
Abstract
This narrative review describes a new approach to navigation in a challenging landscape of clinical drug development in diabetes. Successful outcome studies in recent years have led to new indications and guidelines in type 2 diabetes, yet the number of clinical trials in diabetes is now declining. This is due to many environmental factors acting in concert, including the prioritisation of funding for other diseases, high costs of large randomised clinical trials, increase in regulatory requirements and limited entry of novel candidate drugs. There is a need for novel and cost-effective paradigms of clinical development to meet these and other challenges. The concept of registry-based randomised clinical trials (RRCTs) is an attractive option. In this review we focus on type 2 diabetes and the prevention of cardiovascular and microvascular comorbidities and mortality, using the Swedish SMARTEST trial as an example of an RRCT. We also give some examples from other disease areas. The RRCT concept is a novel, cost-effective and scientifically sound approach for conducting large-scale diabetes trials in a real-world setting.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
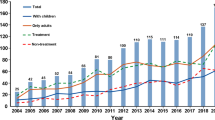
The number of RCTs in diabetes has increased over the last two decades. According to WHO data, most studies between 1999 and 2021 were conducted in the USA (24%), Japan (11%), Germany (9%), India (8.5%), the UK (8%) and China (7%) [1]. There has been unprecedented success in demonstrating long-term improvements in cardiovascular and renal clinical outcomes as well as survival rates in type 2 diabetes with the modern glucose-lowering agents glucagon-like peptide-1 (GLP-1) receptor agonists and sodium–glucose cotransporter 2 (SGLT2) inhibitors. This has been achieved by performing numerous large outcome trials, mainly in type 2 diabetes patients with established cardiovascular or renal disease. However, since 2018 the number of trials has decreased globally, except in South-East Asia (Fig. 1). In Sweden, the number of trials initiated was halved between 2004 and 2016 [2]. The reasons for this declining trend may be related not only to existing healthcare infrastructure and resources and increases in regulatory requirements for safety and efficacy documentation, but also to new legislation and funding environments, such as following Brexit in the UK [3]. Diabetes trials may also be affected by pharmaceutical companies prioritising other disease areas. In the long-term perspective, a decline in clinical trials may preclude patients with diabetes from taking advantage of new effective pharmaceuticals.

Number of registered clinical trials in diabetes worldwide, 2015–2021. Data were obtained from the WHO [1]. This figure is available as part of a downloadable slideset
In most European countries, people with type 2 diabetes are primarily diagnosed, treated and followed up in primary care [4, 5]. Thus, to ensure selection of representative study samples, clinical trials addressing people with type 2 diabetes should ideally take place in primary care settings. However, there are challenges connected to deficiencies in budget, staffing and experience. Incentives to encourage the allocation of time to recruit participants and conduct clinical trials are limited. Most primary care centres lack good clinical practice (GCP)-trained staff, and the turnover of primary care physicians is often high. This may obstruct the participation of people with type 2 diabetes in long-term follow-up studies.
There are still significant gaps in our toolbox for supporting the selection of optimal treatment(s) for each individual with type 2 diabetes, as highlighted by the Lancet Commission on diabetes [6]. Hence, a new paradigm for cost-effective clinical trials in diabetes is warranted. The registry-based randomised clinical trial (RRCT) concept is a novel, pragmatic clinical research option that can substantially cut costs and allow large trials to be conducted in regular healthcare settings. Here, we present the nuts and bolts of the RRCT concept and focus on the potential of RRCTs to evaluate new therapeutic strategies, in particular in type 2 diabetes. Some pioneering examples from various disease areas, including type 2 diabetes, are provided, obtained by searching PubMed for ‘registry-based trial’ and ‘registry-based trial and diabetes’ and screening for diabetes trials at https://clinicaltrials.gov (accessed 31 Jan 2022). This work is not a systematic or complete literature review. Instead, it focuses on recent examples of the use of healthcare registries and the novel RRCT study design. A main purpose is to encourage and stimulate future large-scale clinical trials in the diabetes arena, including in times of increasing societal, financial and regulatory challenges.
Registry-based randomised clinical trials: rationale and scope
Optimally, all questions addressing the relative effects of interventions would be studied using a randomised approach [7, 8]. In lieu of this possibility, methods for observational comparative effectiveness research are rapidly advancing as healthcare databases become increasingly detailed, structured and available. One such data source is quality-of-care registries [9], which typically are infrastructures initiated by healthcare professionals with the purpose of improving adherence to clinical care guidelines. Such registries are usually based on reports of individual data on disease history, treatments and outcomes of all comers in a particular clinical field. In many cases, open comparisons of the delivery and results of clinical care between healthcare units are also provided.
The strengths of observational comparative effectiveness studies within quality-of-care registries include their low costs, generalisability to everyday clinical care, ability to capture interventions as they are actually delivered, broad participant populations and large sample sizes, providing opportunities to identify rare and late events. Weaknesses include the variable and uncertain data quality and risk of bias, There are several examples of observational registry-based studies in Scandinavian and other Western European countries addressing the comparative effectiveness of type 2 diabetes treatments [10, 11], but very few in other regions.
In contrast, RCTs provide unconfounded effect estimates, but their weaknesses include the high or even excessive costs for trial operations, narrowly selected samples and treatment protocols with questionable generalisability to clinical reality. Several large-scale cardiovascular outcome trials in type 2 diabetes have shown low representativeness of participants regarding demographic composition, comorbidities and diabetic complications compared with patients in primary care [12]. This diminishes the generalisability and relevance of results from these trials in relation to real-world settings [13,14,15]. Interestingly, differences in the composition of study populations may become evident when analysing a comparative effectiveness observational study and a traditional RCT in parallel. Such comparisons suggest that traditional RCTs often have less representative study cohorts, and this can also involve the distribution of endpoint measures [10].
Pragmatic trial initiatives to overcome the drawbacks of both observational studies and ordinary RCTs have been proposed for more than half a century [16] but have been implemented only in the last decade following the emergence of quality-of-care registries and the possibility of registering pragmatic trials within them. RRCTs have the advantages of both the generalisability of the study samples and the confounding control of the RCT. While the aim of a traditional RCT is typically to study the efficacy of a treatment in selected participants under ideal circumstances, RRCTs aim to study the effectiveness of treatments in real-world patients under normal everyday circumstances [16]. The quality-of-care registry may support some or most parts of a clinical trial, depending on the maturity of the registry and the nature of the research questions. It can be used to identify suitable study sites and eligible participants, provide and collect consent forms, perform randomisation, collect baseline characteristics and, most importantly, capture clinical endpoints.
The development of pragmatic registry-based randomised clinical trials
RRCTs have been performed in gynaecology, orthopaedic surgery, bariatric surgery and pulmonary medicine [17,18,19,20,21,22]. The pioneers in RRCTs, however, were researchers using the Swedish Web-system for Enhancement and Development of Evidence-based care in Heart disease Evaluated According to Recommended Therapies (SWEDEHEART) [23]. One reason for the initial usefulness of this quality-of-care registry for clinical trials is that the percutaneous coronary intervention part of the registry is used online, that is, staff interact with the registry while the patient and physician are in the same room. Thus, individual patient data entered into the registry are continuously and automatically checked against inclusion criteria for trials. Suitable trials can be proposed in real-time and patients can be easily included. The first RRCT using SWEDEHEART was the Thrombus Aspiration in ST-Elevation Myocardial Infarction in Scandinavia (TASTE) trial [24], which randomised participants to either undergo coronary thrombus aspiration or not. It was extremely cost-effective as only a randomisation module was added to the usual components of the registry. The one-shot nature of the treatment did not demand longer-term safety follow-up and the outcome, all-cause death, could be obtained from the national population registry with no loss to follow-up. Later RRCTs using SWEDEHEART have explored other trial-specific additions needed for out-of-registry inclusion procedures [25] or additional safety follow-up [26]. The ease and speed of inclusion of participants into such trials is evident, as two out of three eligible participants nationwide were rapidly randomised into two of the trials [24, 26]. In contrast, an American trial with a similar research question had to be terminated prematurely because of slow enrolment [27].
Although healthcare registries have been used extensively for observational treatment studies, anchoring an RRCT to one or more registries is a novel approach. However, although pragmatic trial components have been reported historically [28] and recently [29], to date there has been very limited international implementation of the RRCT approach outside the Nordic countries [30, 31]. A prerequisite for RRCTS is timely access to high-quality registry data that can be used to determine if patients meet inclusion/exclusion criteria and/or to obtain efficacy and safety outcomes.
Notably, medical product authorities currently do not view RRCTs differently from ordinary RCTs and the regulatory demands are identical [32]. However, certain literal interpretations of GCP will hamper implementation of RRCTs [33]. An approach to GCP that values trial efficiency and completion, while maintaining adequate risk management and regulatory compliance, is crucial in both sponsor and contract research organisations. It is paramount to use data sources with high coverage that include effect and safety variables that can be reliably validated. Outcomes collected from registers should ideally include safety data with time intervals that are short enough for safe trial conduct. Otherwise, alternative procedures will be needed, for example targeted collection of serious adverse events. When outcome data are to be collected from registries, it is particularly attractive to launch a trial within a healthcare system that covers a representative target population and provides standardised follow-up data. In this regard, data from national healthcare registries that are available for research purposes are very useful. Nonetheless, health maintenance organisation and similar claims databases can also be used. For all healthcare databases, their utility will depend on their data quality and structure, their proximity to patients, healthcare providers and researchers and their coverage over geographical regions and time.
When is a registry-based randomised clinical trial the right tool?
Table 1 summarises the advantages and disadvantages of RRCTs and traditional RCTs. In brief, a traditional RCT is probably preferable when the study involves a small participant group, non-approved investigational treatments, intense safety monitoring and/or the need for frequent on-site follow-up or specialised assessments of endpoint measures (e.g. biochemical, physiological or imaging assessments). RRCTs may be the design of choice for studies involving large patient cohorts, approved and well-documented treatments and simple and established endpoints that are available in registers (e.g. hospitalisations and deaths) or in normal healthcare settings (e.g. anthropometry and routine clinical biochemistry).
Compared with traditional RCTs, RRCTs can be faster, massively cheaper, more decentralised and time-effective. They can be performed over a large geographical area with a high number of participants. This can promote both cost-efficiency and representativeness and increase statistical power. Study treatments in an RRCT are typically, but not necessarily, unmasked to participants and their local healthcare staff (but not to researchers), and this simplifies distribution of study drugs. As prospective studies require individual identifiers for all study participants, RRCTs can most easily be carried out in selected jurisdictions where individual social security numbers provide basic identifying information and are a natural part of society’s administrative system, such as in Nordic countries.
Planning for an RRCT can be even more complex, time-consuming and demanding than planning for an ordinary RCT. Data management procedures are critical, as RRCTs need to use non-traditional trial data sources, and determining the quality and usefulness of such data is key (Fig. 2). To complement the data collection that is possible in quality-of-care registries, there is often a need to set up new tools and processes for effective trial conduct. These can be generic and facilitate multiple trials, or trial-specific; in any case, the costs of and barriers to setting them up need to be factored in early. Digital tools provide multiple benefits [33]. By allowing for remote inclusion visits by video appointment, the participation of underserved and non-traditional study participants and locations is possible, providing a broader and more representative recruitment base. Digital communication also has the potential to maximise participants’ understanding of trials and it allows for remote monitoring of informed consent, saving time and money.

Data capture and flow in an RRCT. Examples of sources of data in an RRCT using multiple registries, here exemplified by the SMARTEST trial [34]. Socialstyrelsen, Swedish National Board of Health and Welfare. This figure is available as part of a downloadable slideset
As the interactions between investigators and study participants are typically sparse in RRCTs, safety surveillance in these trials is a notable focus of regulatory authorities. Ensuring that study participants provide up-to-date contact details to investigators, the development of additional safety follow-up procedures [26] and regular subscription to comprehensive adverse event data [34] may be necessary safety precautions. Ethics authorities have typically not raised higher barriers for RRCTs than for other RCTs, but we have noted a focus on ensuring that integrity issues (e.g. from cross-linking of registries) are sufficiently described in participant information. Privacy legislation in some countries or regions may raise the barriers for effective trial conduct, and exploring the utility of explicit participant consent for such data handling may be important.
Even though the RRCT model can usually easily be launched in daily healthcare settings, including primary care centres, it must be acknowledged that various types of expert support are required. This may include support for central study management, monitoring of sites and participant safety, database development, data capture and analysis, and regulatory matters (Fig. 3). The expert support needed to conduct an RRCT will depend on the objectives, participant population, interventions and outcomes of the specific trial. Hitherto, RRCTs have largely been conducted with academic sponsors; however, such studies in diabetes, as well as other fields, could equally well have a company sponsor, which may reinforce appropriate compliance with GCP guidelines and provide monitoring of and other expert support for inexperienced study sites. A take-home message is that the on-site work can resemble everyday clinical practice, whereas the central study management will be more similar to that in any RCT.

Procedures for data capture and management in an RRCT. This example is from the SMARTEST study [34] and illustrates that data flow can be complex and requires careful planning. The NDR is a quality-of-care register and is used to obtain endpoints. The Swedish Board of Health and Welfare (Socialstyrelsen [SoS]) administers national healthcare registries, used for obtaining endpoints and adverse events. AEs, adverse events; DMC, data monitoring committee; eCRF, electronic case report form; EOS, end of study; f/u, follow-up; IC, informed consent; IPD, individual patient data; PNR, personal ID number; PROMs, patient-reported outcome measures; Rx, randomised treatment; UCR, Uppsala Clinical Research Center (an academic clinical research organisation run jointly by Uppsala University and Uppsala University Hospital); UU, Uppsala University. This figure is available as part of a downloadable slideset
Use of healthcare registries in diabetes trials
Healthcare registries have long been used for observational studies, but carrying out an RCT within a registry is a novel methodology. We are not aware of any RRCTs that are completed, ongoing or being planned in the field of diabetes (https://clinicaltrials.gov; accessed 31 Jan 2022) except for the SGLT2 Inhibitor or Metformin as Standard Treatment of Early Stage Type 2 Diabetes (SMARTEST) trial (EudraCT 2019-001046-17, Clinicaltrials.gov NCT03982381) [34]. This trial can serve as an illustration of a possible design for an RRCT, including for recruitment, documentation and data collection of outcome variables. Crucial to this specific study are the Swedish National Diabetes Register and the nationwide healthcare and population registries, which contain information on medical conditions, drug treatments and deaths, with full national coverage (Fig. 2).
Swedish National Diabetes Register
The St Vincent Declaration recommended several measures to improve the quality of diabetes care and to reduce the burden of complications caused by diabetes. Among these was a need to ‘establish monitoring and control systems using the latest information technology, for quality assurance of diabetes care and for laboratory and technical procedures in diabetes diagnosis, treatment and self-management’ [35]. In Sweden, the legislation also stipulates that the quality of healthcare must be developed and secured systematically and continuously. Taken together, such recommendations and guidelines prompted the initiation of the Swedish National Diabetes Register (NDR) in the first half of the 1990s and its gradual development since then.
The original purpose of the NDR was to enable the follow-up of care units’ treatment results annually, both at individual level and at unit level, providing the possibility to compare results regionally and nationally. Clinical information, risk factor levels, treatments and the presence of diabetic complications have been reported since 1996. Initially, paper forms or computer disks were used to collect data, but presently the clinical data of at least two-thirds of participants are reported directly and securely from electronic medical record systems, and the remainder are entered manually online (https://www.ndr.nu; accessed 8 February 2022). The estimated total coverage of people with diabetes was 88% in 2020.
Several scientific studies based on NDR data have been conducted, The first studies were descriptive, presenting the registry and the presence of risk factors in participants, such as hypertension, microalbuminuria, smoking and obesity [36], but subsequently several studies have addressed the links between risk factors and macrovascular complications [37, 38].
National healthcare registries
The Nordic countries largely share history, culture and political systems as well as the organisation and financing of healthcare. Each citizen has a unique personal identity number that is used in public administration systems. This has recently been reviewed and examples of registry-based research in these countries have been provided [39]. In Sweden, there are numerous official registries and databases that are managed by authorities on behalf of the government. The quality and protection of these data are secured by laws, statutes, anonymity, confidentiality and modern information technology [9]. This includes compliance with the General Data Protection Regulation (GDPR), which has been effective in all EU member states since 25 May 2018.
There are several Swedish registries mandated by the government that have national and complete coverage and may be useful for research in healthcare and medicine. They include the Prescribed Drug Register and the National Patient Register [40, 41]. The former contains data on all prescribed drugs dispensed at pharmacies and the latter contains information on the dates of all hospital visits or inpatient stays, as well as diagnoses and procedures according to ICD-9 (http://www.icd9data.com/2007/Volume1/default.htm) or ICD-10 (http://apps.who.int/classifications/icd10/browse/2016/en) codes. In addition, the Cause of Death Register holds information on all dates and causes of death [42]. Similar registries exist in Norway, Denmark and Finland and have been used in collaborative diabetes research [11, 43].
Everyone who lives in Sweden is automatically reported in many administrative systems, which guarantees a high degree of coverage. However, the data are not primarily intended for research purposes and may be incomplete or incorrect. Nonetheless, the near-complete coverage and very large numbers of people eligible for inclusion in scientific studies present golden opportunities for large-scale studies with long follow-up times and very low dropout rates and superior generalisability.
In RRCTs, linkage to databases from one or more registries is possible with relative simplicity, as all NDR and official databases and registries use the same 12-digit unique individual identifiers. This, in combination with refined statistical methodology, have enabled national and Nordic and European collaborations to be set up to address the associations between diabetes, participant characteristics, risk factors, treatments and procedures, and clinical outcomes [44,45,46,47,48,49].
Using a registry-based randomised clinical trial to provide evidence for first-line medication in type 2 diabetes: the SMARTEST trial
Metformin is currently the recommended first-line medication for most patients with type 2 diabetes according to EASD, ADA and most national treatment guidelines [50, 51].
However, the underlying evidence is surprisingly weak and is mainly based on results from a subset of the UK Prospective Diabetes Study (UKPDS) population. In this study, 1704 participants with newly diagnosed type 2 diabetes and with overweight or obesity were randomly assigned to metformin (n=342), sulfonylureas (n=542), insulin (n=409) or diet treatment (n=411). Metformin displayed some significant or borderline significant benefits regarding diabetes-related clinical outcomes [52].
Other studies and meta-analyses provide meagre support for metformin as a treatment of choice in early type 2 diabetes, and a Cochrane meta-analysis published in 2020 [53] came to the following striking conclusion: ‘There is no clear evidence whether metformin monotherapy compared with no intervention, behaviour changing interventions or other glucose-lowering drugs influences patient-important outcomes’. Nonetheless, there are also obvious benefits of metformin. It has more than 60 years of widespread clinical use. It is effective for short- and long-term glucose lowering and, when used appropriately, it is safe and well tolerated. It can be combined with all other glucose-lowering agents and, importantly, the daily treatment cost is low.
Taken together, there is a need for head-to-head outcome trials comparing metformin with other agents as first-line medication across subgroups of type 2 diabetes. But which other candidates should be included? In fact, several drug classes may be relevant: dipeptidyl peptidase 4 (DPP-4) inhibitors, sulfonylureas, GLP-1 receptor agonists or SGLT2 inhibitors [54]. Several GLP-1 receptor agonists have shown beneficial effects in preventing major adverse cardiovascular events in type 2 diabetes patients with established CVD or very high cardiovascular risk. In such patients, SGLT2 inhibitors prevent heart failure and renal impairment events. Of note, in these trials the experimental drug or placebo was added on top of standard-of-care glucose-lowering medication and, to a large extent, blood pressure- and lipid-lowering agents [55].
Importantly, there is presently no evidence that currently justifies the broad introduction of SGLT2 inhibitors or GLP-1 receptor agonists in early stage type 2 diabetes, although some guidelines, notably from the European Society of Cardiology, have attempted to markedly widen their recommended usage [56].
A route towards evidence-based first-line pharmacological treatment
There is a need to fill the knowledge gaps regarding the efficacy of first-line medication in type 2 diabetes, in particular with respect to the prevention of chronic diabetes complications and premature death. There is some support in observational studies for favourable cardiovascular outcomes of SGLT2 inhibitor use compared with metformin [57, 58]; however, there are no ongoing long-term outcome studies sponsored by the life science industry that will provide head-to-head comparisons between metformin and other glucose-lowering agents. Together with several scientific and clinical experts, we therefore designed a national study programme, the SMARTEST trial, to compare treatment with dapagliflozin and metformin in early stage type 2 diabetes patients. This investigator-sponsored academic study started in late 2019, with Uppsala University Hospital as a pilot study site. It is ongoing across Sweden and includes about 30 study sites, mainly in primary care centres but also at a few hospital-based clinical research centres. This is an RRCT in which national healthcare registries are used for the assessment of all clinical outcomes. National and local registries are also used to identify suitable primary care centres and to enhance the recruitment of study participants. Details on the rationale and study design have been published [34] and a brief summary is presented below and in Figs 2, 3 and 4.

Schematic overview of the SMARTEST trial. Type 2 diabetes patients with less than 4 years since diagnosis are randomly assigned 1:1 to metformin or dapagliflozin treatment. They are followed until 844 events of the primary composite endpoint have occurred, and the time in the study for each participant is estimated to be 2–6 years. Other treatments are according to routine care and glucose-lowering agents can be amended as needed, while avoiding the introduction of either study drug class. PROMs, patient-reported outcome measures; SGLT2i, sodium–glucose cotransporter 2 inhibitors. This figure is available as part of a downloadable slideset
Study population, recruitment, treatments and endpoints
The main eligibility criteria are as follows:
-
adult type 2 diabetes patients without CVD or renal impairment;
-
less than 4 years since diagnosis of type 2 diabetes;
-
taking one oral or no diabetes medication.
Participants are recruited through advertisements or internal NDR or other databases or electronic health records at the individual healthcare centres. The inclusion visit can be performed physically at the nearest study site (often the same as the regular primary care centre) or via a video meeting with a study physician at the coordinating study centre, Uppsala University Hospital. Once informed consent is obtained, eligible patients are randomly allocated to the study treatment (dapagliflozin or metformin) at the inclusion visit or within a maximum of 3 weeks thereafter.
We have developed an electronic informed consent system (https://www.eic.its.uu.se; accessed 1 July 2022) [34] to make trials more cost-effective by also allowing video-based inclusions and further enabling remote monitoring of consent forms. Using digital identifiers that are widely available in Sweden for study participants and trial physicians, the system safeguards many key GCP aspects of the informed consent procedure. Many patients with type 2 diabetes live in rural areas, and video-based inclusion enables people in remote areas to participate. Informed consent can thus be obtained digitally, and screening, including blood sampling and routine clinical examination, is subsequently conducted by local healthcare staff.
Participants are randomly assigned in a 1:1 manner to metformin (1000–3000 mg per day) or dapagliflozin (10 mg once daily) (Fig. 4). Thus, study medication is open label and known to participants, local study site staff and healthcare providers but it is blinded to researchers. Any other ongoing glucose-lowering treatment is discontinued. Routine follow-up is performed in primary care according to clinical guidelines, and clinical and laboratory data are reported to the NDR at least once yearly but usually more frequently.
The composite primary outcome reflects ‘event-free survival’ and is defined as time to the first of any major cardiovascular event or microvascular events (occurrence or progression to the next defined stage) or death. Other endpoints include individual components of the primary outcome (i.e. death, heart failure, myocardial infarction or stroke, retinopathy, nephropathy, lower limb neuropathy or angiopathy); the start of insulin therapy; and cardiovascular risk markers (i.e. HbA1c, lipids, urinary albumin-to-creatinine ratio, blood pressure and BMI). Moreover, data on safety, patient-reported outcome measures and the health economy are obtained. All outcomes are collected through the national healthcare registries described above for a preliminary follow-up time of 2–5 years for each participant. For monitoring of safety and total event rates there are quarterly outputs of key events from registries. These data are blinded, although an independent data safety monitoring committee can access unblinded data as required.
In total, 844 events are required in order to achieve 90% power to detect a 20% reduction of risk with one agent vs the other. An intention-to-treat approach is applied for evaluation of treatment differences with respect to the primary composite endpoint. The initial estimate of the number of participants required was 4300, but because of a higher microvascular event rate than expected this number can probably be markedly reduced. This will improve the feasibility of the trial and is helpful for recruitment, which has been much slower than planned because of the COVID-19 pandemic and its accompanying restrictions on society and healthcare. Study medications are distributed by local pharmacies following electronic prescription and are free of cost to participants and healthcare centres. Treatment adherence is monitored both by annual telephone follow-up and using registry data on prescribed drug use. Currently, about 1100 participants are enrolled in the study (May 2022) and over 95% have started randomised treatment.
In addition to the main study, there are also add-on studies involving specific analyses. Thus, some study sites and participants are involved in detailed registry-based and clinical assessments addressing retinopathy, nephropathy, diabetic foot problems, cardiovascular events, autonomic nerve activity, home blood pressure and treatment satisfaction. In a few study sites, participants are invited to undergo repeated sampling and biobanking for analyses of blood biomarkers and, in a subset of participants, analysis of the faecal microbiome. This may help to identify responders and non-responders and contribute to future precision medicine efforts in type 2 diabetes.
Barriers related to infrastructure and resources in primary care are addressed in the SMARTEST trial by the real-world design [34]. However, to facilitate inclusion further, the SMARTEST trial provides a network connecting primary care with academia, with regular communication and the provision of information through digital and physical seminars. We have also developed a model in which general practitioners are encouraged to refer people fulfilling the inclusion criteria to the nearest clinical trial unit for screening, inclusion and randomisation [34]. These units have the infrastructure required for conducting clinical trials according to GCP guidelines, ensuring high quality of the trial.
Local healthcare databases and digital tools for participant recruitment
In order to facilitate identification of eligible participants and increase recruitment further in the SMARTEST trial, novel digital tools have been used and developed. A software programme (Medrave Software, Sweden) is used to identify patients fulfilling the inclusion criteria from medical records in several primary care centres. Primary care units are encouraged to run the programme to identify eligible patients and to send information by secured email to local recruiting clinical trial units.
As mentioned above, video-based remote visits are a useful option for promoting the inclusion of participants. This requires an electronic informed consent form with validated secure identification and signature functions. Such tools can be generic and facilitate multiple studies or they can be trial-specific. Regardless, the costs of and other barriers to setting them up need to be taken into account early on.
In the SMARTEST trial, monitoring adherence and carrying out follow-up through registers are some of the benefits of the RRCT design, as outlined in Table 1. For example, a head-to-head traditional RCT of 4–5 years’ duration in type 2 diabetes cannot easily be blinded because of the need to add on therapy over time, and frequent travel to sites may hinder participant compliance and increase dropout rates. In this real-world design, follow-up of participants is conducted in primary healthcare and physicians are carefully informed about the study procedures and the treatments allocated to participants. They are further instructed to avoid initiation of metformin or SGLT2 inhibitors (i.e. crossover from one study treatment to the other) unless there is a medical need. Participants are interviewed annually by telephone and adherence to the study medication is reviewed.
Implementation and impact
There is a large unmet need for RCTs in diabetes care, in particular for the evaluation of treatment strategies and specific pharmacological therapies, but also for the assessment of novel devices and electronic disease monitoring systems. Traditional RCTs are often performed in well-defined but narrow patient samples, and therefore ‘real-world’ trials in broader populations are warranted. The costs of traditional RCTs have risen and RRCTs are therefore appealing both because of their cost-effectiveness and because of the opportunity provided to enrol large representative patient groups. According to our experiences and those from other RRCTs, the direct study cost per participant in RRCTs can be reduced by up to 90% compared with traditional RCTs.
National healthcare registries are gradually evolving in many countries and are easily accessible sources for research on outcome data. Thus, RRCTs in diabetes can be launched in both primary and hospital-based care and will provide further evidence on optimal treatment strategies. Emerging results are likely to influence the development or revision of clinical guidelines and practice. Moreover, RRCTs may potentially provide evidence to support the approval of new medical products or new labelling of approved medicines. However, this remains to be explored and agreed with regulatory agencies, as, to our knowledge, there are currently few precedents [59, 60]. Beyond the evaluation of pharmacological treatments, it is also appealing to consider the RRCT concept for broader utility assessments of other intervention types in diabetes as well as other disease areas. These may include diet, behaviour, health service or policy strategies.
We predict that the RRCT concept will become a very useful tool for testing clinically important hypotheses with reliable and robust endpoints in type 2 diabetes. This concept will also prove to be extremely cost-effective and will enable an increasing number of large and representative clinical trials in diabetes and other disease areas to be carried out.
Abbreviations
- GCP:
-
Good clinical practice
- GLP-1:
-
Glucagon-like peptide-1
- NDR:
-
National Diabetes Register
- RRCT:
-
Registry-based randomised clinical trial
- SGLT2:
-
Sodium–glucose cotransporter 2
- SMARTEST:
-
SGLT2 Inhibitor or Metformin as Standard Treatment of Early Stage Type 2 Diabetes
- SWEDEHEART:
-
Swedish Web-system for Enhancement and Development of Evidence-based care in Heart disease Evaluated According to Recommended Therapies
References
World Health Organization (2022) Number of clinical trial registrations by location, disease, phase of development, age and sex of trial participants (1999–2021). Available at: www.who.int/observatories/global-observatory-on-health-research-and-development/monitoring/number-of-trial-registrations-by-year-location-disease-and-phase-of-development. Accessed 28 Apr 2022
Otmani M (2018) New report shows a severe decline in Swedish clinical drug trials. Available at: https://nordiclifescience.org/new-report-shows-a-severe-decline-in-swedish-clinical-drug-trials/. Accessed 10 Jan 2022
DeVito N (2019) UK Trials and Brexit. Oxford DataLab. Available at: www.bennett.ox.ac.uk/blog/2019/08/uk-trials-and-brexit-an-update/. Accessed 10 Jan 2022
Hansen LJ, Siersma V, Beck-Nielsen H, de Fine Olivarius N (2013) Structured personal care of type 2 diabetes: a 19 year follow-up of the study Diabetes Care in General Practice (DCGP). Diabetologia 56(6):1243–1253. https://doi.org/10.1007/s00125-013-2893-1
Sanchez A, Silvestre C, Campo N, Grandes G, Pred DEG (2018) Effective translation of a type-2 diabetes primary prevention programme into routine primary care: The PreDE cluster randomised clinical trial. Diabetes Res Clin Pract 139:32–42. https://doi.org/10.1016/j.diabres.2018.01.006
Chan JCN, Lim LL, Wareham NJ et al (2021) The Lancet Commission on diabetes: using data to transform diabetes care and patient lives. Lancet 396(10267):2019–2082. https://doi.org/10.1016/S0140-6736(20)32374-6
Hernan MA, Robins JM (2006) Estimating causal effects from epidemiological data. J Epidemiol Community Health 60(7):578–586. https://doi.org/10.1136/jech.2004.029496
Gerstein HC, McMurray J, Holman RR (2019) Real-world studies no substitute for RCTs in establishing efficacy. Lancet (British edition) 393(10168):210–211. https://doi.org/10.1016/S0140-6736(18)32840-X
Emilsson L, Lindahl B, Koster M, Lambe M, Ludvigsson JF (2015) Review of 103 Swedish Healthcare Quality Registries. J Intern Med 277(1):94–136. https://doi.org/10.1111/joim.12303
Norhammar A, Bodegard J, Nystrom T, Thuresson M, Nathanson D, Eriksson JW (2019) Dapagliflozin and cardiovascular mortality and disease outcomes in a population with type 2 diabetes similar to that of the DECLARE-TIMI 58 trial: A nationwide observational study. Diabetes Obes Metab 21(5):1136–1145. https://doi.org/10.1111/dom.13627
Kohsaka S, Lam CSP, Kim DJ et al (2020) Risk of cardiovascular events and death associated with initiation of SGLT2 inhibitors compared with DPP-4 inhibitors: an analysis from the CVD-REAL 2 multinational cohort study. Lancet Diabetes Endocrinol 8(7):606–615. https://doi.org/10.1016/S2213-8587(20)30130-3
Hinton W, Feher M, Munro N, Joy M, de Lusignan S (2020) Sodium-glucose co-transporter-2 inhibitor cardiovascular outcome trials and generalizability to English primary care. Diabet Med 37(9):1499–1508. https://doi.org/10.1111/dme.14290
Webb J, Mount J, von Arx LB et al (2022) Cardiovascular risk profiles: A cross-sectional study evaluating the generalizability of the glucagon-like peptide-1 receptor agonist cardiovascular outcome trials REWIND, LEADER and SUSTAIN-6 to the real-world type 2 diabetes population in the United Kingdom. Diabetes Obes Metab 24(2):289–295. https://doi.org/10.1111/dom.14580
Sciannameo V, Berchialla P, Avogaro A, Fadini GP et al (2021) Transposition of cardiovascular outcome trial effects to the real-world population of patients with type 2 diabetes. Cardiovasc Diabetol 20(1):103. https://doi.org/10.1186/s12933-021-01300-y
Sciannameo V, Berchialla P, Orsi E et al (2020) Enrolment criteria for diabetes cardiovascular outcome trials do not inform on generalizability to clinical practice: The case of glucagon-like peptide-1 receptor agonists. Diabetes Obes Metab 22(5):817–827. https://doi.org/10.1111/dom.13962
Schwartz D, Lellouch J (1967) Explanatory and pragmatic attitudes in therapeutical trials. J Chronic Dis 20(8):637–648. https://doi.org/10.1016/0021-9681(67)90041-0
Idahl A, Darelius A, Sundfeldt K, Palsson M, Strandell A (2019) Hysterectomy and opportunistic salpingectomy (HOPPSA): study protocol for a register-based randomized controlled trial. Trials 20(1):10. https://doi.org/10.1186/s13063-018-3083-8
Wolf O, Mukka S, Notini M, Moller M, Hailer NP, Duality G (2020) Study protocol: The DUALITY trial-a register-based, randomized controlled trial to investigate dual mobility cups in hip fracture patients. Acta Orthop 91(5):506–513. https://doi.org/10.1080/17453674.2020.1780059
Leta TH, Gjertsen JE, Dale H et al (2021) Antibiotic-Loaded Bone Cement in Prevention of Periprosthetic Joint Infections in Primary Total Knee Arthroplasty: A Register-based Multicentre Randomised Controlled Non-inferiority Trial (ALBA trial). BMJ Open 11(1):e041096. https://doi.org/10.1136/bmjopen-2020-041096
Blixt S, Mukka S, Forsth P, Westin O, Gerdhem P, SunBurst study group (2022) Study protocol: The SunBurst trial – a register-based, randomized controlled trial on thoracolumbar burst fractures. Acta Orthop 93:256–263. https://doi.org/10.2340/17453674.2022.1614
Sundh J, Bornefalk-Hermansson A, Ahmadi Z et al (2019) REgistry-based randomized controlled trial of treatment and Duration and mortality in long-term OXygen therapy (REDOX) study protocol. BMC Pulm Med 19(1):50. https://doi.org/10.1186/s12890-019-0809-7
Stenberg E, Szabo E, Agren G et al (2016) Closure of mesenteric defects in laparoscopic gastric bypass: a multicentre, randomised, parallel, open-label trial. Lancet 387(10026):1397–1404. https://doi.org/10.1016/S0140-6736(15)01126-5
Jernberg T, Attebring MF, Hambraeus K et al (2010) The Swedish Web-system for enhancement and development of evidence-based care in heart disease evaluated according to recommended therapies (SWEDEHEART). Heart 96(20):1617–1621. https://doi.org/10.1136/hrt.2010.198804
Lagerqvist B, Frobert O, Olivecrona GK et al (2014) Outcomes 1 year after thrombus aspiration for myocardial infarction. N Engl J Med 371(12):1111–1120. https://doi.org/10.1056/NEJMoa1405707
Hofmann R, James SK, Jernberg T et al (2017) Oxygen Therapy in Suspected Acute Myocardial Infarction. N Engl J Med 377(13):1240–1249. https://doi.org/10.1056/NEJMoa1706222
Erlinge D, Omerovic E, Frobert O et al (2017) Bivalirudin versus Heparin Monotherapy in Myocardial Infarction. N Engl J Med 377(12):1132–1142. https://doi.org/10.1056/NEJMoa1706443
Rao SV, Hess CN, Barham B et al (2014) A registry-based randomized trial comparing radial and femoral approaches in women undergoing percutaneous coronary intervention: the SAFE-PCI for Women (Study of Access Site for Enhancement of PCI for Women) trial. JACC Cardiovasc Interv 7(8):857–867. https://doi.org/10.1016/j.jcin.2014.04.007
Ford I, Norrie J (2016) Pragmatic Trials. N Engl J Med 375(5):454–463. https://doi.org/10.1056/NEJMra1510059
Jones WS, Mulder H, Wruck LM et al (2021) Comparative Effectiveness of Aspirin Dosing in Cardiovascular Disease. N Engl J Med 384(21):1981–1990. https://doi.org/10.1056/NEJMoa2102137
Lauer MS, Gordon D, Wei G, Pearson G (2017) Efficient design of clinical trials and epidemiological research: is it possible? Nat Rev Cardiol 14(8):493–501. https://doi.org/10.1038/nrcardio.2017.60
Lund LH, Oldgren J, James S (2017) Registry-Based Pragmatic Trials in Heart Failure: Current Experience and Future Directions. Curr Heart Fail Rep 14(2):59–70. https://doi.org/10.1007/s11897-017-0325-0
Gedeborg R, Cline C, Zethelius B, Salmonson T (2019) Pragmatic clinical trials in the context of regulation of medicines. Ups J Med Sci 124(1):37–41. https://doi.org/10.1080/03009734.2018.1515280
Gesualdo F, Daverio M, Palazzani L et al (2021) Digital tools in the informed consent process: a systematic review. BMC Med Ethics 22(1):18. https://doi.org/10.1186/s12910-021-00585-8
Sundstrom J, Kristofi R, Ostlund O et al (2021) A registry-based randomised trial comparing an SGLT2 inhibitor and metformin as standard treatment of early stage type 2 diabetes (SMARTEST): Rationale, design and protocol. J Diabetes Complicat 35(10):107996. https://doi.org/10.1016/j.jdiacomp.2021.107996
(1990) Diabetes care and research in Europe: the Saint Vincent declaration. Diabet Med 7(4):360. https://doi.org/10.1111/j.1464-5491.1990.tb01405.x
Gudbjornsdottir S, Cederholm J, Nilsson PM, Eliasson B, Steering Committee of the Swedish National Diabetes R (2003) The National Diabetes Register in Sweden: an implementation of the St. Vincent Declaration for Quality Improvement in Diabetes Care. Diabetes Care 26(4):1270–1276. https://doi.org/10.2337/diacare.26.4.1270
Cederholm J, Eeg-Olofsson K, Eliasson B et al (2008) Risk prediction of cardiovascular disease in type 2 diabetes: a risk equation from the Swedish National Diabetes Register. Diabetes Care 31(10):2038–2043. https://doi.org/10.2337/dc08-0662
Gudbjornsdottir S, Eliasson B, Eeg-Olofsson K, Zethelius B, Cederholm J, National Diabetes R (2011) Additive effects of glycaemia and dyslipidaemia on risk of cardiovascular diseases in type 2 diabetes: an observational study from the Swedish National Diabetes Register. Diabetologia 54(10):2544–2551. https://doi.org/10.1007/s00125-011-2218-1
Laugesen K, Ludvigsson JF, Schmidt M et al (2021) Nordic Health Registry-Based Research: A Review of Health Care Systems and Key Registries. Clin Epidemiol 13:533–554. https://doi.org/10.2147/CLEP.S314959
Wettermark B, Hammar N, Fored CM et al (2007) The new Swedish Prescribed Drug Register--opportunities for pharmacoepidemiological research and experience from the first six months. Pharmacoepidemiol Drug Saf 16(7):726–735. https://doi.org/10.1002/pds.1294
Ludvigsson JF, Andersson E, Ekbom A et al (2011) External review and validation of the Swedish national inpatient register. BMC Public Health 11:450. https://doi.org/10.1186/1471-2458-11-450
Brooke HL, Talback M, Hornblad J et al (2017) The Swedish cause of death register. Eur J Epidemiol 32(9):765–773. https://doi.org/10.1007/s10654-017-0316-1
Persson F, Bodegard J, Lahtela JT et al (2018) Different patterns of second-line treatment in type 2 diabetes after metformin monotherapy in Denmark, Finland, Norway and Sweden (D360 Nordic): A multinational observational study. Endocrinol Diabetes Metab 1(4):e00036. https://doi.org/10.1002/edm2.36
Eliasson B, Liakopoulos V, Franzen S et al (2015) Cardiovascular disease and mortality in patients with type 2 diabetes after bariatric surgery in Sweden: a nationwide, matched, observational cohort study. Lancet Diabetes Endocrinol 3(11):847–854. https://doi.org/10.1016/S2213-8587(15)00334-4
Lugner M, Sattar N, Miftaraj M et al (2021) Cardiorenal and other diabetes related outcomes with SGLT-2 inhibitors compared to GLP-1 receptor agonists in type 2 diabetes: nationwide observational study. Cardiovasc Diabetol 20(1):67. https://doi.org/10.1186/s12933-021-01258-x
Rawshani A, Rawshani A, Franzen S et al (2017) Mortality and Cardiovascular Disease in Type 1 and Type 2 Diabetes. N Engl J Med 376(15):1407–1418. https://doi.org/10.1056/NEJMoa1608664
Svanstrom H, Ueda P, Melbye M et al (2019) Use of liraglutide and risk of major cardiovascular events: a register-based cohort study in Denmark and Sweden. Lancet Diabetes Endocrinol 7(2):106–114. https://doi.org/10.1016/S2213-8587(18)30320-6
Ueda P, Svanstrom H, Melbye M et al (2018) Sodium glucose cotransporter 2 inhibitors and risk of serious adverse events: nationwide register based cohort study. BMJ 363:k4365. https://doi.org/10.1136/bmj.k4365
Berkelmans GFN, Gudbjornsdottir S, Visseren FLJ et al (2019) Prediction of individual life-years gained without cardiovascular events from lipid, blood pressure, glucose, and aspirin treatment based on data of more than 500 000 patients with Type 2 diabetes mellitus. Eur Heart J 40(34):2899–2906. https://doi.org/10.1093/eurheartj/ehy839
Buse JB, Wexler DJ, Tsapas A et al (2020) 2019 update to: Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 63(2):221–228. https://doi.org/10.1007/s00125-019-05039-w
American Diabetes Association Professional Practice Committee (2022) Summary of Revisions: Standards of Medical Care in Diabetes—2022. Diabetes Care 45(Supplement_1):S4–S7. https://doi.org/10.2337/dc22-Srev
UK Prospective Diabetes Study (UKPDS) Group (1998) Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 352(9131):854–865. https://doi.org/10.1016/S0140-6736(98)07037-8
Gnesin F, Thuesen ACB, Kahler LKA, Madsbad S, Hemmingsen B (2020) Metformin monotherapy for adults with type 2 diabetes mellitus. Cochrane Database Syst Rev 6:CD012906. https://doi.org/10.1002/14651858.CD012906.pub2
Lin DS, Lee JK, Hung CS, Chen WJ (2021) The efficacy and safety of novel classes of glucose-lowering drugs for cardiovascular outcomes: a network meta-analysis of randomised clinical trials. Diabetologia 64(12):2676–2686. https://doi.org/10.1007/s00125-021-05529-w
North EJ, Newman JD (2019) Review of cardiovascular outcomes trials of sodium-glucose cotransporter-2 inhibitors and glucagon-like peptide-1 receptor agonists. Curr Opin Cardiol 34(6):687–692. https://doi.org/10.1097/HCO.0000000000000673
Cosentino F, Grant PJ, Aboyans V et al (2020) 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J 41(2):255–323. https://doi.org/10.1093/eurheartj/ehz486
Fralick M, Schneeweiss S, Redelmeier DA, Razak F, Gomes T, Patorno E (2021) Comparative effectiveness and safety of sodium-glucose cotransporter-2 inhibitors versus metformin in patients with type 2 diabetes: An observational study using data from routine care. Diabetes Obes Metab 23(10):2320–2328. https://doi.org/10.1111/dom.14474
Shin H, Schneeweiss S, Glynn RJ, Patorno E (2022) Cardiovascular Outcomes in Patients Initiating First-Line Treatment of Type 2 Diabetes With Sodium-Glucose Cotransporter-2 Inhibitors Versus Metformin : A Cohort Study. Ann Intern Med. https://doi.org/10.7326/M21-4012
Baumfeld Andre E, Honig PK (2020) Overcoming Regulatory Aversion to Novel Methods of Evidence Generation. Clin Pharmacol Ther 107(5):1057–1058. https://doi.org/10.1002/cpt.1711
US Food and Drug Administration (2020) Type 2 diabetes mellitus: evaluating the safety of new drugs for improving glycemic control guidance for industry. US Food and Drug Administration, Rockville, MD, USA
Acknowledgements
The authors are grateful for the valuable support and advice received from colleagues in their respective institutions and from all study group members, contributors and participants of the SMARTEST trial [34].
Authors’ relationships and activities
JWE has received research support or honoraria from AstraZeneca, Merck Sharp & Dohme, NovoNordisk, Boehringer Ingelheim and Ilya Pharma. BE reports personal fees from Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Merck Sharp & Dohme, Mundipharma, Navamedic, NovoNordisk and RLS Global, and grants and personal fees from Sanofi, all outside the submitted work. LB has received honoraria from Sanofi. JS reports stock ownership in companies providing services to Amgen, AstraZeneca, Boehringer, Bayer, Eli Lilly, Itrim, Janssen, Novo Nordisk, Pfizer and Takeda, outside the submitted work.
Contribution statement
All authors conceived, wrote and critically reviewed this work and all approved the version to be published.
Funding
Open access funding provided by Uppsala University. The authors’ research in this field is supported by the Swedish Research Council (project KBF-2018-00904), a Linné grant to Lund University Diabetes Centre (LUDC 349-2006-237), EXODIAB, the Swedish Foundation for Strategic Research (LUDC IRC15-0067), Vinnova (the Swedish innovation agency), the Swedish Heart Lung Foundation (project 2019-0403) and ALF grants (government research support for university-affiliated healthcare organisations).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Slideset of figures
(PPTX 367 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eriksson, J.W., Eliasson, B., Bennet, L. et al. Registry-based randomised clinical trials: a remedy for evidence-based diabetes care?. Diabetologia 65, 1575–1586 (2022). https://doi.org/10.1007/s00125-022-05762-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-022-05762-x