
Abstract
The discovery of long-acting incretin receptor agonists represents a major stride forward in tackling the dual epidemic of obesity and diabetes. Here we outline the evolution of incretin-based pharmacotherapy, from exendin-4 to the discovery of the multi-incretin hormone receptor agonists that look set to be our next step toward curing diabetes and obesity. We discuss the multiagonists currently in clinical trials and the improvement in efficacy each new generation of these drugs bring. The success of these agents in preclinical models and clinical trials suggests a promising future for multiagonists in the treatment of metabolic diseases, with the most recent glucose-dependent insulinotropic peptide receptor:glucagon-like peptide 1 receptor:glucagon receptor (GIPR:GLP-1R:GCGR) triagonists rivaling the efficacy of bariatric surgery. However, further research is needed to fully understand how these therapies exert their effect on body weight and in the last section we cover open questions about the potential mechanisms of multiagonist drugs, and the understanding of how gut–brain communication can be leveraged to achieve sustained body weight loss without adverse effects.
Similar content being viewed by others


In this article we underscore need for more effective and safer anti-obesity pharmacotherapies due to the limited efficacy and/or significant adverse effects of drugs prior to the discovery of incretin-based therapies. |
We highlight the significant advances in obesity pharmacotherapy, detailing the role of hormonal regulation, genetic factors, and the central nervous system. |
This review showcases the promising advancements in incretin-based therapies which have demonstrated remarkable improvements in weight management and glucose control, rivaling previous medications and nearing the result of bariatric surgery. |
We identify and discuss ongoing challenges in the field, including the need for a deeper understanding of the mechanisms behind incretin agonists effectiveness, improving their safety profile and the necessity for more research into the central processes affected by these treatments. |
Introduction
The global prevalence of obesity and diabetes continues to surge, contributing significantly to morbidity and mortality through diseases like heart disease and cancer [1]. This is an unsolved problem and the number of people worldwide with diabetes is expected to increase in prevalence by 46% by 2045 [2]. Obesity is a major part of the problem, its complications including chronic kidney disease, hypertensive disease, stroke, and liver disease negatively impacting quality but also quantity of life as life expectancy decreases as body mass index (BMI) increases [3]. The prevalence of obesity is expected to increase from 11% to 18% from 2010 to 2030 [4] paralleling diabetes trends. In the face of this epidemic, while lifestyle modifications are critical, they often prove inadequate for long-term weight management and glycemic control, underscoring the urgent need for research and development of effective and safe anti-obesity pharmacotherapies.
Despite the development, approval and widespread prescription and use of drugs for the treatment of obesity, we have for a long time lacked highly effective anti-obesity drugs [5]. Previous pharmacological attempts to combat obesity have utilized a variety of strategies including (1) anorectics; sympathicomimetics, serotonergic agonists, opioid receptor antagonists; (2) lipase inhibitors (orlistat); (3) increasing basal energy expenditure through mitochondrial uncoupling (2,4-dinitrophenol, DNP); (4) cannabinoid receptor antagonists; and (5) combination therapies of selected gut hormones. However, most of these efforts have been plagued with significant adverse effects and/or limited efficacy which has resulted in the withdrawal and discontinued use for the majority of these medications [5].
The development of effective pharmacotherapies that entail a reduction of adverse effects has hinged on the understanding of the neuroendocrine pathways involved in the homeostatic control of body weight. This new paradigm began with the discovery of leptin, by Jeff Friedman and others in 1994, the first hormone whose primary function is the regulation of body weight, by signaling from the adipose tissue to pro-opiomelanocortin (POMC) neurons [6] in the hypothalamus to reduce food intake. This was followed by the discovery of an opposing hormone, ghrelin, which facilitates signaling from the gut and acts on agouti-related peptide (AgRP) neurons in the hypothalamus to increase food intake [7, 8].
Our understanding of obesity since the discovery of leptin has revolved around the idea that BMI has a genetic basis, with approximately 40–70% of the variation in BMI in the population being attributable to heritable factors [9], and that the majority of genetic variation associated with BMI is in the central nervous system (CNS) [10]. A prominent example of a specific mutation associated with obesity was the discovery that a four base-pair deletion in the melanocortin-4 receptor (MC4R) leads to obesity by inhibiting the leptin-melanocortin pathway [11]. However, the magnitude of the effect of a deficiency in hypothalamic neuroendocrine signaling provides a clear direction for the development of therapeutic strategies. Unfortunately leptin mimetics have failed to provide significant weight loss outside the cases of leptin-replacement therapy for patients with genetic leptin deficiency [12, 13]. Likewise, while ghrelin has the potential to be a useful treatment for cancer cachexia [14], strategies for inhibiting ghrelin signaling have thus far not produced viable pharmacotherapies for obesity, but this is an ongoing area of research [15, 16]. Leptin and ghrelin are only two of many neuroendocrine signaling pathways facilitating peripheral-brain communication regulating metabolism [17, 18]. The obvious question was whether leveraging any one or a combination of these other signaling pathways may provide a more successful path to obesity pharmacotherapy.
Among the many gut hormones implicated in control of glucose metabolism are the incretin hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). Secreted from enteroendocrine cells of the upper (GIP) or lower (GLP-1) intestine in response to food intake, they not only stimulate pancreatic insulin secretion in a glucose-dependent manner but also act in the brain to decrease body weight via inhibition of food intake. Especially in unimolecular formulations with agonism also at the receptor for glucagon, these hormones ultimately turned out to be the most effective obesity and diabetes therapies to date. Glucagon was discovered in 1923 as a hyperglycemic factor in the pancreas [19]. The classical view of the function of glucagon as a counter-regulatory hormone to insulin, whereby it is secreted by pancreatic α-cells in response to hypoglycemia and then acts on the liver to increase glucose production, has not changed [20]. Trying to develop a therapy that increases glucagon signaling would seem to be counterproductive at least in a diabetes context. However, the classical model has evolved into a complex picture: insulin secretion depends on intra-islet glucagon signaling [21] and α-cells secrete glucagon in response to fatty acids, metabolites, paracrine, endocrine, and neuronal signals. Therefore, glucagon acts not only to increase glucose production but also to induce lipolysis, fatty acid oxidation, ketogenesis, increase thermogenesis and energy expenditure, bile acid synthesis, and finally induce satiation and reduce food intake [22, 23]. These are almost all positive things for the treatment of diabetes and obesity, which makes the glucagon signaling pathway an attractive target for an anti-obesity medication despite the induction of glucose production, but only if its hyperglycemic liability can be restrained. Consistent with the observation that glucagon acts on the pancreatic β-cells to stimulate insulin secretion via the GLP-1 receptor (GLP-1R) [24,25,26,27], long-term treatment of diet-induced obese (DIO) mice with a biochemically modified water-soluble glucagon improved glucose metabolism with equal efficacy relative to treatment with exendin-4 [23].
GIP was discovered in the early 1970s by John Brown and colleagues as the gastric-inhibitory polypeptide [28, 29]. Soon thereafter, the peptide was identified as an insulin secretagogue in humans [30]. Since the insulinotropic but not the gastric-inhibitory action of GIP prevailed at physiological concentrations [30,31,32], the peptide was later renamed glucose-dependent insulinotropic polypeptide. Apart from stimulating the secretion of either insulin or glucagon in glucose-dependent manner [33], GIP promotes lipogenesis and lipid deposition under conditions of hyperinsulinema [34,35,36] while stimulating lipolysis and fatty acid oxidation under conditions of normo- or hypoinsulinemia [37,38,39]. GIPR agonism further promotes bone formation [40], has beneficial effects in animal models for atherosclerosis [41,42,43] and neurodegenerative diseases [44, 45], and decreases body weight in obese rodents via inhibition of food intake [46, 47]. GLP-1 was discovered by Svetlana Mojsov and Joel Habener in the early 1980s [48,49,50]. Initially characterized as the second incretin hormone [51], GLP-1 was later found to have pleiotropic action well outside the pancreas, with broad action in the brain and the periphery and which includes decrease of gastric emptying, inhibition of food intake, increase of natriuresis and diuresis, modulation of rodent β-cell proliferation, decrease of inflammation and apoptosis, improvement of cardiovascular function, and modulation of learning and reward behavior [52] (Fig. 1). Consistent with the broad action of the incretins in the brain and the periphery, the receptors for GIP and GLP-1 receptors show widespread distribution that includes also hypothalamic and hindbrain regions implicated in regulation of body weight and food intake [52,53,54,55]. GLP-1R is accordingly also found in the heart, and kidney, while GIPR is also expressed in the brain, adipose tissue, stomach, small intestine, bone, and heart [56]. These incretins share large parts of their amino acid sequences with glucagon, as well as having a short half-life. GIP and GLP-1 are both degraded very quickly by dipeptidyl peptidase 4 (DPP4) [57,58,59], making analogues of these endogenous hormones poor drug candidates. This limitation was first overcome in 1992 when Eng and colleagues discovered exendin-4 in the venom of the Gila monster (Heloderma suspectum) [60]. Exendin-4 shares a large portion of its amino acid sequence and glucose-lowering and insulin-sensitizing effects with GLP-1 [60, 61]. However, when compared to endogenous GLP-1, exendin-4 has a C-terminal extended (CEX) amino acid sequence, along with substitution of the N-terminal alanine-2 residue with glycine, which makes the peptide resistant to degradation by DPP4, resulting in a significantly longer half-life than endogenous GLP-1 [62].

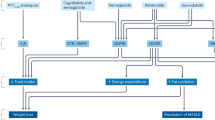
Synergies in mechanisms that regulate body weight and glucose metabolism by incretin agonism across tissues in humans [22, 23, 106]. Different incretin agonists exert complementary effects on key peripheral organs and the brain to both directly and indirectly affect energy and glucose. GIP glucose-dependent insulinotropic polypeptide, GLP-1 glucagon-like protein 1. Created by BioRender.com
Incretin Monoagonists
Exenatide, based on exendin-4, was the first incretin agonist to receive Food and Drug Administration (FDA) approval in the USA, with diabetes indication in 2005. A twice-daily injection of 10 μg yielded placebo-adjusted reductions of hemoglobin A1c (HbA1c) of − 0.8% (5 μg) and − 1.0% (10 μg) after 30 weeks of treatment. Importantly, both arms of the trial showed a significant reduction in body weight [63]. This early success of this first GLP-1R agonist drug has been followed by a number of GLP-1 analogues, including liraglutide [64, 65] and semaglutide, which has completely transformed the landscape of obesity. A once-weekly injection of 2.4 mg of semaglutide resulted in a remarkable reduction in body weight by − 14.9% in adults with obesity, along with substantial improvements in glucose homeostasis and reduced risk of cardiovascular events [66, 67].
As a result of the interrelated nature of different metabolic diseases, and the negative effect on cardiovascular health that prior drugs for the treatment of obesity had, there has long been interest in the effect of GLP-1R agonists on cardiovascular health. Another GLP-1R agonist, lixisenatide, which, like liraglutide and semaglutide, improves glycemic control and was also well tolerated in patients with type 2 diabetes [68], did not have an increase in cardiovascular events in patients with higher risk [69]. It was then found that patients with diabetes and high cardiovascular risk that were treated with liraglutide and semaglutide, but not exenatide, had a lower risk of non-fatal myocardial infarction and stroke [66, 70, 71] and another GLP-1R agonist, dulaglutide, reduced major adverse cardiovascular events [72]. Subsequently, in the SELECT trial, patients with overweight or obesity and established cardiovascular disease, but no diabetes, that were treated with semaglutide had a significantly lower composite of cardiovascular events (death from cardiovascular causes, nonfatal myocardial infarction, and nonfatal stroke) than placebo [73]. These clinical trial results clearly demonstrate the cardiovascular therapeutic usefulness of GLP-1R agonists in addition to their original indications for obesity and diabetes.
Improved patient access and lowering the treatment barrier is another area of active study for GLP-1R agonists. One way to accomplish this is by increasing the treatment interval, a once-monthly injection of efpeglenatide, a novel GLP-1R agonist, maintains a very similar efficacy and safety profile as semaglutide. In a phase II trial, once-monthly efpeglenatide reduces both HbA1c and body weight [74], and in a trial similar to the SELECT trial, patients with obesity, no diabetes, and a history of either cardiovascular or kidney disease that were treated with once-monthly efpeglenatide had a lower risk of cardiovascular events [75]. The recent development of orally administered semaglutide gives patients another treatment option: a daily dose of 50 mg in patients with obesity but not diabetes had a similar efficacy for reduction in body weight (− 15.1%) as the once-weekly injection in a phase 3 trial, with a similar tolerability profile [76]. These developments will contribute to making GLP-1R agonists an excellent treatment option for patients with overweight, diabetes, and cardiovascular risk factors.
Though far exceeding the efficacy of prior anti-obesity medications, GLP-1R monoagonism still falls short of the efficacy of bariatric surgery in treating obesity and diabetes, which resolves type 2 diabetes (T2D) for more than 80% of patients and produces an average of 25–33% reduction in body weight depending on the type of surgery employed [77, 78]. The two most commonly used bariatric surgery techniques are sleeve gastrectomy (61% of procedures in the USA) and Roux-en-Y gastric bypass (17%). The procedures involve the removal of approximately 80% of the stomach or attaching the jejunum directly to the upper portion of the stomach, respectively [79]. They are irreversible, require major lifelong lifestyle changes, and have complication rates of 11–23% [78]. The absence of alternative treatment options for severe obesity and its related diseases has forced many patients to accept the drawbacks of surgery. This situation provides the motivation to build on the effectiveness of these early drugs, aiming to bridge the gap between pharmacotherapies and surgical methods. To do this we need to look beyond GLP-1 alone.
Revisiting Glucagon
Glucagon shares many structural similarities with the incretins, which along with its ability to decrease body weight makes glucagon an interesting target for the treatment of obesity. GCGR is expressed in liver, brain, kidney, preadipocytes, pancreas, and heart [20, 80]. Unfortunately, the solubility of endogenous glucagon at physiological pH is quite low, presenting a challenge for its pharmacological use. However, DiMarchi, Tschöp, and colleagues were able to circumvent this problem by appending the same CEX amino acid sequence from exendin-4 to glucagon which improved solubility. This enabled the direct comparison of the effect of glucagon and GLP-1R agonists using mini-pumps to continuously administer this water-soluble glucagon-CEX, exendin-4, or vehicle to DIO mice. They found that glucagon-CEX and exendin-4 induced a similar reduction in body weight and improvement in glucose tolerance [23]. This finding set the blueprint for GCGR agonism, rather than antagonism, as a therapeutic approach for the treatment of obesity.
GLP-1/Glucagon Dual Agonist
With glucagon and GLP-1, there were now two peptide analogues, each having a similar effect on reducing body weight. The obvious question was whether agonist activity at both receptors simultaneously could have a synergistic effect on weight loss over monotherapies. Day et al. synthesized a peptide based on the shared amino acid sequence between glucagon and GLP-1 and the CEX tail that could act as an agonist at both receptors with nearly equal potency (Fig. 2). In DIO mice treated with the balanced dual-agonist, there was approximately 10% greater reduction in body weight than with the version favoring the GLP-1R. Both versions of the dual agonist had a similar impact on glucose tolerance. When both drugs were given to mice lacking the GLP-1R, the GLP-1-centric dual agonist produced no change in body weight while the balanced dual agonist still produced a significant reduction in body weight [81]. This first dual agonist demonstrated that unimolecular incretin multiagonists can offer synergistic effects on body weight reduction by acting on multiple receptors.

A The amino acid sequence of a GLP-1R/GCGR dual agonist MK1462 [116], B the amino acid sequence of the GLP-1R/GIPR dual agonist tirzepatide [117], and C the amino acid sequence of a triagonist [99]. These multiagonists are an assemblage of parts from each incretin and incorporated lessons learned from prior incretin mimetics: Aib substitute to prevent degradation by DPP4, acylation from liraglutide or semaglutide to increase solubility, and the CEX tail from exendin-4. GCGR glucagon receptor, GIPR GIP receptor, GLP-1R GLP-1 receptor, Aib 2-aminoisobutyric acid, DPP4 dipeptidyl peptidase 4, CEX C-terminal extension
The first GLP-1R/GCGR agonist to be put in clinical trials, cotadutide, produced comparable reductions in body weight and blood sugar levels in adults with overweight or obesity, and T2D as 1.8 mg of liraglutide [82]. While this was not a reciprocation of the magnitude of the effect in mice, cotadutide did induce a dose-dependent 4–8 times greater reduction in triglycerides over liraglutide. Survodutide, a second GLP-1R/GCGR agonist, was more successful and produced up to a − 18.7% reduction in body weight following 46 weeks of treatment in a phase 2 trial in adults with obesity [83] (Table 1). Mazdutide, another GLP-1R–GCGR dual agonist, produced a − 11.7% reduction in body weight in a phase 1b trial after just 12 weeks [84]. This demonstrates that GCGR agonism alongside GLP-1R agonism outperforms GLP-1R monoagonism preclinically and in humans.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
GLP-1/GIP Dual Agonist
The recent discovery of new synthetic GIPR agonists, which exhibit similar effects to endogenous GIP but have longer half-life and improved solubility, present yet another tool for obesity therapy. GIPR is expressed primarily in β-cells in the pancreas, but also in the brain, adipose tissue, stomach, small intestine, bone, and heart [56]. Contrary to early loss of function studies that suggested that GIPR drove an increase in body weight, there is no evidence that GIPR agonism increases body weight [85, 86]. Developing GIPR agonists has an additional challenge compared with glucagon and GLP-1, as the endogenous human and mouse GIPs differ slightly; human GIP is a poor agonist for the mouse GIPR [87, 88]. To solve this problem, a variety of stable and long-acting GIP analogues have been developed and tested for binding efficiency and action on both human and mouse GIPRs [86]. An optimized GIP agonist, exhibiting full potency for the human GIPR that also acts as an agonist at the mouse GIPR, induced a reduction in body weight and blood glucose in GLP-1R knockout mice but not GIPR knockout mice [46, 86]. This indicates that GIP agonism, and not off-target GLP-1R agonism by the GIP analogue, is responsible for the observed body weight loss. Further study found that GIPR agonism in the central nervous system (CNS) alone was sufficient to produce a significant reduction in body weight [46], an effect subsequently shown to result from GIPR signaling in inhibitory γ-aminobutyric acid (GABA)-ergic neurons [47]. This indicates that GIPR agonism offers significant therapeutic potential for the treatment of obesity.
Developed using a similar approach as the GLP-1/glucagon dual agonist, a GLP-1/GIP dual agonist produced greater reductions in body weight than GLP-1 monoagonists alone in DIO mice [46, 47, 89]. This GLP-1/GIP dual agonist was then validated in a short-term obesity study in humans, where it produced a − 2.5% reduction in body weight after 12 weeks compared to − 2% with liraglutide (− 1% for placebo), and a similar reduction in HbA1c levels [90]. Another GLP-1/GIP dual agonist, tirzepatide, was subsequently developed. In its phase 3 trial in adults with obesity, the highest dose of 15 mg once weekly produced an average reduction in body weight of 22.5% after 72 weeks of treatment [91], and a 15.7% reduction in body weight in patients with obesity and diabetes [92]. In the SURPASS trials, tirzepatide consistently outperformed placebo and active comparators in reducing HbA1c across all doses [90, 93,94,95,96]. Critically, the most frequent adverse events with tirzepatide, mild to moderate gastrointestinal events, including vomiting (5–10%), diarrhea (12–16%), and nausea (17–22%), were not higher than semaglutide in SUPRASS-2 [97]. In fact there is evidence in preclinical studies that GIPR agonism attenuates GLP-1R agonist-induced nausea and emesis [98]. Without increasing adverse events, dual agonism of GIPR and GLP-1R significantly outperforms GLP-1R monoagonism preclinically and in humans.
The advent of dual incretin agonists has marked a significant milestone in the management of obesity and T2D. We have begun to harness the synergistic effects of GLP-1 and glucagon, as well as GLP-1 and GIP, to offer superior efficacy in glucose regulation and weight management compared to any prior treatment options other than bariatric surgery.
Triagonists
In 2015, Finan and colleagues designed and characterized in DIO mice the first monomeric triple gut hormone receptor agonist, targeting simultaneously not only GIPR and GLP-1R but also the GCGR [99]. This molecule (Fig. 2) was not only engineered to target the three receptors simultaneously in a high-potency and balanced manner but also to exhibit more optimized pharmacokinetic properties. Indeed, this unimolecular triagonist (1) comprised parts of the native hormone sequences, selected to induce the desired activity at each of the targeted receptors and (2) is based on previous discoveries made during the elaboration of mixed co-agonists. It included protection against DPP4-mediated degradation, the exendin-4-based C-terminal extended sequence, and the addition of a C16 acyl chain to mediate albumin binding [99] (Fig. 1). This triagonist proved to be more efficient at inducing body weight loss than a GIPR/GLP-1R dual agonist (matched potency and equivalent doses) in DIO mice, without further reducing food intake. In the same study, triagonism was associated with greater reduction in fat mass, lower plasma cholesterol levels, and improved liver health. While the triagonist did not induce a stronger reduction in blood sugar levels in a glucose tolerance test, it did produce lower insulin levels when compared to dual agonist-treated DIO mice. In mice deficient for each of the different gut hormone receptors, the improvements in body weight and glucose metabolism seen with the triagonist rely on both GLP-1R and GIPR signaling. However, the agonism of the GCGR is specifically necessary for the supra-improvement observed with the triagonist in regard to body weight loss, fat mass reduction, and decreased food intake [99]. The authors also showed that the additional GCGR agonism enhances energy expenditure without increasing locomotor activity, which suggests an effect on nutrient utilization and fat oxidation and that the triagonist does not act solely by lowering caloric intake [99]. Finan et al. noted the importance of balancing GCGR agonism with GLP-1R/GIPR agonism to prevent any potential diabetogenic effects.
These results motivated the development of similar drugs that have now entered clinical trials, including HM15211 (Hanmi) and retatrutide (aka LY3437943, Eli Lilly). HM15211 has been shown to achieve a reduction in hepatic fat (through effects on energy intake, β-oxidation, and de novo lipogenesis), alongside decreased inflammation and fibrosis in the liver, and its clinical development focused on non-alcoholic steatohepatitis (NASH)/fibrosis treatment [100]. The improvement in liver health with a triagonist treatment is in line with the results reported in DIO mice by Finan et al. in 2015 [99]. Between 2017 and 2021, two studies were conducted, testing HM15211 in humans: a phase 1, single ascending dose trial that confirmed safety and tolerability in otherwise healthy subjects with obesity [100] (NCT03374241; and a phase 1b/2a, multiple ascending dose study, in obese subjects with non-alcoholic fatty liver disease (NCT03744182). HM15211 is currently undergoing phase 2 clinical trial for the treatment of NASH [101].
A second triagonist, retatrutide, is currently in the spotlight as it entered phase 3 clinical trials and has shown, so far, undeniably promising results regarding the treatment of both diabetes and obesity [102, 103]. In the phase 2 study for efficacy for obesity, after 48 weeks of treatment, retatrutide treatment induced a weight reduction of 15% or more in 60%, 75%, and 83% of the participants who received either 4, 8, or 12 mg of retatrutide, respectively, versus 2% of those who received the placebo treatment. The highest dose, 12 mg, of retatrutide resulted in a mean weight reduction of 24.2% in obese participants after 48 weeks [102]. Strikingly, the weight loss was continuous over the 48 weeks of treatment and had not reached a plateau [102]. Of note, at week 48, 72% of the participants beginning the trial with prediabetes had returned to normoglycemia with retatrutide vs 22% in the control placebo group.
The phase 2 trial for retatrutide efficacy in patients with diabetes also compared the safety and tolerability of retatrutide vs a placebo or dulaglutide (GLP-1R agonist)-treated groups [103]. After 24 weeks, reductions in HbA1c were significantly larger than in the placebo group for all doses and protocols of retatrutide, apart from the lowest (0.5 mg). Additionally, 8 and 12 mg of retatrutide produced greater HbA1c reductions than with 1.5 mg dulaglutide [103]. These findings remained consistent at 36 weeks, where body weight dose-dependently decreased with retatrutide treatment (up to 16.94% with 12 mg), compared to 3.00% with placebo and 2.02% with dulaglutide. At doses of 4 mg and greater, retatrutide significantly induced higher body weight loss than placebo or dulaglutide in patients with diabetes [103].
As with other peptide analogues, the most frequent adverse effects with retatrutide affected the gastrointestinal system, with mild to moderate severity, which could be attenuated with a lower starting dose [102, 103].
Taken together, these studies highlight the therapeutical potential of retatrutide for people with T2D and/or with obesity. While the safety and tolerability profile of retatrutide is compellingly similar to that of GLP-1R monoagonism and GLP-1/GIP dual agonism [102, 103], its efficacy, especially regarding obesity and its effect on body weight loss, is again greater, bringing us closer to closing the gap with bariatric surgery.
Open Questions on Mechanism for Weight Loss: Insights from Preclinical Work
Despite the plethora of incretin agonists currently in clinical trials and their impressive efficacy in obesity and T2D management, the precise mechanisms driving the reduction in body weight remain obscure, particularly regarding the enhanced effects observed with multiagonism. Agonism of each incretin receptor has different, often complementary effects on body weight and energy metabolism (Fig. 2) [5, 23, 104,105,106,107].
The most common adverse effect found in clinical trials of incretin agonists is nausea. Is there a way to keep the reduction in body weight and blood glucose while reducing these off-target effects in future generations of incretin agonists? In preclinical models, the addition of GIPR agonism to GLP-1R agonism attenuates emesis while increasing the amount of body weight reduction compared to GLP-1 agonism alone [98]. In human trials thus far, dual and triagonist drugs have shown similar adverse effects as GLP-1R monoagonists, but these preclinical studies suggest that GIPR agonism may hold the key to achieving the weight loss benefits of incretin agonism with milder adverse effects. Thus, there is particular interest in understanding the mechanisms involved in GIPR agonism. So far we know tirzepatide drives insulin secretion primarily via GIPR in human islets [108]. Moreover, GIPR signaling in the hypothalamus regulates appetite and body weight [53]. We see these superior benefits of the dual GLP-1/GIP dual agonist due to action in the CNS [46]. Endogenous GIP is likely degraded before it can reach CNS GIPRs and has its primary effect preferably on adipose tissue where it promotes positive energy balance. It is probable that the GLP-1/GIP dual agonist acts directly on the CNS, inducing satiety and weight loss in a far more potent manner than the GIP-induced promotion of positive energy balance peripherally.
Another key question that remains unanswered is what brain regions and cell types are involved? GLP-1, GCG, and GIP receptors are expressed in several different cell types in the hypothalamus and hindbrain, including endothelial cells, tanycytes, and neurons. In a recent study Liskiewicz and colleagues found that GABAergic neurons expressing the GIPR in the area postrema are activated by long-acting GIPR agonists, and GIPR signaling in GABAergic neurons is required for the body weight-lowering effects of GIP [47].
While this review has primarily focused on unimolecular gut hormone multiagonism, another open question is whether we can more effectively treat diabetes and obesity in specific patient populations by coupling gut hormone agonists with other hormone analogues. This is an active field of research, and there have been preclinical studies where GLP-1R agonists have been conjugated with other molecules. This might be an opportunity to tailor the drug for specific patient subpopulations. For example, it is becoming increasingly clear that high-fat diet feeding and obesity cause alterations in brain circuits that are critical for the control of motivational behavior. In line with this, mice treated with a GLP-1R/dexamethasone conjugate displayed a reduction in food-motivated behavior versus GLP-1R agonism alone, without increasing anxiodepressive behavior and memory [109].
Another pressing question is that of long-term weight management. Patients who discontinue semaglutide therapy and have lost weight tend to regain it fairly quickly [110]. It remains to be determined whether patients with obesity and T2D will need continuous treatment with incretin agonists to maintain glucose homeostasis and body weight, as patients at high risk of heart attack or stroke require continual statin therapy, or if at some point there will be an adaptation to lower body weight and associated benefits which can be maintained without continued treatment. Another issue pertaining to long-term patient health following incretin agonist treatment is that of muscle loss during weight loss. This is a common problem associated with any form of body weight loss, and there is no evidence that incretin agonist treatment leads to a greater proportion of lean mass loss than other methods [111]. However, as low muscle mass is associated with adverse health outcomes and higher all-cause mortality in older adults [112], it is crucial to incorporate interventions aimed at preserving lean mass during weight loss. Strategies such as resistance training and high-protein diets, alongside incretin agonist therapy, are essential for maximizing long-term patient health.
Further research is needed to investigate not only the central mechanisms affected by gut hormone agonists but also the brain areas and cell types affected by and driving obesity. Moreover, it remains unclear what happens downstream of central incretin receptor activation and what circuits are involved. Understanding the mechanisms of these drugs may not only help develop the next generation of anti-obesity medications but also improve our understanding of how body weight is regulated.
Conclusion
The evolution from single incretin agonists to the development of unimolecular agonists has, up to now, culminated in the phase 2 triumph of a retatrutide, underscoring a pivotal shift towards matching the effectiveness of bariatric surgery in treating diabetes and obesity. However, the journey forward necessitates extensive preclinical exploration to elucidate underlying mechanisms and proactive health-policy interventions to democratize patient access to these transformative therapies.
References
Murray CJL, Aravkin AY, Zheng P, et al. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396(10258):1223–49.
IDF Diabetes Atlas 10th edition. International Diabetes Federation; 2021. (IDF Diabetes Atlas). Report no. 10. https://diabetesatlas.org/atlas/tenth-edition/.
Kinlen D, Cody D, O’Shea D. Complications of obesity. QJM Mon J Assoc Physicians. 2018;111(7):437–43.
Lobstein T, Brinsden H, Neveux M. World Obesity Atlas 2022. Ludgate House, 107–111 Fleet Street, London, EC4A 2AB: World Obesity; 2022 May. https://www.worldobesity.org/resources/resource-library/world-obesity-atlas-2022.
Müller TD, Blüher M, Tschöp MH, DiMarchi RD. Anti-obesity drug discovery: advances and challenges. Nat Rev Drug Discov. 2022;21(3):201–23.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–32.
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402(6762):656–60.
Tschöp M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407(6806):908–13.
Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27(4):325–51.
Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206.
Yeo GSH, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20(2):111–2.
Farooqi IS, Jebb SA, Langmack G, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341(12):879–84.
Heymsfield SB, Greenberg AS, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282(16):1568–75.
Khatib MN, Shankar AH, Kirubakaran R, et al. Ghrelin for the management of cachexia associated with cancer. Cochrane Database Syst Rev. 2018;2018(2):CD012229.
Hagemann CA, Jensen MS, Holm S, et al. LEAP2 reduces postprandial glucose excursions and ad libitum food intake in healthy men. Cell Rep Med. 2022;3(4):100582.
Müller TD, Nogueiras R, Andermann ML, et al. Ghrelin. Mol Metab. 2015;4(6):437–60.
Clemmensen C, Müller TD, Woods SC, Berthoud HR, Seeley RJ, Tschöp MH. Gut-brain cross-talk in metabolic control. Cell. 2017;168(5):758–74.
Tschöp MH, Friedman JM. Seeking satiety: from signals to solutions. Sci Transl Med. 2023;15(723):eadh4453.
Murlin JR, Clough HD, Gibbs CBF, Stokes AM. Aqueous extracts of pancreas: I. influence on the carbohydrate metabolism of depancreatized animals. J Biol Chem. 1923;56(1):253–96.
Svoboda M, Tastenoy M, Vertongen P, Robberecht P. Relative quantitative analysis of glucagon receptor mRNA in rat tissues. Mol Cell Endocrinol. 1994;105(2):131–7.
Svendsen B, Larsen O, Gabe MBN, et al. Insulin secretion depends on intra-islet glucagon signaling. Cell Rep. 2018;25(5):1127–34.e2.
Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschöp MH. The metabolic actions of glucagon revisited. Nat Rev Endocrinol. 2010;6(12):689–97.
Müller TD, Finan B, Clemmensen C, DiMarchi RD, Tschöp MH. The new biology and pharmacology of glucagon. Physiol Rev. 2017;97(2):721–66.
Samols E, Marri G, Marks V. Interrelationship of glucagon, insulin and glucose: the insulinogenic effect of glucagon. Diabetes. 1966;15(12):855–66.
D’Alessio DA, Marks V. Glucagon as the first incretin: objects (in the rearview mirror) are closer than they appear. Diabetes. 2023;72(12):1739–40.
Capozzi ME, D’Alessio DA, Campbell JE. The past, present, and future physiology and pharmacology of glucagon. Cell Metab. 2022;34(11):1654–74.
Farahani RA, Egan AM, Welch AA, et al. The effect of glucagon-like peptide 1 receptor blockade on glucagon-induced stimulation of insulin secretion. Diabetes. 2023;72(4):449–54.
Brown JC, Mutt V, Pederson RA. Further purification of a polypeptide demonstrating enterogastrone activity. J Physiol. 1970;209(1):57–64.
Brown JC, Pederson RA, Jorpes E, Mutt V. Preparation of highly active enterogastrone. Can J Physiol Pharmacol. 1969;47(1):113–4.
Dupre J, Ross SA, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab. 1973;37(5):826–8.
Meier JJ, Goetze O, Anstipp J, et al. Gastric inhibitory polypeptide does not inhibit gastric emptying in humans. Am J Physiol Endocrinol Metab. 2004;286(4):E621–5.
Nauck MA, Bartels E, Orskov C, Ebert R, Creutzfeldt W. Lack of effect of synthetic human gastric inhibitory polypeptide and glucagon-like peptide 1 [7-36 amide] infused at near-physiological concentrations on pentagastrin-stimulated gastric acid secretion in normal human subjects. Digestion. 1992;52(3–4):214–21.
Christensen M, Vedtofte L, Holst JJ, Vilsbøll T, Knop FK. Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes. 2011;60(12):3103–9.
Kim SJ, Nian C, McIntosh CHS. Activation of lipoprotein lipase by glucose-dependent insulinotropic polypeptide in adipocytes. A role for a protein kinase B, LKB1, and AMP-activated protein kinase cascade. J Biol Chem. 2007;282(12):8557–67.
Kim SJ, Nian C, McIntosh CHS. Resistin is a key mediator of glucose-dependent insulinotropic polypeptide (GIP) stimulation of lipoprotein lipase (LPL) activity in adipocytes. J Biol Chem. 2007;282(47):34139–47.
Kim SJ, Nian C, McIntosh CHS. GIP increases human adipocyte LPL expression through CREB and TORC2-mediated trans-activation of the LPL gene. J Lipid Res. 2010;51(11):3145–57.
Ebert R, Creutzfeldt W. Metabolic effects of gastric inhibitory polypeptide. In: Blazquez E, editor. Gut regulatory peptides: their role in health and disease. Berlin: S. Karger AG; 1988.
McIntosh CH, Bremsak I, Lynn FC, et al. Glucose-dependent insulinotropic polypeptide stimulation of lipolysis in differentiated 3T3-L1 cells: wortmannin-sensitive inhibition by insulin. Endocrinology. 1999;140(1):398–404.
Heimburger SMN, Nielsen CN, Calanna S, et al. Glucose-dependent insulinotropic polypeptide induces lipolysis during stable basal insulin substitution and hyperglycaemia in men with type 1 diabetes: a randomized, double-blind, placebo-controlled, crossover clinical trial. Diabetes Obes Metab. 2022;24(1):142–7.
Xie D, Zhong Q, Ding KH, et al. Glucose-dependent insulinotropic peptide-overexpressing transgenic mice have increased bone mass. Bone. 2007;40(5):1352–60.
Sachs S, Götz A, Finan B, et al. GIP receptor agonism improves dyslipidemia and atherosclerosis independently of body weight loss in preclinical mouse model for cardio-metabolic disease. Cardiovasc Diabetol. 2023;22(1):217.
Nagashima M, Watanabe T, Terasaki M, et al. Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia. 2011;54(10):2649–59.
Nogi Y, Nagashima M, Terasaki M, Nohtomi K, Watanabe T, Hirano T. Glucose-dependent insulinotropic polypeptide prevents the progression of macrophage-driven atherosclerosis in diabetic apolipoprotein E-null mice. PLoS ONE. 2012;7(4):e35683.
Ji C, Xue GF, Li G, Li D, Hölscher C. Neuroprotective effects of glucose-dependent insulinotropic polypeptide in Alzheimer’s disease. Rev Neurosci. 2016;27(1):61–70.
Holscher C. Incretin analogues that have been developed to treat type 2 diabetes hold promise as a novel treatment strategy for Alzheimer’s disease. Recent Patents CNS Drug Discov. 2010;5(2):109–17.
Zhang Q, Delessa CT, Augustin R, et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. 2021;33(4):833-844.e5.
Liskiewicz A, Khalil A, Liskiewicz D, et al. Glucose-dependent insulinotropic polypeptide regulates body weight and food intake via GABAergic neurons in mice. Nat Metab. 2023;5(12):2075–85.
Lund PK, Goodman RH, Habener JF. Pancreatic pre-proglucagons are encoded by two separate mRNAs. J Biol Chem. 1981;256(13):6515–8.
Lund PK, Goodman RH, Dee PC, Habener JF. Pancreatic preproglucagon cDNA contains two glucagon-related coding sequences arranged in tandem. Proc Natl Acad Sci. 1982;79(2):345–9.
Lund PK, Goodman RH, Montminy MR, Dee PC, Habener JF. Anglerfish islet pre-proglucagon II: nucleotide and corresponding amino acid sequence of the cDNA. J Biol Chem. 1983;258(5):3280–4.
Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7–36: a physiological incretin in man. Lancet. 1987;2(8571):1300–4.
Müller TD, Finan B, Bloom SR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab. 2019;30:72–130.
Adriaenssens AE, Biggs EK, Darwish T, et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. 2019;30(5):987–96.e6.
Dowsett GKC, Lam BYH, Tadross JA, et al. A survey of the mouse hindbrain in the fed and fasted states using single-nucleus RNA sequencing. Mol Metab. 2021;53:101240.
Steuernagel L, Lam BYH, Klemm P, et al. HypoMap—a unified single-cell gene expression atlas of the murine hypothalamus. Nat Metab. 2022;4(10):1402–19.
Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17(6):819–37.
Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab. 1995;80(3):952–7.
Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology. 1995;136(8):3585–96.
Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem. 1993;214(3):829–35.
Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267(11):7402–5.
Gedulin BR, Nikoulina SE, Smith PA, et al. Exenatide (exendin-4) improves insulin sensitivity and {beta}-cell mass in insulin-resistant obese fa/fa Zucker rats independent of glycemia and body weight. Endocrinology. 2005;146(4):2069–76.
Göke R, Fehmann HC, Linn T, et al. Exendin-4 is a high potency agonist and truncated exendin-(9–39)-amide an antagonist at the glucagon-like peptide 1-(7–36)-amide receptor of insulin-secreting beta-cells. J Biol Chem. 1993;268(26):19650–5.
Kendall DM, Riddle MC, Rosenstock J, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care. 2005;28(5):1083–91.
Pi-Sunyer X, Astrup A, Fujioka K, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11–22.
Pratley RE, Nauck M, Bailey T, et al. Liraglutide versus sitagliptin for patients with type 2 diabetes who did not have adequate glycaemic control with metformin: a 26-week, randomised, parallel-group, open-label trial. Lancet. 2010;375(9724):1447–56.
Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375(19):1834–44.
Wilding JPH, Batterham RL, Calanna S, et al. Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med. 2021;384(11):989–1002.
Fonseca VA, Alvarado-Ruiz R, Raccah D, et al. Efficacy and safety of the once-daily GLP-1 receptor agonist lixisenatide in monotherapy: a randomized, double-blind, placebo-controlled trial in patients with type 2 diabetes (GetGoal-Mono). Diabetes Care. 2012;35(6):1225–31.
Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373(23):2247–57.
Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–22.
Holman RR, Bethel MA, Mentz RJ, et al. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2017;377(13):1228–39.
Gerstein HC, Colhoun HM, Dagenais GR, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394(10193):121–30.
Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. Semaglutide and cardiovascular outcomes in obesity without diabetes. N Engl J Med. 2023;389:2221–32.
Del Prato S, Kang J, Trautmann ME, et al. Efficacy and safety of once-monthly efpeglenatide in patients with type 2 diabetes: results of a phase 2 placebo-controlled, 16-week randomized dose-finding study. Diabetes Obes Metab. 2020;22(7):1176–86.
Gerstein HC, Sattar N, Rosenstock J, et al. Cardiovascular and renal outcomes with efpeglenatide in type 2 diabetes. N Engl J Med. 2021;385(10):896–907.
Knop FK, Aroda VR, do Vale RD, et al. Oral semaglutide 50 mg taken once per day in adults with overweight or obesity (OASIS 1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2023;402(10403):705–19.
Buchwald H, Estok R, Fahrbach K, et al. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am J Med. 2009;122(3):248–56.e5.
Chang SH, Stoll CRT, Song J, Varela JE, Eagon CJ, Colditz GA. The effectiveness and risks of bariatric surgery: an updated systematic review and meta-analysis, 2003–2012. JAMA Surg. 2014;149(3):275–87.
Arterburn DE, Telem DA, Kushner RF, Courcoulas AP. Benefits and risks of bariatric surgery in adults: a review. JAMA. 2020;324(9):879–87.
Bomholt AB, Johansen CD, Christensen JB, et al. Evaluation of commercially available glucagon receptor antibodies and glucagon receptor expression. Commun Biol. 2022;5(1):1–13.
Day JW, Ottaway N, Patterson JT, et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5(10):749–57.
Nahra R, Wang T, Gadde KM, et al. Effects of cotadutide on metabolic and hepatic parameters in adults with overweight or obesity and type 2 diabetes: a 54-week randomized phase 2b study. Diabetes Care. 2021;44(6):1433–42.
le Roux CW, Steen O, Lucas KJ, Startseva E, Unseld A, Hennige AM. A phase 2, randomized, double-blind, placebo-controlled, dose-finding study of BI 456906 in people with overweight/obesity, San Diego, CA, USA; 2023 (51-OR).
Ji L, Gao L, Jiang H, et al. Safety and efficacy of a GLP-1 and glucagon receptor dual agonist mazdutide (IBI362) 9 mg and 10 mg in Chinese adults with overweight or obesity: a randomised, placebo-controlled, multiple-ascending-dose phase 1b trial. EClinicalMedicine. 2022;54:101691.
Campbell JE. Targeting the GIPR for obesity: to agonize or antagonize? Potential mechanisms. Mol Metab. 2021;46:101139.
Mroz PA, Finan B, Gelfanov V, et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab. 2019;20:51–62.
Sparre-Ulrich AH, Hansen LS, Svendsen B, et al. Species-specific action of (Pro3)GIP—a full agonist at human GIP receptors, but a partial agonist and competitive antagonist at rat and mouse GIP receptors. Br J Pharmacol. 2016;173(1):27–38.
Ji L, Gao L, Jiang H, et al. Safety and efficacy of a GLP-1 and glucagon receptor dual agonist mazdutide (IBI362) 9 mg and 10 mg in Chinese adults with overweight or obesity: a randomised, placebo-controlled, multiple-ascending-dose phase 1b trial. EClinicalMedicine. 2022;54:101691.
Finan B, Ma T, Ottaway N, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013. https://doi.org/10.1126/scitranslmed.3007218.
Frias JP, Bastyr EJ, Vignati L, et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. 2017;26(2):343–52.e2.
Jastreboff AM, Aronne LJ, Ahmad NN, et al. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. 2022;387(3):205–16.
Garvey WT, Frias JP, Jastreboff AM, et al. Tirzepatide once weekly for the treatment of obesity in people with type 2 diabetes (SURMOUNT-2): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2023;402(10402):613–26.
Dahl D, Onishi Y, Norwood P, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022;327(6):534–45.
Del Prato S, Kahn SE, Pavo I, et al. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. 2021;398(10313):1811–24.
Ludvik B, Giorgino F, Jódar E, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet Lond Engl. 2021;398(10300):583–98.
Rosenstock J, Wysham C, Frías JP, et al. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021;398(10295):143–55.
Frías JP, Davies MJ, Rosenstock J, et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385(6):503–15.
Borner T, Geisler CE, Fortin SM, et al. GIP receptor agonism attenuates GLP-1 receptor agonist-induced nausea and emesis in preclinical models. Diabetes. 2021;70(11):2545–53.
Finan B, Yang B, Ottaway N, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. 2015;21(1):27–36.
Hanmi. Focused pipeline—HM15211(LAPS triple agonist). https://www.hanmipharm.com/ehanmi/handler/Rnd-FocusedPipelineB. Accessed 2023 Nov 11.
Abdelmalek MF, Suzuki A, Sanchez W, et al. A phase 2, adaptive randomized, double-blind, placebo-controlled, multicenter, 52-week study of HM15211 in patients with biopsy-confirmed non-alcoholic steatohepatitis—study design and rationale of HM-TRIA-201 study. Contemp Clin Trials. 2023;130:107176.
Jastreboff AM, Kaplan LM, Frías JP, et al. Triple-hormone-receptor agonist retatrutide for obesity—a phase 2 trial. N Engl J Med. 2023;389(6):514–26.
Rosenstock J, Frias J, Jastreboff AM, et al. Retatrutide, a GIP, GLP-1 and glucagon receptor agonist, for people with type 2 diabetes: a randomised, double-blind, placebo and active-controlled, parallel-group, phase 2 trial conducted in the USA. Lancet. 2023;402(10401):529–44.
Bass J, Tschöp MH, Beutler LR. Dual gut hormone receptor agonists for diabetes and obesity. J Clin Invest. 2023. https://doi.org/10.1172/JCI167952.
Campbell JE, Müller TD, Finan B, DiMarchi RD, Tschöp MH, D’Alessio DA. GIPR/GLP-1R dual agonist therapies for diabetes and weight loss—chemistry, physiology, and clinical applications. Cell Metab. 2023;35(9):1519–29.
Hammoud R, Drucker DJ. Beyond the pancreas: contrasting cardiometabolic actions of GIP and GLP1. Nat Rev Endocrinol. 2023;19(4):201–16.
Nogueiras R, Nauck MA, Tschöp MH. Gut hormone co-agonists for the treatment of obesity: from bench to bedside. Nat Metab. 2023;5(6):933–44.
El K, Douros JD, Willard FS, et al. The incretin co-agonist tirzepatide requires GIPR for hormone secretion from human islets. Nat Metab. 2023;5(6):945–54.
Décarie-Spain L, Fisette A, Zhu Z, et al. GLP-1/dexamethasone inhibits food reward without inducing mood and memory deficits in mice. Neuropharmacology. 2019;151:55–63.
Wilding JPH, Batterham RL, Davies M, et al. Weight regain and cardiometabolic effects after withdrawal of semaglutide: the STEP 1 trial extension. Diabetes Obes Metab. 2022;24(8):1553–64.
Sargeant JA, Henson J, King JA, Yates T, Khunti K, Davies MJ. A review of the effects of glucagon-like peptide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors on lean body mass in humans. Endocrinol Metab (Seoul). 2019;34(3):247–62.
Li R, Xia J, Zhang X, et al. Associations of muscle mass and strength with all-cause mortality among US older adults. Med Sci Sports Exerc. 2018;50(3):458–67.
Ji L, Jiang H, Cheng Z, et al. A phase 2 randomised controlled trial of mazdutide in Chinese overweight adults or adults with obesity. Nat Commun. 2023;14(1):8289.
OPKO Health Inc. Comparison of the oxyntomodulin analog, LY2944876, to once-weekly exenatide and to placebo in patients with type 2 diabetes. OPKO Health Inc. 2014 08-2015. https://www.clinicaltrials.gov/study/NCT02119819?id=NCT02119819&rank=1.
Romero-Gómez M, Lawitz E, Shankar RR, et al. A phase IIa active-comparator-controlled study to evaluate the efficacy and safety of efinopegdutide in patients with non-alcoholic fatty liver disease. J Hepatol. 2023;79(4):888–97.
Palani A, Nawrocki AR, Orvieto F, et al. Discovery of MK-1462: GLP-1 and glucagon receptor dual agonist for the treatment of obesity and diabetes. ACS Med Chem Lett. 2022;13(8):1248–54.
Zhao F, Zhou Q, Cong Z, et al. Structural insights into multiplexed pharmacological actions of tirzepatide and peptide 20 at the GIP, GLP-1 or glucagon receptors. Nat Commun. 2022;13(1):1057.
Medical Writing and Editorial Assistance
This authors did not use any medical writing or editorial assistance for this article.
Funding
This work was funded by the European Union within the scope or the European Research Council ERC-CoG Trusted no.101044445, awarded to TDM. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the awarding authority can be held responsible for them. TDM further received funding from the German Research Foundation (DFG TRR296, TRR152, SFB1123 and GRK 2816/1) and the German Center for Diabetes Research (DZD e.V.).
Author information
Authors and Affiliations
Contributions
Robert Gutgesell, Ruben Nogueiras, Matthias Hans Tschop, and Timo Dirk Muller contributed to the manuscript conception and design. The first draft of the manuscript was written by Robert M. Gutgesell and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Robert M. Gutgesell and Rubén Nogueiras declare that they have nothing to disclose. MHT is a member of the scientific advisory board of ERX Pharmaceuticals, Cambridge, Mass. He was a member of the Research Cluster Advisory Panel (ReCAP) of the Novo Nordisk Foundation between 2017 and 2019. He attended a scientific advisory board meeting of the Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, in 2016. He received funding for his research projects by Novo Nordisk (2016–2020) and Sanofi-Aventis (2012–2019). He was a consultant for Bionorica SE (2013–2017), Menarini Ricerche S.p.A. (2016), and Bayer Pharma AG Berlin (2016). As former Director of the Helmholtz Diabetes Center and the Institute for Diabetes and Obesity at Helmholtz Zentrum München (2011–2018), and since 2018, as CEO of Helmholtz Zentrum München, he has been responsible for collaborations with a multitude of companies and institutions, worldwide. In this capacity, he discussed potential projects with and has signed/signs contracts for his institute(s) and for the staff for research funding and/or collaborations with industry and academia, worldwide, including but not limited to pharmaceutical corporations like Boehringer Ingelheim, Eli Lilly, Novo Nordisk, Medigene, Arbormed, BioSyngen, and others. In this role, he was/is further responsible for commercial technology transfer activities of his institute(s), including diabetes-related patent portfolios of Helmholtz Zentrum München as, e.g., WO/2016/188932 A2 or WO/2017/194499 A1. MHT confirms that to the best of his knowledge none of the above funding sources were involved in the preparation of this paper. TDM receives research funding from Novo Nordisk but these funds are unrelated to the work described here. TDM received speaking fees within the last 3 years from Novo Nordisk, Eli Lilly, AstraZeneca, Merck, Berlin Chemie AG, and Mercodia.
Ethics Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Gutgesell, R.M., Nogueiras, R., Tschöp, M.H. et al. Dual and Triple Incretin-Based Co-agonists: Novel Therapeutics for Obesity and Diabetes. Diabetes Ther 15, 1069–1084 (2024). https://doi.org/10.1007/s13300-024-01566-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-024-01566-x