/10.1186/s13023-019-1102-9
Padhye B, Dalla-Pozza L, Little D, Munns C (2016) Incidence and outcome of osteonecrosis in children and adolescents after intensive therapy for acute lymphoblastic leukemia (ALL). Cancer Med. https://doi.org/10.1002/cam4.645
De Franceschi L, Gabbiani D, Giusti A et al (2020) Development of algorithm for clinical management of sickle cell bone disease: evidence for a role of vertebral fractures in patient follow-up. J Clin Med 9:1601. https://doi.org/10.3390/jcm9051601
BackaCico T, Kullolli S, Harizi I et al (2013) SAT0381 The use of bisphosphonates in the treatment of low bone mineral density in adults with sickle cell anemia. Ann Rheum Dis 71(600):3–601. https://doi.org/10.1136/annrheumdis-2012-eular.3327
Munns C, Shaw N, Kiely M et al (2016) Global consensus recommendations on prevention and management of nutrition rickets. Pediatriya 56:60–63. https://doi.org/10.1016/j.yped.2016.08.050
Ward LM, Konji VN, Ma J (2016) The management of osteoporosis in children. Osteoporos Int 27:2147–2179. https://doi.org/10.1007/s00198-016-3515-9
Nasomyont N, Hornung LN, Gordon CM, Wasserman H (2019) Outcomes following intravenous bisphosphonate infusion in pediatric patients: a 7-year retrospective chart review. Bone 121:60–67. https://doi.org/10.1016/j.bone.2019.01.003
Halton J, Gaboury I, Grant R et al (2009) Advanced vertebral fracture among newly diagnosed children with acute lymphoblastic leukemia: results of the Canadian Steroid-Associated Osteoporosis in the Pediatric Population (STOPP) research program. J Bone Miner Res 24:1326–1334. https://doi.org/10.1359/jbmr.090202
Zemel BS, Kalkwarf HJ, Gilsanz V et al (2011) Revised reference curves for bone mineral content and areal bone mineral density according to age and sex for black and non-black children: results of the bone mineral density in childhood study. J Clin Endocrinol Metab 96:3160–3169. https://doi.org/10.1210/jc.2011-1111
Zemel BS, Leonard MB, Kelly A et al (2010) Height adjustment in assessing dual energy x-ray absorptiometry measurements of bone mass and density in children. J Clin Endocrinol Metab 95:1265–1273. https://doi.org/10.1210/jc.2009-2057
Glorieux FH, Travers R, Taylor A et al (2000) Normative data for iliac bone histomorphometry in growing children. Bone 26:103–109. https://doi.org/10.1016/S8756-3282(99)00257-4
Adekile AD, Gupta R, Yacoub F et al (2001) Avascular necrosis of the hip in children with sickle cell disease and high Hb F: magnetic resonance imaging findings and influence of α-thalassemia trait. Acta Haematol 105:27–31. https://doi.org/10.1159/000046529
Aguilar CM, Neumayr LD, Eggleston BE et al (2005) Clinical evaluation of avascular necrosis in patients with sickle cell disease: children’s hospital oakland hip evaluation scale - a modification of the harris hip score. Arch Phys Med Rehabil 86:1369–1375. https://doi.org/10.1016/j.apmr.2005.01.008
Mesleh Shayeb A, Smeltzer MP, Kaste SC, et al (2018) Vaso-occlusive crisis as a predictor of symptomatic avascular necrosis in children with sickle cell disease. Pediatr Blood Cancer 1–4https://doi.org/10.1002/pbc.27435
Roland-Billecart T, Maes JM, Vieillard MH et al (2021) Avascular necrosis of the jaw resulting from sickle cell disease. J Oral Med Oral Surg 27:2020–2022. https://doi.org/10.1051/mbcb/2020039
Khan AA, Morrison A, Hanley DA et al (2015) Diagnosis and management of osteonecrosis of the jaw: a systematic review and international consensus. J Bone Miner Res 30:3–23. https://doi.org/10.1002/jbmr.2405
Rauch F, Travers R, Plotkin H, Glorieux FH (2002) The effects of intravenous pamidronate on the bone tissue of children and adolescents with osteogenesis imperfecta. J Clin Invest 110:1293–1299. https://doi.org/10.1172/JCI0215952
Sbrocchi AM, Rauch F, Jacob P et al (2012) The use of intravenous bisphosphonate therapy to treat vertebral fractures due to osteoporosis among boys with Duchenne muscular dystrophy. Osteoporos Int 23:2703–2711. https://doi.org/10.1007/s00198-012-1911-3
Palomo T, Fassier F, Ouellet J et al (2015) Intravenous bisphosphonate therapy of young children with osteogenesis imperfecta: skeletal findings during follow up throughout the growing years. J Bone Miner Res 30:2150–2157. https://doi.org/10.1002/jbmr.2567
Glorieux FH, Bishop NJ, Plotkin H et al (1998) Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med 339:947–952. https://doi.org/10.1056/NEJM199810013391402
Itzep NP, Jadhav SP, Kanne CK, Sheehan VA (2019) Spontaneous healing of avascular necrosis of the femoral head in sickle cell disease. Am J Hematol 94:E160–E162. https://doi.org/10.1002/ajh.25453
Osunkwo I, O’Connor HF, Saah E (2020) Optimizing the management of chronic pain in sickle cell disease. Hematol Am Soc Hematol Educ Progr 2020:562–569. https://doi.org/10.1182/hematology.2020000143
Iolascon G, Moretti A (2019) Pharmacotherapeutic options for complex regional pain syndrome. Expert Opin Pharmacother 20:1377–1386. https://doi.org/10.1080/14656566.2019.1612367
Figura N, Smith J, Yu HHM (2018) Mechanisms of, and adjuvants for, bone pain. Hematol Oncol Clin North Am 32:447–458. https://doi.org/10.1016/j.hoc.2018.01.006
Taddio A, Zennaro F, Pastore S, Cimaz R (2017) An update on the pathogenesis and treatment of chronic recurrent multifocal osteomyelitis in children. Pediatr Drugs 19:165–172. https://doi.org/10.1007/s40272-017-0226-4
Hofmann SR, Kapplusch F, Girschick HJ et al (2017) Chronic recurrent multifocal osteomyelitis (CRMO): presentation, pathogenesis, and treatment. Curr Osteoporos Rep 15:542–554. https://doi.org/10.1007/s11914-017-0405-9
Kuźnik A, Październiok-Holewa A, Jewula P, Kuźnik N (2020) Bisphosphonates—much more than only drugs for bone diseases. Eur J Pharmacol 866https://doi.org/10.1016/j.ejphar.2019.172773
Barbu EA, Mendelsohn L, Samsel L, Thein SL (2020) Pro-inflammatory cytokines associate with NETosis during sickle cell vaso-occlusive crises. Cytokine 127:154933. https://doi.org/10.1016/j.cyto.2019.154933
Dalle Carbonare L, Matte A, Teresa Valenti M, et al (2015) Hypoxia-reperfusion affects osteogenic lineage and promotes sickle cell bone diseasehttps://doi.org/10.1182/blood-2015-04
Giordano P, Urbano F, Lassandro G, Faienza MF (2021) Mechanisms of bone impairment in sickle bone disease. Int J Environ Res Public Health 18:1–11. https://doi.org/10.3390/ijerph18041832
Mahadeo KM, Oyeku S, Taragin B et al (2011) Increased prevalence of osteonecrosis of the femoral head in children and adolescents with sickle-cell disease. Am J Hematol 86:806–808. https://doi.org/10.1002/ajh.22103
Adekile AD, Gupta R, Al-Khayat A, et al (2019) Risk of avascular necrosis of the femoral head in children with sickle cell disease on hydroxyurea: MRI evaluation. Pediatr Blood Cancer 66https://doi.org/10.1002/pbc.27503
Ware RE, de Montalembert M, Tshilolo L, Abboud MR (2017) Sickle cell disease. Lancet 390:311–323
Acknowledgements
MER was supported by a Junior Clinical Research Chair from the University of Ottawa. LMW was supported by a Tier 1 Clinical Research Chair from the University of Ottawa and the Children’s Hospital of Eastern Ontario Research Institute.
Funding
NA has received funding support from the Sickle Cell Disease Association of Canada.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
The study was approved by the University of Alberta, CHEO, and CHU St. Justine Research Ethics boards. The study has been performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Conflicts of interest
CG has been a consultant to Ultragenyx and IPSEN. LMW has been a consultant to, and participated in clinical trials with, Ultragenyx, Novartis, and Amgen (with funds to Dr. Ward’s institution). NA has participated in clinical trials with Novartis, Amgen, Europharm, and Levo therapeutics. MER has been a consultant to Ultragenyx and IPSEN and has received a research grant from Ascendis Biopharma. YDP has been a consultant to and participated in clinical trials with Novartis, Agios, Forma Pharmaceutic, and Principia. RG has been a consultant to IPSEN and Pfizer and has received research grants from Novo-Nordisk and Ascendis. PDE, JLJ, AB, VNK, MP, MS, and ES declare that they have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Grimbly, C., Escagedo, P.D., Jaremko, J.L. et al. Sickle cell bone disease and response to intravenous bisphosphonates in children. Osteoporos Int 33, 2397–2408 (2022). https://doi.org/10.1007/s00198-022-06455-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-022-06455-2