
Abstract
Incretin hormones, principally glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1(GLP-1), potentiate meal-stimulated insulin secretion through direct (GIP + GLP-1) and indirect (GLP-1) actions on islet β-cells. GIP and GLP-1 also regulate glucagon secretion, through direct and indirect pathways. The incretin hormone receptors (GIPR and GLP-1R) are widely distributed beyond the pancreas, principally in the brain, cardiovascular and immune systems, gut and kidney, consistent with a broad array of extrapancreatic incretin actions. Notably, the glucoregulatory and anorectic activities of GIP and GLP-1 have supported development of incretin-based therapies for the treatment of type 2 diabetes and obesity. Here we review evolving concepts of incretin action, focusing predominantly on GLP-1, from discovery, to clinical proof of concept, to therapeutic outcomes. We identify established vs uncertain mechanisms of action, highlighting biology conserved across species, while illuminating areas of active investigation and uncertainty that require additional clarification.
Graphical abstract

Similar content being viewed by others


History and discovery of incretin action
In 1902, the first hormone to regulate pancreatic exocrine secretion, secretin, was revealed [1]. It was suspected that there would also be hormonal regulation of metabolism, and after the discovery of insulin, researchers began to think about incretins, substances that could regulate insulin secretion similar to the regulation of exocrine function by excretins [2]. Since insulin could not yet be measured, these studies required complex cross-circulation experiments [3], and incretin research hardly advanced. In 1964, the radioimmunoassay for insulin enabled demonstration that oral intake of glucose resulted in greater insulin secretion than i.v. infusion (i.e. the incretin effect) [4, 5], presumably because of intestinal release of incretin hormone(s), and the hunt to identify the(se) hormone(s) began. By 1971, John Brown, who trained in the laboratory of Victor Mutt in Stockholm, had isolated a candidate hormone from porcine intestine, a peptide of 42 amino acids, which was first named as gastric inhibitory polypeptide (GIP) [6]. In 1973, Brown and John Dupré tested its possible incretin activity in humans using a purified porcine GIP preparation [7]. Indeed, GIP cause a marked potentiation of glucose-stimulated insulin secretion, and it was suggested that the peptide should be renamed glucose-dependent insulinotropic polypeptide (allowing the acronym to be retained). Subsequent careful ‘mimicry’ experiments (where levels of endogenous hormone concentrations are mimicked by exogenous infusion) established GIP as an incretin hormone, which was able to virtually fully explain the incretin effect [8].
The interest in the new incretin hormone fostered hope that incretins could promote insulin secretion in people with diabetes; it was therefore disappointing when Krarup and colleagues demonstrated in 1987 that porcine GIP did not stimulate insulin secretion in people with type 2 diabetes [9], a finding that extinguished interest in the incretin concept for many. In related studies, Michael Nauck observed in 1986 that the incretin effect was lost or severely reduced in people with type 2 diabetes [10], making it unlikely that incretins would be useful therapeutically. However, it was suspected early on that incretin action was exerted by more than a single hormone. Indeed, numerous new peptides capable of stimulating insulin secretion were isolated from gut extracts in the laboratory of Erik Jorpes and Victor Mutt in Stockholm, and the wealth of new peptides inspired Werner Creutzfeldt to establish a set of criteria fulfilled by incretin hormone activity.
A candidate incretin would have to be secreted into the circulation upon glucose ingestion; neuropeptides (many of the insulinotropic peptides isolated in Mutt’s laboratory and elsewhere turned out to be neuropeptides), therefore, would not qualify. It would also have to stimulate insulin secretion in relevant concentrations and at relevant plasma glucose concentrations, defined as those observed in relation to glucose intake [11]. Simultaneously, evidence was accumulating that GIP was not the only incretin, as immunoneutralisation of circulating GIP only partially reduced the incretin effect [12]. Moreover, Kjeld Lauritsen and colleagues [13] showed that the incretin effect (determined as the difference between insulin responses to isoglycaemic oral and i.v. glucose challenges) in individuals with small intestinal resections of various lengths did not correlate with GIP responses after oral glucose, but rather with the length of distal small intestine still in continuity. Thus, there had to be another incretin.
Interest focused on the products of the intestinal endocrine L cells [14], which react with antibodies towards glucagon, and glucagon was known to stimulate insulin secretion [15]. It turned out that the L cells produced a peptide of 69 amino acids, initially named glicentin, [16] which was assumed to represent at least part of ‘proglucagon’ [17] because it contains the full glucagon sequence at positions 33–61. Thus, glicentin (now known to be a partial form of a proglucagon precursor) (Fig. 1) was shown to be cleaved differentially in the alpha cells of the pancreatic islets (releasing glucagon) and in the L cells (releasing glicentin and a fragment corresponding to proglucagon 33–69 [designated ‘oxyntomodulin’]) [18]. Glicentin did not stimulate insulin secretion; oxyntomodulin did but with low potency [19] and the circulating concentrations were too low to cause stimulation of insulin secretion. At the same time, molecular biology experiments with cell-free translation of islet mRNA indicated that the real proglucagon precursor was much larger than the 69 amino acids that make up glicentin [20], engendering the question: what peptide sequences were encoded in the rest of proglucagon, designated the so called ‘major proglucagon fragment’ (MPGF)?

(a) A schematic of the structure of proglucagon is shown. Processing of proglucagon-derived peptides occurs in a tissue-specific manner, with glucagon and MPGF generated in the pancreas, and glicentin, oxyntomodulin, GLP-1, GLP-2 and intervening peptide (IP)-2 generated in the intestine and brain. (b) The incretin effect is defined as the augmentation of insulin secretion when nutrients or glucose is administered into the gut, resulting in a greater increment in insulin secretion, relative to an isoglycaemic exposure achieved through parenteral or i.v. glucose infusion. The incretin effect is diminished in people with type 2 diabetes, largely reflecting impairment of beta cell function. Asterisks denote significant difference (p≤0.05). The original conversion factor used was 1 mU/l insulin = 7.3 pmol/l. Adapted from Nauck et al [10] with permission. This figure is available as part of a downloadable slideset
By the early 1980s, Joel Habener and colleagues deduced, based on mRNA extracted from the endocrine islet organ (Brockmann bodies) of the anglerfish, the full structure of anglerfish proglucagon and found that a glicentin-like sequence was located at the N-terminal part, but an additional glucagon-like sequence was identified in the carboxy-terminal region [21,22,23]. This exciting finding was followed by identification of the mammalian proglucagon sequence in 1983. Graeme Bell and colleagues identified the sequences of hamster and human proglucagon [24, 25], with other labs rapidly elucidating the bovine [26] and rat [27] proglucagon sequences. Unexpectedly, mammalian MPGF contained two glucagon-like peptide sequences (Fig. 1). However, the deduced human peptides, corresponding to proglucagon 72–107 and 126–160, were inactive with respect to insulin secretion. In contrast, native endogenous glucagon-like peptide-1 (GLP-1), extracted from human and porcine gut on the basis of reactivity with antibodies, turned out to be potently insulinotropic in a perfused pancreas preparation [28]. The endogenous intestinal GLP-1 sequence corresponded to a truncated form of GLP-1, formed by a monobasic cleavage between residues 77 and 78 in proglucagon, resulting in an amidated peptide with the full sequence of PG 78–106 amide [29]. At the same time, studies in Boston demonstrated that a similarly truncated peptide, GLP-1(7–37), stimulated glucose-dependent insulin release directly from islet cells [30] and the perfused rat pancreas [31]. A formal ‘incretin analysis’ in humans was published in December of 1987 from Steven Bloom’s laboratory [32], clearly demonstrating the incretin-like insulinotropic potential of GLP-1, which was actually greater than that of GIP studied in the same experiments. Comparative analyses based on radioimmunoassay measurements revealed that both peptides, GLP-1 and GIP, stimulated insulin secretion at the beginning of the meal, before any appreciable increase in plasma glucose. Furthermore, their insulinotropic action was potentiated by glucose elevations corresponding to those induced by the intake of a meal [33].
Excitingly, and in contrast to GIP, GLP-1 was able to stimulate insulin secretion to virtually normal levels during a hyperglycaemic clamp in individuals with type 2 diabetes [34]. Another feature of GLP-1 was its action to inhibit glucagon secretion [35], whereas GIP stimulated glucagon secretion [36]. These properties of GLP-1 addressed both the inadequate insulin secretion and the hyperglucagonaemia characteristic of type 2 diabetes, known to underlie the pathophysiology of hyperglycaemia. Therapeutic proof of concept was provided by the observation that 4 h of GLP-1 infusion normalised blood glucose levels in people with long-standing type 2 diabetes, associated with increased insulin and decreased glucagon levels; importantly, these actions of GLP-1 were attenuated as glucose concentrations approached 5 mmol/l [37].
Physiology of GIP and GLP-1 in islet cells and the incretin effect
The incretin effect is of major importance for normal glucose tolerance. This was demonstrated in experiments where increasing amounts of glucose were given to volunteers either orally or (on a separate day) by i.v. infusions, yielding identical glucose excursions [38]. In these experiments, plasma glucose excursions were virtually identical despite the much larger oral vs i.v. doses, revealing a mechanism that would prevent hyperglycaemia despite the greater amounts of ingested oral glucose. The incretin effect, therefore, maintains physiologically normal glycaemic excursions, regardless of the doses of glucose or carbohydrates ingested. The underlying mechanism was revealed to be greater insulin secretion with oral vs i.v. glucose [38]. Subsequent studies showed that the secretion of the incretin hormones followed the same pattern in healthy control participants and in people with type 2 diabetes, supporting the comparatively greater stimulation of insulin secretion with ingested glucose [39].
Elucidation of the physiology of endogenous GLP-1 was enabled by use of exendin(9–39), a selective antagonist of the mammalian GLP-1 receptor [40]. Similarly, an amidated fragment of GIP, GIP(3–30)NH2, was demonstrated to be a selective and potent GIP receptor antagonist in humans [41]. Blocking both receptors by co-infusion of the incretin receptor antagonists deteriorated glucose tolerance [42], illustrating the importance of the incretin system in humans. From the measurements of insulin secretion rates, it could be calculated that GIP was responsible for about half of the insulin responses to oral glucose, while GLP-1 and glucose alone were responsible for the remaining 30% and 20%, respectively [43].
The receptors for GIP and GLP-1 are both expressed in islet beta cells, explaining their direct insulinotropic effects. GIP receptors (GIPR) and GLP-1 receptors (GLP-1R) are also expressed in subsets of alpha cells (but with GLP-1R at very low levels) [44]. Intra-islet glucagon, acting through the beta cell GLP-1R and glucagon receptors, may also contribute to the incretin effect, perhaps reconciling findings of low circulating levels of GLP-1 yet substantial meal-stimulated GLP-1R-dependent potentiation of insulin secretion [45, 46]. Similarly, incretin-like actions of GIP may also proceed indirectly through amino acids and potentiation of GIPR-dependent stimulation of glucagon secretion from alpha cells [47]. Despite structural and functional similarity of the incretin receptors, it remains unclear why GIPR signalling is defective in type 2 diabetes; the lack of GIP responsivity has been ascribed to hyperglycaemia-associated switching of G protein signalling in beta cells [48].
Circulating levels of GIP and GLP-1 in healthy individuals and people with insulin resistance, type 2 diabetes or obesity
The majority of GLP-1-producing L cells are located in the distal gut, whereas GIP is synthesised within K cells predominantly localised to the duodenum, with a small subset of enteroendocrine cells producing both hormones [49]. GIP and GLP-1 are secreted at low levels in the fasted or interprandial state, and circulating levels rise briskly following a meal or glucose ingestion [50]. Glucose activates enteroendocrine hormone secretion via the sodium–glucose cotransporter 1 (SGLT1) [51]. Lipids and fatty acids also stimulate incretin secretion via fatty acid receptors such as G-protein-coupled receptor (GPR) 40 and GPR119 [52, 53]. The transporters and receptors coupling protein and amino acids to GIP and GLP-1 secretion are less well understood, and are mediated through diverse mechanisms, including activation of the calcium sensing receptor [54]. Oral administration of amino acids stimulates GIP as well as GLP-1 secretion in healthy humans [55].
The presence or absence of type 2 diabetes does not meaningfully impair the secretion of either hormone. In large population studies of people with type 2 diabetes, a modest impairment of GLP-1 secretion was identified [56]. Obesity, on the other hand, is frequently associated with decreased GLP-1 secretion. In animals, obesity does not impair GIP or GLP-1 secretion from isolated perfused preparations of small intestine, and weight loss does not influence GLP-1 secretion [57]. In humans with obesity, levels of GLP-1, and to a lesser extent GIP, increased after diet-induced weight loss in some [58, 59], but not all [60], studies.
GIP and GLP-1 glucoregulatory actions in type 2 diabetes
Insulin responses to both GIP and GLP-1 infused to mimic normal postprandial concentrations during a hyperglycaemic clamp were markedly diminished in people with long-standing type 2 diabetes, and were improved, but not normalised, after 4 weeks of insulin therapy [61]. Supraphysiological levels of GLP-1, but not GIP, achieved via infusion restored insulin responses to normal levels [62]. In experiments administering GIP and GLP-1 individually or together, only GLP-1 infusions lowered plasma glucose and suppressed glucagon, whereas simultaneous infusion of GIP abolished the inhibition of glucagon observed with GLP-1 [63].
Short term studies employing a 5 h infusion of GIP in individuals with type 2 diabetes who had already been treated with long-acting GLP-1R agonists (GLP-1RAs) had little acute effect on appetite or energy expenditure, and plasma glucose actually increased due to enhanced glucagon levels [64]. Similarly, continuous subcutaneous administration of native GIP for 6 days in men with type 1 diabetes increased hepatic steatosis but had little impact on a wide range of metabolic variables, including markers of bone resorption, biomarkers of inflammation, and body weight [65]. Nevertheless, unimolecular GIP–GLP-1 co-agonism is an effective therapy for type 2 diabetes, associated with weight loss and reduction of fat mass, in rats, mice, monkeys and humans [66].
A balanced GIP–GLP-1 co-agonist (equal GIP and GLP-1 potency) developed by DiMarchi and colleagues was studied in people with type 2 diabetes over 12 weeks but did not demonstrate beneficial metabolic actions beyond those achieved in an open label control arm of people treated with liraglutide [67]. In contrast, in 2018, the long-acting GIP–GLP-1 co-agonist tirzepatide was found to have substantial glucose-lowering and weight loss properties, to a greater extent than that achieved by GLP-1R agonism with dulaglutide [68], findings replicated in Phase III trials comparing tirzepatide with semaglutide in people with type 2 diabetes [69]. Tirzepatide is a biased agonist favouring cAMP production over β-arrestin recruitment, which may reduce receptor internalisation and potentially enhance GLP-1 signalling [70]
GLP-1 and control of gastric emptying: normal physiology and diabetes
GLP-1 inhibits gastrointestinal motility and gastric secretion (Fig. 2). During GLP-1 infusions, the stomach completely loses its tone and becomes flaccid [71]. In pig experiments, GLP-1 inhibited efferent vagal activity [72]. In humans, vagotomy abolished the inhibitory effects of GLP-1 on gastric emptying, as well as the inhibition of the vagal marker, pancreatic polypeptide [73]. Consistent with these findings, genetic attenuation of mouse GLP-1R expression within autonomic (including vagal) neurons reveals a role for physiological GLP-1R signalling in the basal control of gastric emptying [74]. The inhibitory effect of native GLP-1 on gastric emptying shows rapid tachyphylaxis (within hours) and disappears upon continued stimulation [75]. The inhibitory actions of long-acting GLP-1RAs on gastric emptying in people with type 2 diabetes or obesity is more variable, with substantial tachyphylaxis evident in some [76], but not all [77], studies. Reduction of gastric emptying contributes substantially to the postprandial glucose control achieved with the short acting GLP-1RAs such as exenatide twice daily (exenatide BID) and lixisenatide. GLP-1 also attenuates small bowel motility, enhancing time for enzymatic nutrient digestion and absorption. Following gastrectomy, a higher rate of nutrient transit with energy-dense liquid meals produces a large GLP-1 secretory response, resulting in diarrhoea and, in some cases, hypoglycaemia [78]. In contrast, GIP does not impact gut motility or gastric emptying.

Physiological vs pharmacological actions of GLP-1. Nutrients stimulate GLP-1 secretion from gut L cells, where it may engage local GLP-1R+ sensory afferent neurons within the gut originating in the nodose ganglion, which then activate neurons within the nucleus of the solitary tract (also known as the nucleus tractus solitarii; NTS). Alternatively, intrahepatic GLP-1R+ neurons, including a subset within the portal system, may be activated by circulating GLP-1and communicate ascending GLP-1R-dependent signals to the brain. Ascending fibres from solitary tract neurons may generate reflexes in the hypothalamus, and descending signals from the brainstem or hypothalamus may activate vagal motor neurons that send stimulatory or inhibitory impulses to the pancreas and the gastrointestinal tract. Exogenously administered or endogenous circulating GLP-1 may act independently of the gut–brain axis to directly engage GLP-1Rs in the brain to reduce food intake and body weight, and in the pancreas to stimulate insulin and inhibit glucagon secretion. This figure is available as part of a downloadable slideset
GIP, GLP-1 and appetite: physiology and central nervous system mechanisms
The approval of liraglutide 3 mg once daily for people with obesity was followed by the development of semaglutide 2.4 mg once weekly [79], resulting in up to 18% weight loss after 68 weeks [80]. GLP-1Rs are widely distributed in the central nervous system (CNS) (Fig. 3) [81], and the use of site-specific brain injections, together with chemogenetics targeting distinct GLP-1R+ populations, illustrates considerable redundancy in GLP-1R+ sites coupled to reduction of food intake. Although autonomic and peripheral GLP-1R+ neurons, including those within the nodose ganglion, may partially contribute to the weight loss actions of peripherally administered GLP-1RA [74, 82], ablation of CNS neurons targeted by nestin-Cre or Wnt1-Cre abolishes the anorectic actions of peripherally administered GLP-1RA in mice [74, 83]. Whether GLP-1RAs simply engage CNS GLP-1Rs accessible via circumventricular organs or are actively transported to brain regions via GLP-1Rs within the blood brain barrier, such as those expressed in tanycytes [84], remains a topic of ongoing investigation (Fig. 2).

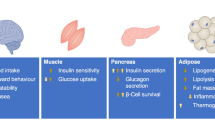
The metabolic actions of GLP-1 in different organs and cell types. The actions shown are those with translational relevance predominantly conserved across species. For comparison of the actions of GIP vs GLP-1, please see review by Hammoud and Drucker [174]. Adapted from Drucker [100] with permission from Elsevier. This figure is available as part of a downloadable slideset
The physiological effects of endogenous intestinal GLP-1 on food intake and body weight are comparatively modest. Although GLP-1 released from the gut may interact with local GLP-1R+ sensory nerve fibres communicating signals to the parabrachial nucleus that enable meal termination [85], the importance of these circuits for long-term control of body weight has not been established. Transient interruption of GLP-1R signalling using antagonists of the GLP-1R increases food intake and weight gain in animals [86] and humans [87]. However, GLP-1 receptor knockout mice do not become obese [88], and deletion of the mouse Gcg gene (encoding GLP-1) from the entire intestine, resulting in a 90% reduction of circulating GLP-1, does not perturb food intake or weight gain [89].
Chemogenetic activation of hypothalamic GIPR+ neurons reduces food intake in mice [90], and GIP reduces food intake when given peripherally (in mice) or via intracerebroventricular injection [91, 92]. However, its anorectic effects and impact on weight loss are comparatively modest relative to those observed with GLP-1R agonism. GIP is not known to interact with the peripheral autonomic nervous system, but its appetite inhibiting effect in rodents requires interaction with cerebral GIPR [92]. Paradoxically, reduction of intestinal GIP expression [93], GIPR antagonism [94] or genetic elimination of GIPR [95], also reduces food intake and promotes resistance to diet-induced obesity and weight loss in multiple species. GIPR agonism may indirectly potentiate the tolerability of co-administered GLP-1RA through attenuation of CNS GLP-1-activated aversive circuits [96].
GLP-1 and GIP in the cardiovascular system
GLP1 action in the cardiovascular system
The widespread distribution of GLP-1R expression within the heart, blood vessels, immune system and brain regions (Fig. 3) controlling autonomic function [97,98,99] has sparked considerable interest in the cardiovascular biology of native GLP-1 and GLP-1RA. Both native GLP-1 and GLP-1RA exert multiple actions in the cardiovascular system, including reduction of blood pressure in hypertensive individuals, inhibition of postprandial chylomicron secretion, attenuation of inflammation in the heart and blood vessels, and reduction of ischaemic cardiac injury, but also increases in heart rate (Fig. 3) [100, 101]. Multiple long-acting GLP-1RAs reduce rates of major adverse cardiovascular events (MACE) in outcome trials of people with type 2 diabetes [102], heightening interest in understanding the mechanisms linking GLP-1R activation to cardioprotection.
Interpretation of the cardiovascular actions of native GLP-1 requires consideration that carboxy- terminal fragments, such as GLP-1(9–36) and GLP-1(28–36), the latter generated from neutral endopeptidase-mediated proteolytic cleavage, retain biological activity in the cardiovascular system through mechanisms independent of the canonical GLP-1R. These actions may be mediated through regulation of soluble adenylate cyclase 10 and mitochondrial activity controlling glycolysis and glucose oxidation [103, 104]. Nevertheless, multiple degradation-resistant and structurally distinct GLP-1RAs do not generate the same GLP-1 metabolites yet reduce blood pressure, increase heart rate, inhibit enterocyte chylomicron secretion and reduce the extent of myocardial infarction in rats and mice through mechanisms requiring the canonical GLP-1R [99, 105,106,107,108].
Delineation of mechanisms linking GLP-1R activation to cardioprotection and reduction of myocardial injury is difficult in part due to low levels of GLP-1R expression in target organs such as the heart, as well as the species-specific differences in the distribution of cardiac GLP-1Rs [98, 109]. For example, single cell RNA-seq analyses reveal that cardiac GLP-1Rs are expressed in some atrial and ventricular endothelial cells, and localised to the ventricular endocardium in mice; genetic ablation of murine GLP-1Rs within this endothelial cell population attenuates the acute cardioprotective actions of liraglutide in the setting of myocardial ischaemic injury [99]. In contrast, GLP-1Rs are relatively more abundant in human ventricles than mouse ventricles [109], and single cell RNA-seq interrogation of the normal and ischaemic human heart localises cardiac GLP-1Rs to subpopulations of atrial and ventricular cardiomyocytes [99].
GLP-1RAs also reduce the rates of stroke in cardiovascular outcome trials (CVOTs) in people with type 2 diabetes [110, 111], and are neuroprotective in experimental models of stroke and cerebral infarction (Fig. 3) [112]. Putative mechanisms linking GLP-1R activation to reduced rates of stroke are still emerging. Liraglutide attenuates thromboxane-induced platelet aggregation in people with obesity after several weeks of treatment [113], findings associated with platelet binding of LUXendin645, a fluorescent GLP-1RA, to platelets ex vivo. Moreover, the binding of LUXendin645 and the reduction of platelet aggregation by liraglutide was attenuated by the GLP-1R antagonist exendin(9–39) [113]. Nevertheless, a more careful analysis is required to determine whether human platelets express a functional canonical GLP-1R that can transduce a sustained reduction in platelet aggregation [114]. This analysis would ideally include the demonstration of full length GLP1R mRNA in platelets or platelet precursors.
SGLT-2 inhibitors reduce the rates of heart failure and MACE within weeks of administration [115]. However, the temporal reduction of MACE events in people with type 2 diabetes treated with GLP-1RA takes longer, becoming evident from 12 to 18 months from trial initiation [115, 116]. GLP-1RAs reduce hospitalisation for heart failure events in people with type 2 diabetes by ~11% in CVOTs [102]. However, a subset of individuals with type 2 diabetes and a history of hospitalisation for heart failure and/or impaired ventricular function (ejection fraction of <25% [117] or <35% [118]) do not exhibit improvements in heart failure and are not ideal candidates for GLP-1RA therapy. Given the association of weight loss with improved functional status in people with heart failure and preserved ejection fraction (HFpEF), the potential benefits of semaglutide are being examined in people with HFpEF (Fig. 4) and a BMI >30 with (ClinicalTrials.gov registration no. NCT04916470) or without type 2 diabetes (NCT04788511).

Timeline of GLP-1 discovery and clinical development. (a) Key events in the discovery and development of GLP-1-based therapies. (b) Key areas of clinical investigation in late stage clinical trials. DKD, diabetic kidney disease; DPP-4, dipeptidyl peptidase-4; PAD, peripheral artery disease; T2D, type 2 diabetes. This figure is available as part of a downloadable slideset
The time to reduction in MACE observed in the GLP-1RA CVOTs is consistent with a more chronic process, such as a reduction in atherosclerosis. GLP-1RAs reduce experimental atherosclerosis in genetically sensitised mouse models, findings associated with reduction of systemic and aortic inflammation [119, 120]. Endothelial cells are the major cellular site of GLP-1R expression in the mouse aorta; however, genetic elimination of endothelial and hematopoietic GLP-1Rs in mice with atherosclerosis did not diminish the anti-atherogenic actions of semaglutide [120]. Whether GLP-1RAs reduce vascular inflammation and atherosclerosis in people with type 2 diabetes remains uncertain. Imaging of the carotid arteries and aorta using [18F]fluorodeoxyglucose positron emission tomography did not detect reduction of inflammation after 26 weeks of daily liraglutide therapy [121]. The potential benefits of semaglutide 1 mg once weekly in people with atherosclerosis and peripheral artery disease (Figs 3, 4) are being examined over 52 weeks, with the primary outcomes of treadmill walking distance and secondary outcomes of changes in quality of life and pain free walking distance (NCT04560998).
GIP action in the cardiovascular system
The development of the GLP-1R–GIPR co-agonist tirzepatide for type 2 diabetes has sparked resurgent interest in the cardiovascular actions of GIP. GIPR is expressed in a subset of vascular endothelial cells, and in preclinical studies, activation of GIPR signalling attenuates, whereas loss of GIPR signalling exacerbates, the development of aortic inflammation and atherosclerosis [122, 123]. GIPRs are expressed at low levels in the mouse [124] and human heart [109] within populations of atrial and ventricular cardiomyocytes, adipocytes and pericytes [99]. Activation of GIPR signalling attenuates cardiac hypertrophy and fibrosis in mice with experimental hypertension [125], whereas loss of cardiomyocyte GIPR signalling is cardioprotective in mice with ischaemic cardiac injury secondary to coronary artery occlusion [124]. A meta-analysis of incident MACE in the Phase III tirzepatide clinical trial programme for type 2 diabetes revealed evidence for cardiovascular safety and numerically fewer MACE events in people randomised to tirzepatide therapy [126]. The cardiovascular safety of tirzepatide is also being studied in two larger dedicated CVOTs in people with type 2 diabetes, and in people living with overweight and obesity.
GLP-1 and GIP in the immune system
GLP-1-producing enteroendocrine L cells sense sterile or microbial inflammation and respond with an increase in GLP-1 secretion [101], findings reported to be mimicked by administration of cytokines [127] or lipopolysaccharide (LPS) [128]. Furthermore, blockade of the IL-6 receptor with tocilizumab reduces meal-stimulated levels of GLP-1 in people with type 2 diabetes or obesity [129]. The extent of systemic inflammation and the magnitude of increase in circulating levels of GLP-1 correlates with outcomes in people with sepsis or myocardial infarction [130, 131], although levels of GLP-1 are not universally elevated in people with acute or chronic inflammation [132]. Conversely, GLP-1RAs reduce inflammation in animals and humans with or without type 2 diabetes, independent of weight loss, with systemic anti-inflammatory actions evident within minutes to hours following acute administration of native GLP-1 [133] or GLP-1RAs [134,135,136].
The most abundant cellular site of GLP-1R expression in the immune system is the intestinal intraepithelial lymphocyte (IEL) (Fig. 3), and loss of Glp1r enhances the expression of proinflammatory biomarkers in the mouse intestine [137]. Surprisingly, despite the broad systemic anti-inflammatory actions of GLP-1RAs, much (100–1000 fold) lower levels of GLP-1R mRNA transcripts are detected in spleen, thymus, lymph nodes and hematopoietic cell lineages, relative to levels of GLP-1R expression in gut IELs [138, 139], suggesting that a substantial proportion of GLP-1 action on immune cells may be indirect. The anti-inflammatory pathways engaged by GLP-1RA to suppress T cell driven gut- and systemic inflammation, as exemplified in studies using anti-CD3, require signalling through the IEL GLP-1R [136]. In contrast, the actions of GLP-1RAs to reduce systemic inflammation, as modelled by administration of LPS, are independent of the IEL GLP-1R [136]. It seems likely that inter-organ communication, perhaps facilitated by neural pathways, contributes to the widespread anti-inflammatory actions of GLP-1 in tissues that do not contain GLP-1R+ immune cells [98].
GLP-1RAs also reduce experimental inflammation in the heart [140], islets [141], blood vessels [119], kidney [142, 143], lung [144, 145] and brain [146]. These anti-inflammatory actions may contribute to reduction of diabetes-associated complications in people treated with chronic GLP-1RA therapy. The GLP-1R is also expressed in astrocytes, and genetic loss of GLP-1Rs in mouse astrocytes increases hypothalamic inflammation and gliosis in high-fat diet-fed germ-free mice [147].
The direct and indirect anti-inflammatory actions of GLP-1RA may also contribute to resolution of hepatic inflammation in people with metabolic liver disease and non-alcoholic steatohepatitis (NASH) (Figs 3, 4) [148]. Both liraglutide and semaglutide suppress hepatic inflammation (Fig. 3) while preventing progression of fibrosis in people with NASH [149, 150]. Nevertheless, semaglutide 2.4 mg once weekly was less effective in achieving resolution of NASH when administered to individuals with both NASH and compensated cirrhosis [151]. The GLP-1R is not expressed in hepatocytes, hence the hepatic anti-inflammatory actions of GLP-1RAs are thought to be primarily indirect, perhaps secondary to weight loss [148]. The GLP-1R is expressed on a small subset of murine intrahepatic endothelial and γδ T cells, and genetic elimination of Glp1r in these cells attenuated the anti-inflammatory actions of semaglutide in the livers of HFD-fed mice [120]. The therapeutic potential and anti-inflammatory actions of semaglutide 2.4 mg once weekly in people with NASH (Fig. 4) is being assessed in the Phase III ESSENCE trial (NCT04822181).
GIPR is expressed at low levels in some immune cells, predominantly in subsets of T cells and macrophages [139]. Within the bone marrow, GIPR is expressed in myeloid precursors, and loss of GIPR in Gipr−/− mice, or more selective loss of the bone marrow Gipr, impairs formation of myeloid lineage cells [152, 153]. Conversely, administration of GIP receptor agonists to normal mice or mice exposed to chemotherapy, LPS or the TLR1/TLR2 agonist Pam3CysSerLys4 had little effect on circulating or bone marrow hematopoietic cell populations [153]. Activation of GIPR signalling reduced white adipose tissue (WAT) inflammation, characterised by reduced accumulation of monocytes and macrophages in WAT from HFD-fed mice [154]. In contrast, chronic administration of GIP to obese mice [155], or short term GIP infusion within WAT of humans [156], augments adipose tissue inflammation, characterised by increased infiltration of mononuclear cells and enhanced cytokine expression. Genetic elimination or marked reduction of GIPR in murine bone marrow or myeloid cells results in enhanced WAT and macrophage-driven inflammation, associated with upregulation of the alarmin S100A8/9 in myeloid cells [153, 157]. Although human data examining the effects of continuous GIP administration are limited, subcutaneous infusion of GIP for 6 days in men with type 1 diabetes did not alter biomarkers of inflammation in the circulation or in WAT [65].
The safety of incretin-based therapies
The widespread extrapancreatic expression of GLP-1 and GIPR raises questions surrounding the long-term consequences of incretin-based therapies for type 2 diabetes and obesity. Interpretation of reports of GLP-1R expression in some cancers is challenged by problems with accurate receptor ascertainment, reflecting use of poorly characterised antibodies with insufficient sensitivity and specificity [98, 109, 158, 159]. Interrogation of GLP-1R expression in human cancers has also been carried out indirectly using radiolabelled GLP-1R agonist and antagonist peptides. GLP-1R binding sites were detected in islet and neuroendocrine tumours, as well as in a subset of brain tumours [160]. Binding sites were also localised to a minority of ovarian and prostate cancers, although GLP-1R binding sites were not detected in colorectal, lung, liver, stomach or pancreatic cancers [160, 161]. A combination of immunohistochemistry and in situ autoradiography failed to detect GLP-1R expression in ductal pancreatic cancer or in well-differentiated thyroid cancer, although several medullary thyroid cancers contained GLP-1R+ cells [162].
The incidence of malignancy in people with type 2 diabetes treated with GLP-1RA has been scrutinised in CVOTs, as well as in real-world data. A meta-analysis of 45 trials (94,063 participants), including people with type 2 diabetes enrolled in CVOTs, did not reveal an imbalance between use of GLP-1RA and the incidence of benign or malignant thyroid disease [163]. Interrogation of real-world data sets using the Explorys system to assess cancer rates in 300 different healthcare systems in the USA following initiation of GLP-1RA therapy in 64,230 individuals with type 2 diabetes revealed lower rates of prostate, lung and colon cancer, but higher rates of thyroid cancer [164]. GLP-1RAs are contraindicated in people with a family history of medullary thyroid cancer (MTC) or multiple endocrine neoplasia type 2, and an ongoing registry of MTC cases has been established for surveillance purposes [165].
GIPR agonists have not been utilised in the clinic, hence less is known about the potential effects of manipulating GIPR signalling in the context of cancer. GIPR is upregulated in a subset of people with bilateral macronodular adrenal hyperplasia and food-induced Cushing’s syndrome [166]. GIPR expression has also been detected, using in situ ligand binding, in a wide range of human neuroendocrine tumours [167]. However, the functional implications, if any, of these findings for long-term manipulation of GIPR signalling in the clinic is not known.
GLP-1, GIP and neurodegenerative disorders
Physiological and pharmacological GLP-1R signalling regulates learning, behaviour, neuronal integrity and resistance to experimental brain injury in animals [112]. Similarly, rates of stroke [110, 111], and new diagnoses and progression of cognitive impairment [168] are reduced in post hoc analyses of secondary endpoints in CVOTs of GLP-1RA in people with type 2 diabetes. Moreover, GLP-1RAs suppress neuroinflammation in preclinical studies [101, 146], and exenatide, given either twice daily or once weekly, improved disease activity scores in people with Parkinson’s disease [169, 170]. Substantial preclinical data demonstrate the therapeutic potential of GLP-1RA and GIP–GLP-1RA co-agonists in mouse models of neurodegeneration, findings associated with preservation of brain structure and function, and reduction of neuroinflammation [171]. The therapeutic potential of oral semaglutide once daily is being explored in two clinical trials, in populations with and without co-existing vascular disease, studying people at risk of developing Alzheimer’s disease (NCT04777396 and NCT04777409).
Summary
Substantial clinical trial and real-world data has demonstrated the efficacy and long-term safety of GLP-1RAs in people with type 2 diabetes. However, much less long-term data are available for these agents in people with obesity. Ongoing outcome trials (Fig. 4) will ascertain the risks vs benefits in people living with obesity. GLP-1RAs are also being explored in ongoing trials in people at risk for diabetic kidney disease, people with HFpEF, people with NASH and individuals with peripheral artery disease (Fig. 4). The results of these trials will further refine, and may expand, the clinical utility of GLP-1RAs in important subpopulations with metabolic disorders. Progress in precision medicine approaches using genetics and biomarkers may identify subgroups of people that are ideally suited (or less responsive) to incretin-based therapies, enabling more targeted use of different therapeutic agents [172]. The development of tirzepatide and ongoing investigation of GLP-1-based multi-agonists has opened up an exciting new chapter in GLP-1 pharmacology [173], with an expanding range of molecules producing impressive results in early clinical trials. Each one of these agents will need to be carefully scrutinised to ensure they preserve or exceed the benefits and safety profile of GLP-1RA alone, without introduction of unanticipated new liabilities impacting therapeutic safety. Taken together, the clinical impact of GLP-1RA over 2 decades has been substantial and seems likely to be expanded, based on forthcoming clinical trial data and investigational drug development activity, in the years to come.
Abbreviations
- CNS:
-
Central nervous system
- CVOT:
-
Cardiovascular outcome trial
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GIPR:
-
Glucose-dependent insulinotropic polypeptide receptor
- GLP-1:
-
Glucagon-like peptide-1
- GLP-1R:
-
Glucagon-like peptide-1 receptor
- GLP-1RA:
-
Glucagon-like peptide-1 receptor agonist
- GPR:
-
G-protein-coupled receptor
- HFpEF:
-
Heart failure and preserved ejection fraction
- IEL:
-
Intraepithelial lymphocyte
- LPS:
-
Lipopolysaccharide
- MACE:
-
Major adverse cardiovascular events
- MPGF:
-
Major proglucagon fragment
- MTC:
-
Medullary thyroid cancer
- NASH:
-
Non-alcoholic steatohepatitis
- SGLT:
-
Sodium–glucose cotransporter
- WAT:
-
White adipose tissue
References
Bayliss WM, Starling EH (1902) The mechanism of pancreatic secretion. J Physiol 28(5):325–353. https://doi.org/10.1113/jphysiol.1902.sp000920
Zunz E, Barre JL (1929) Contributions A L'Étude des Variations Physiologiques De la Sécrétion Interne Du Pancréas. Arch Int Physiol 31(2):162–179. https://doi.org/10.3109/13813452909145169
Foa PP, Galansino G, Pozza G (1957) Glucagon, a second pancreatic hormone. Recent Prog Horm Res 13:473–503 discussion 503-410
McIntyre N, Holdsworth CD, Turner DS (1964) New interpretation of oral glucose tolerance. Lancet 2(7349):20–21. https://doi.org/10.1016/s0140-6736(64)90011-x
Elrick H, Stimmler L, Hlad CJ Jr, Arai Y (1964) Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab 24:1076–1082. https://doi.org/10.1210/jcem-24-10-1076
Brown JC, Dryburgh JR (1971) A gastric inhibitory polypeptide. II. The complete amino acid sequence. Can J Biochem 49(8):867–872. https://doi.org/10.1139/o71-122
Dupre J, Ross SA, Watson D, Brown JC (1973) Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab 37(5):826–828. https://doi.org/10.1210/jcem-37-5-826
Nauck M, Schmidt WE, Ebert R et al (1989) Insulinotropic properties of synthetic human gastric inhibitory polypeptide in man: interactions with glucose, phenylalanine, and cholecystokinin-8. J Clin Endocrinol Metab 69(3):654–662. https://doi.org/10.1210/jcem-69-3-654
Krarup T, Saurbrey N, Moody AJ, Kuhl C, Madsbad S (1987) Effect of porcine gastric inhibitory polypeptide on beta-cell function in type I and type II diabetes mellitus. Metab Clin Exp 36(7):677–682. https://doi.org/10.1016/0026-0495(87)90153-3
Nauck M, Stockmann F, Ebert R, Creutzfeldt W (1986) Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 29:46–52. https://doi.org/10.1007/BF02427280
Creutzfeldt W (1979) The incretin concept today. Diabetologia 16(2):75–85. https://doi.org/10.1007/BF01225454
Ebert R, Creutzfeldt W (1982) Influence of gastric inhibitory polypeptide antiserum on glucose- induced insulin secretion in rats. Endocrinology 111(5):1601–1606. https://doi.org/10.1210/endo-111-5-1601
Lauritsen KB, Moody AJ, Christensen KC, Lindkaer Jensen S (1980) Gastric inhibitory polypeptide (GIP) and insulin release after small-bowel resection in man. Scand J Gastroenterol 15(7):833–840. https://doi.org/10.3109/00365528009181538
Solcia E, Capella C, Buffa R et al (1981) The diffuse endocrine-paracrine system of the gut in health and disease: ultrastructural features. Scand J Gastroenterol Suppl 70:25–36
Samols E, Marri G, Marks V (1965) Promotion of insulin secretion by glucagon. Lancet 2(7409):415–416. https://doi.org/10.1016/s0140-6736(65)90761-0
Thim L, Moody AJ (1981) The primary structure of porcine glicentin (proglucagon). Regul Pept 2(2):139–150. https://doi.org/10.1016/0167-0115(81)90007-0
Moody AJ, Holst JJ, Thim L, Lindkaer Jensen SL (1981) Relationship of glicentin to proglucagon and glucagon in the porcine pancreas. Nature 289:514–516. https://doi.org/10.1038/289514a0
Holst JJ (1983) Gut glucagon, enteroglucagon, gut glucagonlike immunoreactivity, glicentin -- Current status. Gastroenterology 84:1602–1613. https://doi.org/10.1016/0016-5085(83)90388-8
Baldissera FG, Holst JJ, Knuhtsen S, Hilsted L, Nielsen OV (1988) Oxyntomodulin (glicentin-(33-69)): pharmacokinetics, binding to liver cell membranes, effects on isolated perfused pig pancreas, and secretion from isolated perfused lower small intestine of pigs. Regul Pept 21(1-2):151–166. https://doi.org/10.1016/0167-0115(88)90099-7
Patzelt C, Schiltz E (1984) Conversion of proglucagon in pancreatic alpha cells: The major endproducts are glucagon and a single peptide, the major proglucagon fragment, that contains two glucagon-like sequences. Proc Natl Acad Sci USA 81:5007–5011. https://doi.org/10.1073/pnas.81.16.5007
Lund PK, Goodman RH, Dee PC, Habener JF (1982) Pancreatic preproglucagon cDNA contains two glucagon-related coding sequences arranged in tandem. Proc Natl Acad Sci USA 79(2):345–349. https://doi.org/10.1073/pnas.79.2.345
Lund PK, Goodman RH, Habener JF (1981) Pancreatic pre-proglucagons are encoded by two separate mRNAs. J Biol Chem 256:6515–6518. https://doi.org/10.1016/S0021-9258(19)69015-0
Lund PK, Goodman RH, Jacobs JW, Habener JF (1980) Glucagon precursors indentified by immunoprecipitation of products of cell-free translation of messenger RNA. Diabetes 29:583–586. https://doi.org/10.2337/diab.29.7.583
Bell GI, Sanchez-Pescador R, Laybourn PJ, Najarian RC (1983) Exon duplication and divergence in the human preproglucagon gene. Nature 304:368–371. https://doi.org/10.1038/304368a0
Bell GI, Santerre RF, Mullenbach GT (1983) Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature 302:716–718. https://doi.org/10.1038/302716a0
Lopez LC, Frazier ML, Su CJ, Kumar A, Saunders GF (1983) Mammalian pancreatic preproglucagon contains three glucagon-related peptides. Proc Natl Acad Sci USA 80:5485–5489. https://doi.org/10.1073/pnas.80.18.5485
Heinrich G, Gros P, Habener JF (1984) Glucagon gene sequence. Four of six exons encode separate functional domains of rat pre-proglucagon. J Biol Chem 259(22):14082–14087. https://doi.org/10.1016/S0021-9258(18)89859-3
Holst JJ, Orskov C, Nielsen OV, Schwartz TW (1987) Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett 211:169–174. https://doi.org/10.1016/0014-5793(87)81430-8
Orskov C, Bersani M, Johnsen AH, Hojrup P, Holst JJ (1989) Complete sequences of glucagon-like peptide-1 from human and pig small intestine. J Biol Chem 264(22):12826–12829. https://doi.org/10.1016/S0021-9258(18)51561-1
Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF (1987) Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci USA 84:3434–3438. https://doi.org/10.1073/pnas.84.10.3434
Mojsov S, Weir GC, Habener JF (1987) Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Investig 79:616–619. https://doi.org/10.1172/JCI112855
Kreymann B, Williams G, Ghatei MA, Bloom SR (1987) Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 2(8571):1300–1304. https://doi.org/10.1016/s0140-6736(87)91194-9
Vilsboll T, Krarup T, Madsbad S, Holst JJ (2003) Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept 114(2-3):115–121. https://doi.org/10.1016/s0167-0115(03)00111-3
Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W (1993) Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Investig 91:301–307. https://doi.org/10.1172/JCI116186
Orskov C, Holst JJ, Nielsen OV (1988) Effect of truncated glucagon-like peptide-1 [proglucagon-(78-107) amide] on endocrine secretion from pig pancreas, antrum, and nonantral stomach. Endocrinology 123(4):2009–2013. https://doi.org/10.1210/endo-123-4-2009
Jensen SL, Holst JJ, Nielsen OV, Lauritsen KB (1981) Secretory effects of gastric inhibitory polypeptide on the isolated perfused porcine pancreas. Acta Physiol Scand 111(3):233–238. https://doi.org/10.1111/j.1748-1716.1981.tb06731.x
Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W (1993) Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7-36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 36(8):741–744. https://doi.org/10.1007/BF00401145
Nauck MA, Homberger E, Siegel EG et al (1986) Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab 63(2):492–498. https://doi.org/10.1210/jcem-63-2-492
Bagger JI, Knop FK, Lund A, Vestergaard H, Holst JJ, Vilsboll T (2011) Impaired regulation of the incretin effect in patients with type 2 diabetes. J Clin Endocrinol Metab 96(3):737–745. https://doi.org/10.1210/jc.2010-2435
Raufman J-P, Singh L, Singh G, Eng J (1992) Truncated glucagon-like peptide-1 interacts with exendin receptors on disperced acini from guinea pig pancreas. Identification of a mammalian homolgue of the reptilian peptide exendin-4. J Biol Chem 267:21432–21437. https://doi.org/10.1016/S0021-9258(19)36628-1
Sparre-Ulrich AH, Gabe MN, Gasbjerg LS et al (2017) GIP(3-30)NH2 is a potent competitive antagonist of the GIP receptor and effectively inhibits GIP-mediated insulin, glucagon, and somatostatin release. Biochem Pharmacol 131:78–88. https://doi.org/10.1016/j.bcp.2017.02.012
Gasbjerg LS, Helsted MM, Hartmann B et al (2019) Separate and combined glucometabolic effects of endogenous glucose-dependent insulinotropic polypeptide and glucagon-like peptide 1 in healthy individuals. Diabetes 68(5):906–917. https://doi.org/10.2337/db18-1123
Gasbjerg LS, Bergmann NC, Stensen S et al (2020) Evaluation of the incretin effect in humans using GIP and GLP-1 receptor antagonists. Peptides 125:170183. https://doi.org/10.1016/j.peptides.2019.170183
Segerstolpe A, Palasantza A, Eliasson P et al (2016) Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 24(4):593–607. https://doi.org/10.1016/j.cmet.2016.08.020
Svendsen B, Larsen O, Gabe MBN et al (2018) Insulin secretion depends on intra-islet glucagon signaling. Cell Rep 25(5):1127–1134 e1122. https://doi.org/10.1016/j.celrep.2018.10.018
Capozzi ME, Wait JB, Koech J et al (2019) Glucagon lowers glycemia when beta-cells are active. JCI Insight 5. https://doi.org/10.1172/jci.insight.129954
El K, Gray SM, Capozzi ME et al (2021) GIP mediates the incretin effect and glucose tolerance by dual actions on alpha cells and beta cells. Sci Adv 7(11):eabf1948. https://doi.org/10.1126/sciadv.abf1948
Oduori OS, Murao N, Shimomura K et al (2020) Gs/Gq signaling switch in beta cells defines incretin effectiveness in diabetes. J Clin Investig 130(12):6639–6655. https://doi.org/10.1172/JCI140046
Mortensen K, Christensen LL, Holst JJ, Orskov C (2003) GLP-1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regul Pept 114(2-3):189–196. https://doi.org/10.1016/S0167-0115(03)00125-3
Gribble FM, Reimann F (2016) Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu Rev Physiol 78:277–299. https://doi.org/10.1146/annurev-physiol-021115-105439
Gorboulev V, Schurmann A, Vallon V et al (2012) Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 61(1):187–196. https://doi.org/10.2337/db11-1029
Edfalk S, Steneberg P, Edlund H (2008) Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57(9):2280–2287. https://doi.org/10.2337/db08-0307
Chu ZL, Carroll C, Alfonso J et al (2008) A role for intestinal endocrine cell-expressed GPR119 in glycemic control by enhancing GLP-1 and GIP release. Endocrinology 149(5):2038–2047. https://doi.org/10.1210/en.2007-0966
Modvig IM, Kuhre RE, Jepsen SL et al (2021) Amino acids differ in their capacity to stimulate GLP-1 release from the perfused rat small intestine and stimulate secretion by different sensing mechanisms. Am J Physiol Endocrinol Metab 320(5):E874–E885. https://doi.org/10.1152/ajpendo.00026.2021
Lindgren O, Pacini G, Tura A, Holst JJ, Deacon CF, Ahren B (2015) Incretin effect after oral amino acid ingestion in humans. J Clin Endocrinol Metab 100(3):1172–1176. https://doi.org/10.1210/jc.2014-3865
Faerch K, Torekov SS, Vistisen D et al (2015) GLP-1 response to oral glucose is reduced in prediabetes, screen-detected type 2 diabetes, and obesity and influenced by sex: the ADDITION-PRO study. Diabetes 64(7):2513–2525. https://doi.org/10.2337/db14-1751
Hunt JE, Holst JJ, Jepsen SL (2022) Glucose- and bile acid-stimulated secretion of gut hormones in the isolated perfused intestine is not impaired in diet-induced obese mice. Front Endocrinol (Lausanne) 13:884501. https://doi.org/10.3389/fendo.2022.884501
Iepsen EW, Lundgren J, Holst JJ, Madsbad S, Torekov SS (2016) Successful weight loss maintenance includes long-term increased meal responses of GLP-1 and PYY3-36. Eur J Endocrinol 174(6):775–784. https://doi.org/10.1530/EJE-15-1116
Otten J, Ryberg M, Mellberg C et al (2019) Postprandial levels of GLP-1, GIP and glucagon after 2 years of weight loss with a Paleolithic diet: a randomised controlled trial in healthy obese women. Eur J Endocrinol 180(6):417–427. https://doi.org/10.1530/EJE-19-0082
Li L, Decker AM, Stobaus N et al (2022) Weight loss did not modify macronutrient specific response of hormones and satiety in overweight and obese people without metabolic disease - results from a clinical trial. Clin Nutr 41(4):948–957. https://doi.org/10.1016/j.clnu.2022.02.004
Hojberg PV, Vilsboll T, Rabol R et al (2009) Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 52(2):199–207. https://doi.org/10.1007/s00125-008-1195-5
Vilsboll T, Krarup T, Madsbad S, Holst JJ (2002) Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia 45(8):1111–1119. https://doi.org/10.1007/s00125-002-0878-6
Mentis N, Vardarli I, Kothe LD et al (2011) GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes. Diabetes 60(4):1270–1276. https://doi.org/10.2337/db10-1332
Bergmann NC, Gasbjerg LS, Heimburger SM et al (2020) No acute effects of exogenous glucose-dependent insulinotropic polypeptide on energy intake, appetite, or energy expenditure when added to treatment with a long-acting glucagon-like peptide 1 receptor agonist in men with type 2 diabetes. Diabetes Care 43(3):588–596. https://doi.org/10.2337/dc19-0578
Heimburger SMN, Hoe B, Nielsen CN et al (2022) GIP affects hepatic fat and brown adipose tissue thermogenesis but not white adipose tissue transcriptome in type 1 diabetes. J Clin Endocrinol Metab 107(12):3261–3274. https://doi.org/10.1210/clinem/dgac542
Finan B, Ma T, Ottaway N et al (2013) Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med 5(209):209ra151. https://doi.org/10.1126/scitranslmed.3007218
Frias JP, Bastyr EJ 3rd, Vignati L et al (2017) The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab 26(2):343–352 e342. https://doi.org/10.1016/j.cmet.2017.07.011
Frias JP, Nauck MA, Van J et al (2018) Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 392(10160):2180–2193. https://doi.org/10.1016/S0140-6736(18)32260-8
Frias JP, Davies MJ, Rosenstock J et al (2021) Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med 385(6):503–515. https://doi.org/10.1056/NEJMoa2107519
Willard FS, Douros JD, Gabe MB et al (2020) Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight 5(17):e140532. https://doi.org/10.1172/jci.insight.140532
Nagell CF, Wettergren A, Pedersen JF, Mortensen D, Holst JJ (2004) Glucagon-like peptide-2 inhibits antral emptying in man, but is not as potent as glucagon-like peptide-1. Scand J Gastroenterol 39(4):353–358. https://doi.org/10.1080/00365520410004424
Wettergren A, Wojdemann M, Holst JJ (1998) Glucagon-like peptide-1 inhibits gastropancreatic function by inhibiting central parasympathetic outflow. Am J Physiol 275(5):G984–G992. https://doi.org/10.1152/ajpgi.1998.275.5.G984
Plamboeck A, Veedfald S, Deacon CF et al (2013) The effect of exogenous GLP-1 on food intake is lost in male truncally vagotomized subjects with pyloroplasty. Am J Physiol Gastrointest Liver Physiol 304(12):G1117–G1127. https://doi.org/10.1152/ajpgi.00035.2013
Varin EM, Mulvihill EE, Baggio LL et al (2019) Distinct neural sites of GLP-1R expression mediate physiological versus pharmacological control of incretin action. Cell Rep 27(11):3371–3384 e3373. https://doi.org/10.1016/j.celrep.2019.05.055
Nauck MA, Kemmeries G, Holst JJ, Meier JJ (2011) Rapid tachyphylaxis of the glucagon-like peptide 1-induced deceleration of gastric emptying in humans. Diabetes 60(5):1561–1565. https://doi.org/10.2337/db10-0474
Urva S, Coskun T, Loghin C et al (2020) The novel dual glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 (GLP-1) receptor agonist tirzepatide transiently delays gastric emptying similarly to selective long-acting GLP-1 receptor agonists. Diabetes Obes Metab 22(10):1886–1891. https://doi.org/10.1111/dom.14110
Dahl K, Brooks A, Almazedi F, Hoff ST, Boschini C, Baekdal TA (2021) Oral semaglutide improves postprandial glucose and lipid metabolism, and delays gastric emptying, in subjects with type 2 diabetes. Diabetes Obes Metab 23(7):1594–1603. https://doi.org/10.1111/dom.14373
Ohrstrom CC, Worm D, Kielgast UL, Holst JJ, Hansen DL (2020) Evidence for relationship between early dumping and postprandial hypoglycemia after Roux-en-Y gastric bypass. Obes Surg 30(3):1038–1045. https://doi.org/10.1007/s11695-020-04387-6
Drucker DJ (2022) GLP-1 physiology informs the pharmacotherapy of obesity. Mol Metab 57:101351. https://doi.org/10.1016/j.molmet.2021.101351
Wilding JPH, Batterham RL, Calanna S et al (2021) Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med 384(11):989. https://doi.org/10.1056/NEJMoa2032183
Gabery S, Salinas CG, Paulsen SJ et al (2020) Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight 5(6):e133429. https://doi.org/10.1172/jci.insight.133429
Brierley DI, Holt MK, Singh A et al (2021) Central and peripheral GLP-1 systems independently suppress eating. Nat Metab 3(2):258–273. https://doi.org/10.1038/s42255-021-00344-4
Sisley S, Gutierrez-Aguilar R, Scott M, D’Alessio DA, Sandoval DA, Seeley RJ (2014) Neuronal GLP1R mediates liraglutide's anorectic but not glucose-lowering effect. J Clin Investig 124(6):2456–2463. https://doi.org/10.1172/JCI72434
Imbernon M, Saponaro C, Helms HCC et al (2022) Tanycytes control hypothalamic liraglutide uptake and its anti-obesity actions. Cell Metab 34(7):1054–1063 e1057. https://doi.org/10.1016/j.cmet.2022.06.002
Borgmann D, Ciglieri E, Biglari N et al (2021) Gut-brain communication by distinct sensory neurons differently controls feeding and glucose metabolism. Cell Metab 33(7):1466–1482 e1467. https://doi.org/10.1016/j.cmet.2021.05.002
Patterson JT, Ottaway N, Gelfanov VM et al (2011) A novel human-based receptor antagonist of sustained action reveals body weight control by endogenous GLP-1. ACS Chem Biol 6(2):135–145. https://doi.org/10.1021/cb1002015
Svane MS, Jorgensen NB, Bojsen-Moller KN et al (2016) Peptide YY and glucagon-like peptide-1 contribute to decreased food intake after Roux-en-Y gastric bypass surgery. Int J Obes 40(11):1699–1706. https://doi.org/10.1038/ijo.2016.121
Scrocchi LA, Brown TJ, MacLusky N et al (1996) Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide receptor gene. Nat Med 2:1254–1258. https://doi.org/10.1038/nm1196-1254
Song Y, Koehler JA, Baggio LL, Powers AC, Sandoval DA, Drucker DJ (2019) Gut-proglucagon-derived peptides are essential for regulating glucose homeostasis in mice. Cell Metab 30(5):976–986 e973. https://doi.org/10.1016/j.cmet.2019.08.009
Adriaenssens AE, Biggs EK, Darwish T et al (2019) Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab 30(5):987–996 e986. https://doi.org/10.1016/j.cmet.2019.07.013
Mroz PA, Finan B, Gelfanov V et al (2019) Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab 20:51–62. https://doi.org/10.1016/j.molmet.2018.12.001
Zhang Q, Delessa CT, Augustin R et al (2021) The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab 33(4):833–844 e835. https://doi.org/10.1016/j.cmet.2021.01.015
Althage MC, Ford EL, Wang S, Tso P, Polonsky KS, Wice BM (2008) Targeted ablation of glucose-dependent insulinotropic polypeptide-producing cells in transgenic mice reduces obesity and insulin resistance induced by a high fat diet. J Biol Chem 283(26):18365–18376. https://doi.org/10.1074/jbc.M710466200
Killion EA, Wang J, Yie J et al (2018) Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med 10(472):eaat3392. https://doi.org/10.1126/scitranslmed.aat3392
Miyawaki K, Yamada Y, Ban N et al (2002) Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 8(7):738–742. https://doi.org/10.1038/nm727
Borner T, Geisler CE, Fortin SM et al (2021) GIP receptor agonism attenuates GLP-1 receptor agonist-induced nausea and emesis in preclinical models. Diabetes 70(11):2545–2553. https://doi.org/10.2337/db21-0459
Yamamoto H, Kishi T, Lee CE et al (2003) Glucagon-like peptide-1-responsive catecholamine neurons in the area postrema link peripheral glucagon-like peptide-1 with central autonomic control sites. J Neurosci 23(7):2939–2946. https://doi.org/10.1523/JNEUROSCI.23-07-02939.2003
McLean BA, Wong CK, Campbell JE, Hodson DJ, Trapp S, Drucker DJ (2021) Revisiting the complexity of GLP-1 action from sites of synthesis to receptor activation. Endocr Rev 42(2):101–132. https://doi.org/10.1210/endrev/bnaa032
McLean BA, Wong CK, Kabir MG, Drucker DJ (2022) Glucagon-like Peptide-1 receptor Tie2+ cells are essential for the cardioprotective actions of liraglutide in mice with experimental myocardial infarction. Mol Metab 66:101641. https://doi.org/10.1016/j.molmet.2022.101641
Drucker DJ (2016) The cardiovascular biology of glucagon-like peptide-1. Cell Metab 24(1):15–30. https://doi.org/10.1016/j.cmet.2016.06.009
Drucker DJ (2018) Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab 27(4):740–756. https://doi.org/10.1016/j.cmet.2018.03.001
Sattar N, Lee MMY, Kristensen SL et al (2021) Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol 9(10):653–662. https://doi.org/10.1016/S2213-8587(21)00203-5
Ban K, Kim H, Cho J et al (2010) GLP-1(9-36) protects cardiomyocytes and endothelial cells from ischemia-reperfusion injury via cytoprotective pathways independent of the GLP-1 receptor. Endocrinology 151(4):1520–1531. https://doi.org/10.1210/en.2009-1197
Siraj MA, Mundil D, Beca S et al (2020) Cardioprotective GLP-1 metabolite prevents ischemic cardiac injury by inhibiting mitochondrial trifunctional protein-alpha. J Clin Investig 130(3):1392–1404. https://doi.org/10.1172/JCI99934
Kim M, Platt M, Shibasaki T et al (2013) GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat Med 19(5):567–575. https://doi.org/10.1038/nm.3128
Baggio LL, Ussher JR, McLean BA et al (2017) The autonomic nervous system and cardiac GLP-1 receptors control heart rate in mice. Mol Metab 6(11):1339–1349. https://doi.org/10.1016/j.molmet.2017.08.010
Hsieh J, Longuet C, Baker CL et al (2010) The glucagon-like peptide 1 receptor is essential for postprandial lipoprotein synthesis and secretion. Diabetologia 53(3):552–561. https://doi.org/10.1007/s00125-009-1611-5
Noyan-Ashraf MH, Momen MA, Ban K et al (2009) GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 58(4):975–983. https://doi.org/10.2337/db08-1193
Baggio LL, Yusta B, Mulvihill EE et al (2018) GLP-1 receptor expression within the human heart. Endocrinology 159(4):1570–1584. https://doi.org/10.1210/en.2018-00004
Gerstein HC, Hart R, Colhoun HM et al (2020) The effect of dulaglutide on stroke: an exploratory analysis of the REWIND trial. Lancet Diabetes Endocrinol 8(2):106–114. https://doi.org/10.1016/S2213-8587(19)30423-1
Strain WD, Frenkel O, James MA et al (2022) Effects of semaglutide on stroke subtypes in type 2 diabetes: post hoc analysis of the randomized SUSTAIN 6 and PIONEER 6. Stroke 53(9):2749–2757. https://doi.org/10.1161/STROKEAHA.121.037775
During MJ, Cao L, Zuzga DS et al (2003) Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med 9(9):1173–1179. https://doi.org/10.1038/nm919
Cahill KN, Amin T, Boutaud O et al (2022) Glucagon-like peptide-1 receptor regulates thromboxane-induced human platelet activation. JACC Basic Transl Sci 7(7):713–715. https://doi.org/10.1016/j.jacbts.2022.04.004
Loganathan J, Cohen AC, Kaloupis GM et al (2022) A pilot clinical study to evaluate liraglutide-mediated anti-platelet activity in patients with type-2 diabetes (ELAID study). J Diabetes Complicat 36(5):108188. https://doi.org/10.1016/j.jdiacomp.2022.108188
Cherney DZI, Udell JA, Drucker DJ (2021) Cardiorenal mechanisms of action of glucagon-like-peptide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors. Med (N Y) 2(11):1203–1230. https://doi.org/10.1016/j.medj.2021.10.004
Marso SP, Daniels GH, Brown-Frandsen K et al (2016) Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 375:311–322. https://doi.org/10.1056/NEJMoa1603827
Margulies KB, Hernandez AF, Redfield MM et al (2016) Effects of liraglutide on clinical stability among patients with advanced heart failure and reduced ejection fraction: a randomized clinical trial. JAMA 316(5):500–508. https://doi.org/10.1001/jama.2016.10260
Jorsal A, Kistorp C, Holmager P et al (2017) Effect of liraglutide, a glucagon-like peptide-1 analogue, on left ventricular function in stable chronic heart failure patients with and without diabetes (LIVE)-a multicentre, double-blind, randomised, placebo-controlled trial. Eur J Heart Fail 19(1):69–77. https://doi.org/10.1002/ejhf.657
Rakipovski G, Rolin B, Nohr J et al (2018) The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE(-/-) and LDLr(-/-) mice by a mechanism that includes inflammatory pathways. JACC Basic Transl Sci 3(6):844–857. https://doi.org/10.1016/j.jacbts.2018.09.004
McLean BA, Wong CK, Kaur KD, Seeley RJ, Drucker DJ (2021) Differential importance of endothelial and hematopoietic cell GLP-1Rs for cardiometabolic versus hepatic actions of semaglutide. JCI Insight 6(22):e153732. https://doi.org/10.1172/jci.insight.153732
Ripa RS, Zobel EH, von Scholten BJ et al (2021) Effect of liraglutide on arterial inflammation assessed as [(18)F]FDG uptake in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled trial. Circ Cardiovasc Imaging 14(7):e012174. https://doi.org/10.1161/CIRCIMAGING.120.012174
Nagashima M, Watanabe T, Terasaki M et al (2011) Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia 54(10):2649–2659. https://doi.org/10.1007/s00125-011-2241-2
Pujadas G, Baggio LL, Kaur KD, McLean BA, Cao X, Drucker DJ (2022) Genetic disruption of the Gipr in Apoe(-/-) mice promotes atherosclerosis. Mol Metab 65:101586. https://doi.org/10.1016/j.molmet.2022.101586
Ussher JR, Campbell JE, Mulvihill EE et al (2018) Inactivation of the glucose-dependent insulinotropic polypeptide receptor improves outcomes following experimental myocardial infarction. Cell Metab 27(2):450–460. https://doi.org/10.1016/j.cmet.2017.11.003
Hiromura M, Mori Y, Kohashi K et al (2016) Suppressive effects of glucose-dependent insulinotropic polypeptide on cardiac hypertrophy and fibrosis in angiotensin ii-infused mouse models. Circ J 80(9):1988–1997. https://doi.org/10.1253/circj.CJ-16-0152
Sattar N, McGuire DK, Pavo I et al (2022) Tirzepatide cardiovascular event risk assessment: a pre-specified meta-analysis. Nat Med 28(3):591–598. https://doi.org/10.1038/s41591-022-01707-4
Ellingsgaard H, Hauselmann I, Schuler B et al (2011) Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 17(11):1481–1489. https://doi.org/10.1038/nm.2513
Nguyen AT, Mandard S, Dray C et al (2014) Lipopolysaccharides-mediated increase in glucose-stimulated insulin secretion: involvement of the GLP-1 pathway. Diabetes 63(2):471–482. https://doi.org/10.2337/db13-0903
Ellingsgaard H, Seelig E, Timper K et al (2020) GLP-1 secretion is regulated by IL-6 signalling: a randomised, placebo-controlled study. Diabetologia 63(2):362–373. https://doi.org/10.1007/s00125-019-05045-y
Kahles F, Ruckbeil MV, Mertens RW et al (2020) Glucagon-like peptide 1 levels predict cardiovascular risk in patients with acute myocardial infarction. Eur Heart J 41(7):882–889. https://doi.org/10.1093/eurheartj/ehz728
Lebherz C, Schlieper G, Mollmann J et al (2017) GLP-1 levels predict mortality in patients with critical illness as well as end-stage renal disease. Am J Med 130(7):833–841 e833. https://doi.org/10.1016/j.amjmed.2017.03.010
Modrzynska J, Klein CF, Iversen K et al (2021) Plasma levels of glucagon but not GLP-1 are elevated in response to inflammation in humans. Endocr Connect 10(2):205–213. https://doi.org/10.1530/EC-20-0590
Daousi C, Pinkney JH, Cleator J, Wilding JP, Ranganath LR (2013) Acute peripheral administration of synthetic human GLP-1 (7-36 amide) decreases circulating IL-6 in obese patients with type 2 diabetes mellitus: a potential role for GLP-1 in modulation of the diabetic pro-inflammatory state? Regul Pept 183:54–61. https://doi.org/10.1016/j.regpep.2013.03.004
Chaudhuri A, Ghanim H, Vora M et al (2012) Exenatide exerts a potent antiinflammatory effect. J Clin Endocrinol Metab 97(1):198–207. https://doi.org/10.1210/jc.2011-1508
Lebrun LJ, Lenaerts K, Kiers D et al (2017) Enteroendocrine L cells sense LPS after gut barrier injury to enhance GLP-1 secretion. Cell Rep 21(5):1160–1168. https://doi.org/10.1016/j.celrep.2017.10.008
Wong CK, Yusta B, Koehler JA et al (2022) Divergent roles for the gut intraepithelial lymphocyte GLP-1R in control of metabolism, microbiota, and T cell-induced inflammation. Cell Metab 34(10):1514–1531 e1517. https://doi.org/10.1016/j.cmet.2022.08.003
Yusta B, Baggio LL, Koehler J et al (2015) GLP-1 receptor (GLP-1R) agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte (IEL) GLP-1R. Diabetes 64(7):2537–2549. https://doi.org/10.2337/db14-1577
Hadjiyanni I, Siminovitch KA, Danska JS, Drucker DJ (2010) Glucagon-like peptide-1 receptor signalling selectively regulates murine lymphocyte proliferation and maintenance of peripheral regulatory T cells. Diabetologia 53(4):730–740. https://doi.org/10.1007/s00125-009-1643-x
Heng TS, Painter MW, Immunological Genome Project C (2008) The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9(10):1091–1094. https://doi.org/10.1038/ni1008-1091
Noyan-Ashraf MH, Shikatani EA, Schuiki I et al (2013) A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation 127(1):74–85. https://doi.org/10.1161/CIRCULATIONAHA.112.091215
Pugazhenthi U, Velmurugan K, Tran A, Mahaffey G, Pugazhenthi S (2010) Anti-inflammatory action of exendin-4 in human islets is enhanced by phosphodiesterase inhibitors: potential therapeutic benefits in diabetic patients. Diabetologia 53(11):2357–2368. https://doi.org/10.1007/s00125-010-1849-y
Bisgaard LS, Bosteen MH, Fink LN et al (2016) Liraglutide reduces both atherosclerosis and kidney inflammation in moderately uremic LDLr-/- mice. PloS One 11(12):e0168396. https://doi.org/10.1371/journal.pone.0168396
Moschovaki Filippidou F, Kirsch AH, Thelen M et al (2020) Glucagon-like peptide-1 receptor agonism improves nephrotoxic serum nephritis by inhibiting T-cell proliferation. Am J Pathol 190(2):400–411. https://doi.org/10.1016/j.ajpath.2019.10.008
Sato T, Shimizu T, Fujita H et al (2020) GLP-1 receptor signaling differentially modifies the outcomes of sterile vs viral pulmonary inflammation in male mice. Endocrinology 161(12):bqaa201. https://doi.org/10.1210/endocr/bqaa201
Toki S, Newcomb DC, Printz RL et al (2021) Glucagon-like peptide-1 receptor agonist inhibits aeroallergen-induced activation of ILC2 and neutrophilic airway inflammation in obese mice. Allergy 76(11):3433–3445. https://doi.org/10.1111/all.14879
Kopp KO, Glotfelty EJ, Li Y, Greig NH (2022) Glucagon-like peptide-1 (GLP-1) receptor agonists and neuroinflammation: Implications for neurodegenerative disease treatment. Pharmacol Res 186:106550. https://doi.org/10.1016/j.phrs.2022.106550
Heiss CN, Manneras-Holm L, Lee YS et al (2021) The gut microbiota regulates hypothalamic inflammation and leptin sensitivity in Western diet-fed mice via a GLP-1R-dependent mechanism. Cell Rep 35(8):109163. https://doi.org/10.1016/j.celrep.2021.109163
Yabut JM, Drucker DJ (2023) Glucagon-like peptide-1 receptor-based therapeutics for metabolic liver disease. Endocr Rev 44(1):14–32. https://doi.org/10.1210/endrev/bnac018
Armstrong MJ, Gaunt P, Aithal GP et al (2016) Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 387(10019):679–690. https://doi.org/10.1016/S0140-6736(15)00803-X
Newsome PN, Buchholtz K, Cusi K et al (2021) A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med 384(12):1113–1124. https://doi.org/10.1056/NEJMoa2028395
Loomba R, Abdelmalek MF, Armstrong MF et al (2022) Semaglutide 2.4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: a randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol Hepatol https://doi.org/10.1016/S2468-1253(23)00068-7
Mantelmacher FD, Fishman S, Cohen K et al (2017) Glucose-dependent insulinotropic polypeptide receptor deficiency leads to impaired bone marrow hematopoiesis. J Immunol 198(8):3089–3098. https://doi.org/10.4049/jimmunol.1601441
Pujadas G, Varin EM, Baggio LL et al (2020) The gut hormone receptor GIPR links energy availability to the control of hematopoiesis. Mol Metab 39:101008. https://doi.org/10.1016/j.molmet.2020.101008
Varol C, Zvibel I, Spektor L et al (2014) Long-acting glucose-dependent insulinotropic polypeptide ameliorates obesity-induced adipose tissue inflammation. J Immunol 193(8):4002–4009. https://doi.org/10.4049/jimmunol.1401149
Chen S, Okahara F, Osaki N, Shimotoyodome A (2015) Increased GIP signaling induces adipose inflammation via a HIF-1alpha-dependent pathway and impairs insulin sensitivity in mice. Am J Physiol Endocrinol Metab 308(5):E414–E425. https://doi.org/10.1152/ajpendo.00418.2014
Gogebakan O, Osterhoff MA, Schuler R et al (2015) GIP increases adipose tissue expression and blood levels of MCP-1 in humans and links high energy diets to inflammation: a randomised trial. Diabetologia 58(8):1759–1768. https://doi.org/10.1007/s00125-015-3618-4
Mantelmacher FD, Zvibel I, Cohen K et al (2019) GIP regulates inflammation and body weight by restraining myeloid-cell-derived S100A8/A9. Nat Metab 1(1):58–69. https://doi.org/10.1038/s42255-018-0001-z
Panjwani N, Mulvihill EE, Longuet C et al (2013) GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE-/- mice. Endocrinology 154(1):127–139. https://doi.org/10.1210/en.2012-1937
Ast J, Broichhagen J, Hodson DJ (2021) Reagents and models for detecting endogenous GLP1R and GIPR. EBioMedicine 74:103739. https://doi.org/10.1016/j.ebiom.2021.103739
Korner M, Stockli M, Waser B, Reubi JC (2007) GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J Nucl Med 48(5):736–743. https://doi.org/10.2967/jnumed.106.038679
Korner M, Rehmann R, Reubi JC (2012) GLP-2 receptors in human disease: high expression in gastrointestinal stromal tumors and Crohn's disease. Mol Cell Endocrinol 364(1-2):46–53. https://doi.org/10.1016/j.mce.2012.08.008
Waser B, Blank A, Karamitopoulou E, Perren A, Reubi JC (2015) Glucagon-like-peptide-1 receptor expression in normal and diseased human thyroid and pancreas. Mod Pathol 28(3):391–402. https://doi.org/10.1038/modpathol.2014.113
Hu W, Song R, Cheng R et al (2022) Use of GLP-1 receptor agonists and occurrence of thyroid disorders: a meta-analysis of randomized controlled trials. Front Endocrinol (Lausanne) 13:927859. https://doi.org/10.3389/fendo.2022.927859
Wang J, Kim CH (2022) Differential risk of cancer associated with glucagon-like peptide-1 receptor agonists: analysis of real-world databases. Endocr Res 47(1):18–25. https://doi.org/10.1080/07435800.2021.1955255
Hale PM, Ali AK, Buse JB et al (2020) Medullary thyroid carcinoma surveillance study: a case-series registry. Thyroid 30(10):1397–1398. https://doi.org/10.1089/thy.2019.0591
Chasseloup F, Bourdeau I, Tabarin A et al (2021) Loss of KDM1A in GIP-dependent primary bilateral macronodular adrenal hyperplasia with Cushing's syndrome: a multicentre, retrospective, cohort study. Lancet Diabetes Endocrinol 9(12):813–824. https://doi.org/10.1016/S2213-8587(21)00236-9
Waser B, Rehmann R, Sanchez C, Fourmy D, Reubi JC (2012) Glucose-dependent insulinotropic polypeptide receptors in most gastroenteropancreatic and bronchial neuroendocrine tumors. J Clin Endocrinol Metab 97(2):482–488. https://doi.org/10.1210/jc.2011-2454
Cukierman-Yaffe T, Gerstein HC, Colhoun HM et al (2020) Effect of dulaglutide on cognitive impairment in type 2 diabetes: an exploratory analysis of the REWIND trial. Lancet Neurol 19(7):582–590. https://doi.org/10.1016/S1474-4422(20)30173-3
Aviles-Olmos I, Dickson J, Kefalopoulou Z et al (2013) Exenatide and the treatment of patients with Parkinson's disease. J Clin Investig 123(6):2730–2736. https://doi.org/10.1172/JCI68295
Athauda D, Maclagan K, Skene SS et al (2017) Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet 390(10103):1664–1675. https://doi.org/10.1016/S0140-6736(17)31585-4
Reich N, Holscher C (2022) The neuroprotective effects of glucagon-like peptide 1 in Alzheimer's and Parkinson's disease: an in-depth review. Front Neurosci 16:970925. https://doi.org/10.3389/fnins.2022.970925
Dawed AY, Mari A, Brown A et al (2023) Pharmacogenomics of GLP-1 receptor agonists: a genome-wide analysis of observational data and large randomised controlled trials. Lancet Diabetes Endocrinol 11(1):33–41. https://doi.org/10.1016/S2213-8587(22)00340-0
Baggio LL, Drucker DJ (2021) Glucagon-like peptide-1 receptor co-agonists for treating metabolic disease. Mol Metab 46:101090. https://doi.org/10.1016/j.molmet.2020.101090
Hammoud R, Drucker DJ (2022) Beyond the pancreas: contrasting cardiometabolic actions of GIP and GLP1. Nat Rev Endocrinol. https://doi.org/10.1038/s41574-022-00783-3
Authors’ relationships and activities
DJD has served as a consultant or speaker within the past 12 months to Altimmune, Amgen, Kallyope, Merck Research Laboratories, Novo Nordisk and Pfizer. Neither DJD or his family members hold issued stock directly or indirectly in any of these companies. DJD holds non-exercised options in Kallyope. JJH has served as a consultant or speaker within the past 12 months to Kallyope, Merck Research Laboratories and Novo Nordisk. Neither JJH or his family members hold issued stock directly or indirectly in any of these companies. JJH is supported by the Nordisk Foundation. JJH is one of the founders of Bainan Biotech and Antag Therapeutics.
Contribution statement
Both authors wrote and edited the manuscript and approved the final version.
Funding
DJD is supported by a Banting and Best Diabetes Centre Chair in Incretin Biology, the Sinai Health Novo Nordisk Foundation Fund in Regulatory Peptides, and CIHR Foundation grant 154321. JJH is supported by grants from the Novo Nordisk Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Slideset of figures
(PPTX 1.87 MB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Drucker, D.J., Holst, J.J. The expanding incretin universe: from basic biology to clinical translation. Diabetologia 66, 1765–1779 (2023). https://doi.org/10.1007/s00125-023-05906-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-023-05906-7