
Abstract
Type 1 diabetes results from defects in immune self-tolerance that lead to inflammatory infiltrate in pancreatic islets, beta cell dysfunction and T cell-mediated killing of beta cells. Although therapies that broadly inhibit immunity show promise to mitigate autoinflammatory damage caused by effector T cells, these are unlikely to permanently reset tolerance or promote regeneration of the already diminished pool of beta cells. An emerging concept is that certain populations of immune cells may have the capacity to both promote tolerance and support the restoration of beta cells by supporting proliferation, differentiation and/or regeneration. Here we will highlight three immune cell types—macrophages, regulatory T cells and innate lymphoid cells—for which there is evidence of dual roles of immune regulation and tissue regeneration. We explore how findings in this area from other fields might be extrapolated to type 1 diabetes and highlight recent discoveries in the context of type 1 diabetes. We also discuss technological advances that are supporting this area of research and contextualise new therapeutic avenues to consider for type 1 diabetes.
Graphical abstract

Similar content being viewed by others


Introduction
Type 1 (autoimmune) diabetes is caused by T cell-mediated destruction of beta cells and characterised by a stepwise progression from autoantibody detection to impaired glucose tolerance to clinical disease [1]. In addition to autoantibodies, pancreatic abnormalities may also be detectable pre-diagnosis, including declining levels of C-peptide and altered expression of key metabolic factors varying by endotype [2, 3]. During this multi-stage process, islets become progressively infiltrated with autoreactive CD8+ T cells, and beta cells are gradually depleted [4]. Evidence that non-diabetic pancreases also have islet autoantigen-specific CD8+ T cells suggests that the breakdown of peripheral tolerance occurs at the local, rather than systemic, level [5,6,7,8]. Factors driving the development of local autoimmunity could include upregulation of MHC expression and a change in local immunoregulation within individual islets that would normally keep pathogenic T cells at bay [9, 10].
Despite the development of clinical autoimmunity, T cell-mediated beta cell destruction may not be complete, and even people with long-standing type 1 diabetes can have detectable residual beta cells [11]. This finding opens the possibility of developing therapies that could enhance the function of these preserved beta cells [12]. Immune therapies for type 1 diabetes have traditionally focused on suppressing autoreactive T cells, but as immune therapies such as teplizumab (an anti-CD3 monoclonal antibody) show promise in type 1 diabetes prevention, there is an opportunity to layer approaches that not only protect beta cells from T cell-directed attack, but also promote immunoregulation and beta cell proliferation and regeneration [13,14,15,16]. In this review we will discuss evidence supporting the concept that the properties of specialised subsets of immunoregulatory cells could be harnessed to achieve the parallel effects of immune regulation and beta cell regeneration. We focus on three cell types—macrophages (Mϕs), regulatory T cells (Tregs) and innate lymphoid cells (ILCs)—for which there is mounting evidence of dual functions of regulation and regeneration [17,18,19,20,21,22].
Is endogenous beta cell regeneration possible?
Traditional disease models postulated that beta cells were fully depleted at the time of clinical presentation, but more recent findings support the notion that even in advanced disease states, it is possible that residual reservoirs of insulin-producing beta cell mass could persist [11, 23] and might have the potential to re-gain function, even post-diagnosis [24]. The replicative quiescence of adult beta cells is thought to result from several interrelated mechanisms, including the functional potential of cell cycle regulators and epigenetic factors [25], but nevertheless there may be opportunities to intervene and restore or enhance their regenerative potential [12, 25, 26]. For example, enhancement of beta cell proliferation and regeneration has been achieved through pharmacological targeting of pathways such as the glucagon-like peptide-1 receptor (GLP-1R) and TGF-β family members [27]. To date, the highest levels of human beta cell proliferation have been induced by inhibiting the harmine-sensitive dual-specificity tyrosine phosphorylation-regulated kinase (DYRK1A) pathway. DYRK1A inhibitors are thought to work by reversing the inhibitory effect of DYRK1A on the nuclear factor activated in T cells (NFAT) family of transcription factors, which are required for cell cycle activation in beta cells. Exposure to DYRK1A inhibitors on their own increases beta cell proliferation to ~1–3% [25]; although the effect size is small, it is sufficient to improve glucose tolerance in immunodeficient mice transplanted with human islets [18, 25]. These pro-proliferative effects can be enhanced in combination with GLP-1R agonists, stimulating up to 6% of beta cells to proliferate [28]. It is encouraging that even a very small amount of endogenous beta cell regeneration and/or proliferation can be highly beneficial. It is important to note that since NFAT transcription factors are ubiquitously expressed, DYRK1A inhibitors are not beta cell specific, with reported effects in the central nervous system (CNS) and on proliferation of islet alpha and ductal cells, highlighting the need to develop beta cell-specific drug targeting strategies. Other strategies such as islet transplantation [29] or stem cell replacement also hold promise as a way to increase beta cell mass, but, as with endogenous cells subject to autoimmunity, must be coupled with approaches that limit immune attack [30]. Thus, combined with increasing evidence that specific subsets of immune cells can promote tissue repair and regeneration in a variety of disease contexts [30,31,32], research to understand how immune cells could function to both restore loss of beta function and maintain immune homeostasis post-regeneration is an exciting area for exploration.
Mϕs
Mϕs are a heterogeneous population of cells arising from different progenitors, existing within lymphoid and non-lymphoid tissues. They contribute to tissue homeostasis and inflammation through phagocytosis and the production of cytokines and growth factors [33]. States of Mϕ activation span a wide spectrum ranging from proinflammatory to regenerative phenotypes. Classically activated (M1-like) Mϕs are characterised by the expression of proinflammatory cytokines, including TNF-α, IL-1β, IL-6 and nitric oxide, and they are important for defence against pathogens [33, 34]. Regenerative or alternatively activated (M2-like) Mϕs are characterised by the production of IL-10 and TGF-β, as well as the tumour growth-promoting factor ornithine [34]. Ornithine is a precursor in the synthesis of polyamine and proline, which are important for cellular proliferation and tissue repair [35]. M2-like Mϕs are involved in anti-inflammatory responses, wound healing, tissue remodelling and immune regulation [33]. The phenotypes of tissue-resident Mϕs are plastic and can shift depending on the local environment. For example, drug-induced demyelination in the CNS causes CNS-resident Mϕs to shift from an inflammatory to a regulatory phenotype and mediate remyelination [36, 37]. Promotion of an M2-like Mϕ phenotype is similarly beneficial in angiogenesis and cardiovascular disorders [38].
Regenerative potential of Mϕs in diabetes
Mϕs constitute the majority of leucocytes detected in the islets of non-diabetic humans [39] and NOD mice before the onset of inflammation [40]. In NOD mice, Mϕs are in close contact with beta cells, sampling and presenting the contents of islet secretory granules, such as insulin peptides, to T cells [41]. Unlike Mϕs in the gut and lung, which are noninflammatory during homeostasis, mouse islet Mϕs have a classically M1-like activated phenotype and express Tnfa and Il1b even under steady state conditions [42]. In the early stages of diabetes in mice, this proinflammatory gene expression (e.g. Tnf and Il1b) in islet Mϕs is further increased [42]. Indeed, these cells are the major source of proinflammatory cytokines in response to signals such as glucose and islet amyloid polypeptide (IAPP), which are elevated with diabetes progression [43]. This increased expression of IL-1β from islet Mϕs contributes to enhanced immune responses and is associated with beta cell dysfunction and impaired glucose tolerance in a transgenic mouse model of human IAPP expression and type 2 diabetes [44]. Although an IL-1 receptor antagonist has shown some promise in improving beta cell function in type 2 diabetes [45], IL-1β signalling blockade did not preserve beta cell function in a type 1 diabetes clinical trial [46]. By contrast, C-peptide production in new-onset type 1 diabetes was preserved by administration of golimumab, a biologic that targets TNF-α, which is highly expressed by proinflammatory islet macrophages [47].
Emerging evidence suggests a previously unappreciated role for Mϕs in beta cell regeneration and proliferation in both late fetal and adult life [48]. Paradoxically, there is also evidence that islet IL-1β production can stimulate beta cell proliferation [49] and enhance beta cell function [50]. Alternatively activated Mϕs secrete molecules involved in tissue remodelling and beta cell regeneration, for example TGF-β, Wnts, IGF-1, and platelet-derived growth factor (PDGF) [22, 48]. In one study, replication of beta cells was induced by Mϕs recruited by endothelial cells upon vascular endothelial growth factor (VEGF) induction [51]. In another study, the beneficial effect of mesenchymal stem cell therapy on beta cell replication in streptozotocin (STZ)-treated mice was found to be mediated by stromal cell-derived factor 1 recruitment of Mϕs [52]. We found that in mice with STZ-induced diabetes, beta cell death is associated with marked upregulation of IGF-1 expression in islet Mϕs, and depletion of Mϕs led to impaired glucose tolerance [22]. We also showed that, similar to STZ-treated mice, islet Mϕs in db/db mice (a rodent model of type 2 diabetes) expressed lower concentrations of inflammatory cytokines (e.g. IL-6) and higher Igf1 mRNA compared with wild-type mice, supporting the idea that in models of beta cell loss and diabetes, islet Mϕs are skewed towards a tissue regenerative phenotype [22]. Several other signalling pathways may be involved in Mϕ-mediated beta cell regeneration following islet injury. One pathway is likely mediated by secretion of Wnt ligand by alternatively activated Mϕs; this pathway induces expression of activated β-catenin, leading to beta cell expansion [53, 54]. Other pathways include alternatively activated Mϕ-mediated release of TGF-β, leading to upregulation of SMAD7 and beta cell proliferation via induction of cyclin Ds and cytoplasmic translocation of p27 [17]. Beta cell proliferation can also be induced by a Mϕ-mediated PDGF-PDGF receptor (PDGFR) signalling-dependent mechanism [55].
Overall, these data suggest that islet Mϕs hold therapeutic potential to promote regeneration and proliferation of either endogenous or transplanted beta cells. Challenges to harnessing this strategy include Mϕ plasticity and methods for islet-specific targeting. With respect to plasticity, the beneficial pro-regenerative M2 phenotype could be programmed by strategies to manipulate signalling pathways to re-educate M2 Mϕs [56], or through phagocytosis of anti-inflammatory drug-loaded microparticles to enhance gene expression related to the M2 phenotype [57]. In terms of islet-specific targeting, if used with encapsulated islet replacement strategies, one could envision a co-encapsulation approach. One of the caveats associated with islet encapsulation is the poor survival of encapsulated islets caused by pericapsular fibrotic overgrowth and allograft rejection. Co-transplantation of mesenchymal stem cells (MSC) with anti-inflammatory properties together with encapsulated islets successfully improves graft survival [58], an effect possibly mediated by MSC-mediated reprogramming of Mϕs to an M2 phenotype [59]. Thus, co-transplantation of M2 macrophages with encapsulated islets may overcome the challenges associated with the long-term survival of encapsulated insulin-producing cells.
Tregs
Forkhead box P3 protein (FOXP3)+ Tregs maintain immune homeostasis and promote tolerance by suppressing the effector activity of conventional CD4+ and CD8+ T cells, as well as numerous other immune cell subsets [60]. In the NOD mouse, insulin-specific Tregs are present in inflamed islets [61], but their suppressive activity wanes with time, possibly due to inadequate IL-2 [62]. In humans, clear evidence for islet-localised Tregs is lacking, but the presence of islet-specific Tregs in blood is associated with disease protection [8]. Most children born with dysfunctional Tregs due to FOXP3 mutations have early onset type 1 diabetes [63], and numerous changes in the gene expression, phenotype and/or function of circulating Tregs in people with type 1 diabetes are reported [64]. Thus, in both mice and humans, defects in islet-specific Treg activity likely contribute to disease pathogenesis. A phase I clinical trial using polyclonal Tregs to reverse autoimmunity in patients with type 1 diabetes was found to be well-tolerated, highlighting their feasibility as a therapeutic intervention [65]. Evidence that their immune-inhibitory effect can be significantly enhanced by delivery of autoantigen-specific cells is now fuelling attempts to re-direct Treg specificity using transgenic T cell receptors or chimeric antigen receptors specific for islet-related antigens [66].
Role of Tregs in tissue regeneration
An emerging concept in Treg biology is that these cells seem to not only regulate immune homeostasis but also have the potential to promote the repair of damaged tissue. Highly activated Tregs accumulate at sites of wounded epithelia and suppress IFN-γ production and proinflammatory M1-like Mϕs, while inducing expression of pro-regenerative epidermal growth factor receptor (EGFR) [67, 68]. This phenomenon was first reported in the context of a mouse skeletal muscle injury model in which muscle-localised Tregs were found to expand post-injury via an IL-33-dependent mechanism [69] and produce amphiregulin (AREG)—an EGFR ligand—which increased satellite cell function and muscle repair. Subsequent studies in mice found evidence for Treg tissue reparative functions in other tissues, including the lung, skin and heart [67, 68, 70]. A consistent observation in mice is that tissue-localised, activated Tregs express high levels of ST2, the IL-33 receptor, and respond to IL-33 by proliferating and producing AREG [67, 71]. By stimulating EGFR signalling, AREG then mediates a variety of wound healing and tissue repair functions. In humans, we found that Tregs from blood and multiple tissues can produce AREG, but that its production negatively correlated with T cell receptor (TCR) activation [72]. Notably, at least in the tissue sources examined, human Tregs did not express ST2, so IL-33-stimulated AREG production was only observed if the cells were genetically modified to express ST2. However, consistent with mouse studies, IL-33-stimulated human ST2+ Tregs promoted the induction of a regenerative phenotype in Mϕs [72], supporting the notion that human Tregs may have both direct and indirect ways to promote tissue regeneration. Recent data suggest that human tissue-repair Tregs can be identified as CCR8+HLA-DR+ cells which express the basic leucine zipper ATF-like transcription factor (BATF) [70]. Evidence that this phenotype can be induced in peripheral Tregs by culture with TGF-β, IL-12, IL-21 and IL-23, raises the possibility of modulating therapeutic Tregs to enhance tissue repair capacity.
In addition to AREG production, Tregs can also support wound healing by expressing anti-inflammatory cytokines such as IL-10 and TGF-β [69, 73, 74]. These cytokines suppress effector T cell activation, promote the differentiation of additional Tregs at sites of damage through the process of infectious tolerance [75], and facilitate the development of a pro-repair environment. Although the mechanisms of action are yet-to-be identified, other tissue reparative functions of Tregs include enhancing the proliferation and/or differentiation of non-lymphoid cell precursors and dampening of fibrosis [71]. Beyond FOXP3+ Tregs, other types of regulatory T cells may also promote wound healing, as we recently showed for type 1 regulatory T (Tr1) cells, which promoted the repair of damaged intestinal epithelial cells via an IL-22-dependent mechanism [76].
There is also evidence that Tregs promote angiogenesis under both homeostatic and pathological conditions [32, 77]. For example, in studies of cancer, where angiogenesis is undesirable, there are associations between increased Tregs, VEGF and deleterious outcomes [77]. Tregs are thought to promote angiogenesis both by producing VEGF and via their regulatory function on other immune cells which release proangiogenic cytokines [77]. In a VEGF-independent pathway, Tregs promote angiogenesis via upregulation of TGF-β in CD4+ effector T cells [78]. In a zebrafish model of cardiac tissue injury, damaged myocardial cells stimulated peripheral FOXP3+ Tregs to infiltrate the damaged area 3 to 7 days after injury, where they regulated inflammation and promoted regeneration via the IL-10 cytokine pathway [79].
In terms of direct effects of Tregs on beta cell regeneration and proliferation, to date there is limited evidence. One study of STZ-induced diabetes found that Tregs expressing AREG promoted beta cell regeneration [80], and since human Tregs also express AREG [72], this may be a relevant pathway contributing to the success of Treg-targeted therapies. However, there is a larger body of evidence suggesting the beneficial effect of Tregs on beta cells operates via indirect mechanisms—i.e. via their well-known anti-inflammatory function and their ability to promote pro-regenerative Mϕs.
ILCs
ILCs are a family of innate lymphocytes which develop from a common lymphoid progenitor and lack antigen-specific receptors, such as those expressed by B and T lymphocytes [20]. The ILC family includes natural killer (NK) cells and non-cytotoxic ILC1s, ILC2s and ILC3s. Like NK cells, ILC1s secrete IFN-γ and express the transcription factor T-bet but are differentiated from NK cells by the lack of expression of Eomesodermin (EOMES), killer-cell immunoglobulin-like receptors (KIRs), and the CD94/NKG2 family of C-lectin receptors. ILC2s express the transcription factors GATA3 and RAR-related orphan receptor α (RORα) and secrete type 2 cytokines, including IL-4, IL-5, IL-9 and IL-13. ILC3s are subdivided based on expression of natural cytotoxicity receptors (NCR) NKp44 (human only) or NKp46 (human and mouse). Both NCR+ and NCR− ILC3s express RORγt and IL-22, alone or in combination with GM-CSF and IL-17A. Beyond functions in host defence, tissue-resident ILCs influence a broad range of biological processes including tissue repair, thermogenesis and neurogenesis [20]. Dysregulated ILC responses are also linked to a variety of autoimmune, fibrotic and rheumatoid diseases [81], making defining factors that direct ILC tissue-reparative vs proinflammatory programmes a major focus of ILC research. Complicating our understanding of ILCs further, several ‘regulatory’ populations of ILCs have been described, including regulatory NK-like cells that have cytokine profiles distinct from those of conventional NK cells [82,83,84], ILCregs which produce IL-10 and TGF-β and are defined by DNA-binding protein inhibitor ID3 expression [85], and IL-10-producing ILC2s that secrete IL-10 downstream of retinoic acid or IL-2 [85,86,87]. These regulatory ILC populations can limit autologous or allogeneic immune responses in diverse contexts [20, 81, 88], yet whether they have applications for type 1 diabetes is a relatively unexplored area of research.
Role of ILCs in tissue regeneration
Similar to Tregs, ILC2s secrete the tissue-reparative molecule AREG [20, 31, 81] and contribute to regeneration within multiple tissues. Mouse ILC2s promote epithelial cell proliferation in the intestine and lung to repair tissue damage caused by acute or chronic inflammation [31, 89, 90] and contribute to wound healing following cutaneous injury [91]. Showing direct ability to promote repair, adoptive transfer of ILC2s ameliorated ischaemia reperfusion injury—an effect linked to promotion of M2-like Mϕs in the kidney and dependent on ILC2-derived AREG [92]. Beyond AREG-driven reparative functions, type 2 cytokines from ILCs can also impact parenchymal cells and promote a regenerative programme. For example, helminth infection triggers tuft cells to secrete IL-25, which in turn induces ILC2 secretion of IL-13. ILC2-derived IL-13 then acts on epithelial crypt stem cells to differentiate and remodel the intestine after helminth expulsion [93].
ILC3s also have important functions in tissue regeneration, often through the production of IL-22. IL-22-producing ILCs stimulate epithelial cells in mucosa-associated lymphoid tissue to proliferate, secrete IL-10, and express a variety of mitogenic and anti-apoptotic molecules [94]. In the liver, IL-22-producing ILCs are central to regeneration [95]. Using a mouse model in which adaptive lymphocytes were depleted, ILC-derived IL-22 promoted liver regeneration after partial hepatectomy, and depletion of IL-22 lead to delayed hepatocellular proliferation and increased injury [95]. ILC3-derived IL-22 also allows tissue regeneration through the preservation of intestinal stem cells [96]. Intestinal IL-22 is increased following bone marrow transplant, and recipient deficiency of ILC3s leads to increased crypt apoptosis, depletion of intestinal stem cells and loss of epithelial integrity [96].
ILC1 and NK cell secretion of type 1 cytokine responses is best known for driving proinflammatory immune responses, but paradoxically these same functions are necessary to initiate tissue repair [20]. Both cell types orchestrate proinflammatory immune responses which induce organised killing and elimination of infected cells, pathogens and damaged tissue. These responses are essential for tissue repair, as repair of pathogen-induced tissue damage requires the local site be free of microbes, dying cells and debris. NK cells also participate in tissue regeneration directly, as some studies have reported induction of reparative factors by external signals. For example, mesenchymal stromal cells inhibit the proinflammatory effector functions of NK cells and promote a regulatory, senescent-like phenotype with increased expression of VEGF [97]. NK cell-derived VEGF can induce tube formation in human microvascular endothelial cells [97].
Current understanding of the role of ILCs in islet function and islet transplantation
NK cells and ILCs have been found in pancreases at steady state and in type 1 diabetic conditions in mice [14, 98, 99], and some evidence points to NK cell aberrations in type 1 diabetic patients [100, 101], but how they contribute to pancreatic homeostasis, or how ILC responses are dysregulated in type 1 diabetes is relatively unknown [102]. There is no consensus at this point for whether NK cells are protective or harmful in type 1 diabetes, as most studies examined bulk NK cells and did not distinguish between CD56bright NK cells, CD56dim NK cells or regulatory NK-like cells [103]. While studies are limited, both ILC2 and ILC3s have been linked to cellular circuits that promote beta cell function and islet homeostasis [14, 98]. For example, IL-33 production by pancreatic mesenchymal cells promoted IL-13 and colony stimulating factor (CSF)-2 secretion by ILC2s, which in turn stimulated dendritic cell and Mϕ production of retinoic acid that acted on beta cells to increase insulin production [14]. Additionally, ILC2s were protective in type 2 diabetes: ILC2-derived IL-5 was indispensable for recruitment of eosinophils and M2-like Mϕs in visceral adipose tissue, and loss of ILC2s resulted in increased insulin resistance [104]. An ILC3-dependent network was also shown to maintain islet homeostasis [98]. Specifically, gut microbiota stimulated ILC3s to produce IL-22, which induced expression of mouse β-defensin 14 (mBD14) by pancreatic endocrine cells. mBD14 then induced regulatory B cells, which in turn induced regulatory macrophages and Tregs, limiting autoimmune diabetes in NOD mice [98]. ILC3-derived IL-22 may be particularly important in the pancreas, as both exogenous and endogenous IL-22 protects beta cells from oxidation and endoplasmic reticulum stress, thereby improving insulin production [105]. These findings remain controversial, however, as exogenous IL-22 was separately shown to have no effect on the onset of diabetes in NOD mice [106]. While data on human ILC3s in healthy pancreas or in type 1 diabetes are limited, in the duodenum of individuals with type 1 diabetes, ILC1s were increased in number and frequency, with a concurrent decrease in the proportion of ILC3s [107], supporting the possibility that ILC dysregulation may occur in type 1 diabetes.
While we are still deciphering the role of ILCs in the pancreas at steady state and in type 1 diabetes, two studies reported that ILC populations can limit allogeneic immune responses to transplanted islets. Following islet transplant, NK1.1+ mouse ILCs were required for induction of tolerance to islet allografts [108]. Since the mechanism for protection was perforin dependent, it was most likely mediated by NK cells rather than ILC1s or ILC3s, which also express NK1.1. More recently, mouse IL-10-producing ILC2s were shown to prevent islet allograft rejection by increasing Treg proliferation and inhibiting inflammatory immune cells [109]. Administration of IL-33 prior to allogeneic islet transplant promoted ILC2 expansion, increased graft survival and improved blood glucose control during i.p. GTTs.
With this foundation of knowledge, it will now be important to untangle how human ILCs interact with other immune and parenchymal cells in the pancreas at steady state, contribute to type 1 diabetes pathology, or could be harnessed as therapies.
Technological advances enabling the study of immune cell–islet cross talk
Particularly in the context of human cells, past studies of natural islet–immune cell interactions have been restricted by the rarity of samples and the complexity of methods needed to untangle phenotypic heterogeneity and cell interaction complexity [10]. Single-cell technologies now enable in-depth insight into the range of immune cells found in proximity to islets as well as molecular pathways that may be active or repressed. Novel platforms may also address long-standing questions as to why beta cells are resistant to regeneration, and hence inform strategies that could be used to overcome this state [25, 102]. Spatial sequencing [110] and 3D imaging and modelling techniques [111] offer further promise for developing an accurate and reproducible map of islet–immune cell microenvironments. Single cell-omics technologies such as assay of transposable-accessible chromatin sequencing (ATAC-Seq) and cellular indexing of transcriptomes and epitopes by sequencing (CITE-Seq) are being leveraged to characterise rare cell subpopulations in pancreatic tissue and address key questions about the anatomical features and functions of pancreatic tissue [112]. Exciting technologies such as long-term pancreatic cell culture using pancreas slices or organoids [113, 114] as well as ‘organ on chip’ bioengineering methods will enable studies to develop more sophisticated, longitudinal cell network analyses to provide data on real-time lineage tracing and pathophysiological development [113]. These are already being used to model type 1 diabetes to expand the scope and relevance of in vitro methods that can be used to dissect mechanistic pathways and model microenvironment conditions [115, 116]. Finally, various online databases, such as the Human Pancreas Analysis Program, are connecting various, often extensive, pipelines of data together and will enable detailed, multi-faceted characterisation of pancreatic tissue in tandem with the expansion of data science evaluations. Overall, these new methods will enable sophisticated approaches to unravel networks of immunoregulatory–beta cell interactions so that their pro-regenerative effects can be harnessed therapeutically.
Towards a network model of immune cells and islets in type 1 diabetes
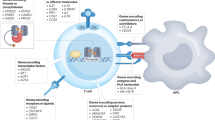
Defining how pancreas-relevant immunoregulatory and pro-regenerative functions of immune cell subsets can be harnessed in type 1 diabetes requires a detailed understanding of the normal function of protective vs pathogenic subsets so that strategies to marry immune therapies with beta cell replacement or regeneration can be developed [10, 117,118,119]. It is notable that much of the evidence for immune-mediated regenerative function seems to involve networks of immune cells, often culminating with Mϕs mediating the final effector function (Fig. 1). As research in this area evolves, it will be important to consider whether, and how, islet cells might feed back and mediate effects on the immune cells themselves. As an example, insulin can promote T cell-driven inflammation [120] and inhibit Treg suppression [121], raising the possibility that the biology of pancreatic tissue may directly contribute to autoimmune attack.

Proposed model for how beta cells interact with a network of Mϕs, Tregs and ILCs. The crosstalk among Mϕs, Tregs and ILCs with beta cells during diabetes induces beta cell regeneration. This process is dominated by Mϕs, which are influenced by Tregs and ILCs. Two major subsets of islet Mϕs may influence beta cells. Islets normally contain resident Mϕs with an M1-like phenotype that produce low levels of inflammatory cytokines. As diabetes develops, these Mϕs increase in number and produce more inflammatory cytokines, which contribute to beta cell death and dysfunction and enhanced autoimmunity. Emerging data suggest that properties of M2-like macrophages, Tregs and ILCs may be harnessed to slow or reverse this process. During beta cell recovery, islet Mϕs polarise to the M2 phenotype, and expression of proinflammatory cytokines is decreased, while the secretion of several growth factors that promote beta cell regeneration, including retinoic acid, TGF-β, IGF, PDGF and Wnt, is increased. SMAD7 overexpression in beta cells enhances proliferation by increasing cyclin Ds and inducing nuclear export of p27. Tregs and ILCs influence factors such as AREG, IL-22 and mBD14 which can directly or indirectly support beta cell function, with mechanisms often ultimately mediated by the islet-resident Mϕs. In addition to direct effects on beta cells, secretion of VEGF by Mϕs is critical for inducing angiogenesis, a process important for beta cell regeneration and pancreatic development [122, 123]. Additional signalling pathways that have not yet been identified likely exist among Tregs, ILCs, Mϕs and beta cells. This figure is available as a downloadable slide
Despite the divergent origins and immune functions of Mϕs, Tregs and ILCs, their tissue reparative properties seem to converge on numerous common pathways/mechanisms—specifically secretion of ligands for receptor tyrosine kinases expressed on beta cells, or cytokines with anti-inflammatory properties (Table 1). However, this convergence upon common pathways may also arise from a bias toward investigating previously known mechanisms. A more complete understanding of how networks of immunoregulatory cells work together to promote beta cell regeneration will require a more holistic investigation of relevant immune cells and an unbiased investigation of mechanistic pathways. These approaches are now being facilitated by numerous technological advances outlined above. Overall, the potential to harness parallel functions of immune suppression and beta cell regeneration is an exciting new area of research that promises to lead to new insight into disease pathogenesis and new therapeutic strategies for type 1 diabetes.
Abbreviations
- AREG:
-
Amphiregulin
- CNS:
-
Central nervous system
- DYRK1A:
-
Dual-specificity tyrosine phosphorylation-regulated kinase
- EGFR:
-
Epidermal growth factor receptor
- FOXP3:
-
Forkhead box P3 protein
- GLP-1R:
-
Glucagon-like peptide-1 receptor
- IAPP:
-
Islet amyloid polypeptide
- ILC:
-
Innate lymphoid cell
- Mϕ:
-
Macrophage
- mBD14:
-
Mouse β-defensin 14
- MSC:
-
Mesenchymal stem cell
- NCR:
-
Natural cytotoxicity receptor
- NFAT:
-
Nuclear factor activated in T cells
- NK:
-
Natural killer
- PDGF:
-
Platelet-derived growth factor
- ROR:
-
RAR-related orphan receptor
- STZ:
-
Streptozotocin
- Treg:
-
Regulatory T cell
- VEGF:
-
Vascular endothelial growth factor
References
Wherrett DK, Chiang JL, Delamater AM et al (2015) Defining pathways forDevelopment of disease-modifying therapies in children with type 1 diabetes: a consensus report. Diabetes Care 38(10):1975–1985. https://doi.org/10.2337/dc15-1429
Bogun MM, Bundy BN, Goland RS, Greenbaum CJ (2020) C-peptide levels in subjects followed longitudinally before and after type 1 diabetes diagnosis in TrialNet. Diabetes Care 43(8):1836–1842. https://doi.org/10.2337/dc19-2288
Battaglia M, Ahmed S, Anderson MS et al (2020) Introducing the Endotype concept to address the challenge of disease heterogeneity in type 1 diabetes. Diabetes Care 43(1):5–12. https://doi.org/10.2337/dc19-0880
Tree TIM, Peakman M (2004) Autoreactive T cells in human type 1 diabetes. Endocrinol Metab Clin N Am 33(1):113–133. https://doi.org/10.1016/s0889-8529(03)00081-1
Bender C, Rajendran S, von Herrath MG (2021) New insights into the role of autoreactive CD8 T cells and cytokines in human type 1 diabetes. Front Endocrinol 11. https://doi.org/10.3389/fendo.2020.606434
Bender C, Rodriguez-Calvo T, Amirian N, Coppieters KT, von Herrath MG (2020) The healthy exocrine pancreas contains preproinsulin-specific CD8 T cells that attack islets in type 1 diabetes. Sci Adv 6(42). https://doi.org/10.1126/sciadv.abc5586
Rodriguez-Calvo T, Krogvold L, Amirian N, Dahl-Jørgensen K, von Herrath M (2021) One in ten CD8+ cells in the pancreas of living individuals with recent onset type 1 diabetes recognizes the preproinsulin epitope PPI 15–24. Diabetes db200908-db200908. https://doi.org/10.2337/db20-0908
Wen X, Yang J, James E, Chow IT, Reijonen H, Kwok WW (2020) Increased islet antigen-specific regulatory and effector CD4+ T cells in healthy individuals with the type 1 diabetes-protective haplotype. Sci Immunol 5(44). https://doi.org/10.1126/sciimmunol.aax8767
Richardson SJ, Rodriguez-Calvo T, Gerling IC et al (2016) Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia 59(11):2448–2458. https://doi.org/10.1007/s00125-016-4067-4
Peters L, Posgai A, Brusko TM (2019) Islet–immune interactions in type 1 diabetes: the nexus of beta cell destruction. Clin Exp Immunol 198(3):326–340. https://doi.org/10.1111/cei.13349
Coppieters KT, Dotta F, Amirian N et al (2012) Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 209(1):51–60. https://doi.org/10.1084/jem.20111187
Oram RA, Sims EK, Evans-Molina C (2019) Beta cells in type 1 diabetes: mass and function; sleeping or dead? Diabetologia 62(4):567–577. https://doi.org/10.1007/s00125-019-4822-4
Herold KC, Bundy BN, Long SA et al (2019) An anti-CD3 antibody, Teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med 381(7):603–613. https://doi.org/10.1056/nejmoa1902226
Dalmas E, Lehmann FM, Dror E et al (2017) Interleukin-33-activated islet-resident innate lymphoid cells promote insulin secretion through myeloid cell retinoic acid production. Immunity 47(5):928–942.e927. https://doi.org/10.1016/j.immuni.2017.10.015
Dirice E, Kahraman S, De Jesus DF et al (2019) Increased β-cell proliferation before immune cell invasion prevents progression of type 1 diabetes. Nat Metab 1(5):509–518. https://doi.org/10.1038/s42255-019-0061-8
Molofsky AB, Van Gool F, Liang HE et al (2015) InterleuKin-33 and interferon-Γ counter-regulate group 2 innate lymphoid cell activation during immune perturbation. Immunity 43(1):161–174. https://doi.org/10.1016/j.immuni.2015.05.019
Xiao X, Gaffar I, Guo P, et al. (2014) M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc Natl Acad Sci U S A 111(13). https://doi.org/10.1073/pnas.1321347111
Wang P, Alvarez-Perez JC, Felsenfeld DP et al (2015) A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med 21(4):383–388. https://doi.org/10.1038/nm.3820
Wang YJ, Traum D, Schug J et al (2019) Multiplexed in situ imaging mass cytometry analysis of the human endocrine pancreas and immune system in type 1 diabetes. Cell Metab 29(3):769–783.e764. https://doi.org/10.1016/j.cmet.2019.01.003
Vivier E, Artis D, Colonna M et al (2018) Innate lymphoid cells: 10 years on. Cell 174(5):1054–1066. https://doi.org/10.1016/j.cell.2018.07.017
Panduro M, Benoist C, Mathis D (2016) Tissue Tregs. Annu Rev Immunol 34:609–633. https://doi.org/10.1146/annurev-immunol-032712-095948
Nackiewicz D, Dan M, Speck M et al (2020) Islet macrophages shift to a reparative state following pancreatic Beta-cell death and are a major source of islet insulin-like growth Factor-1. iScience 23(1):100775–100775. https://doi.org/10.1016/j.isci.2019.100775
Keenan HA, Sun JK, Levine J et al (2010) Residual insulin production and pancreatic β-cell turnover after 50 years of diabetes: Joslin medalist study. Diabetes 59(11):2846–2853. https://doi.org/10.2337/db10-0676
Rodriguez-Calvo T, Atkinson M, von Herrath M (2017) Beta-cell mass versus function in type 1 diabetes mellitus: truth or dare? Nat Rev Endocrinol 13(9):1. https://doi.org/10.1038/nrendo.2017.83
Karakose E, Ackeifi C, Wang P, Stewart AF (2018) Advances in drug discovery for human beta cell regeneration. Diabetologia 61(8):1693–1699. https://doi.org/10.1007/s00125-018-4639-6
Tang S, Zhang M, Zeng S et al (2020) Reversal of autoimmunity by mixed chimerism enables reactivation of β cells and transdifferentiation of α cells in diabetic NOD mice. Proc Natl Acad Sci U S A 117(49):31219–31230. https://doi.org/10.1073/pnas.2012389117
Wang P, Karakose E, Liu H et al (2019) Combined inhibition of DYRK1A, SMAD, and Trithorax pathways synergizes to induce robust replication in adult human Beta cells. Cell Metab 29(3):638–652.e635. https://doi.org/10.1016/j.cmet.2018.12.005
Ackeifi C, Wang P, Karakose E et al (2020) GLP-1 receptor agonists synergize with DYRK1A inhibitors to potentiate functional human β cell regeneration. Sci Transl Med 12(530):eaaw9996. https://doi.org/10.1126/scitranslmed.aaw9996
Shapiro AM, Pokrywczynska M, Ricordi C (2017) Clinical pancreatic islet transplantation. Nat Rev Endocrinol 13(5):268–277. https://doi.org/10.1038/nrendo.2016.178
Chen S, Du K, Zou C (2020) Current progress in stem cell therapy for type 1 diabetes mellitus. Stem Cell Res Ther 11(1):275. https://doi.org/10.1186/s13287-020-01793-6
Zaiss DMW, Gause WC, Osborne LC, Artis D (2015) Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 42(2):216–226. https://doi.org/10.1016/j.immuni.2015.01.020
Do Valle Duraes F, Lafont A, Beibel M et al (2020) Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight 5(3). https://doi.org/10.1172/jci.insight.130651
Italiani P, Boraschi D (2014) From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol 5:514. https://doi.org/10.3389/fimmu.2014.00514
Mills CD, Lenz LL, Ley K (2015) Macrophages at the fork in the road to health or disease. Front Immunol 6. https://doi.org/10.3389/fimmu.2015.00059
Clemente SG, van Waarde A, Antunes IF, Domling A, Elsinga PH (2020) Arginase as a potential biomarker of disease progression: a molecular imaging perspective. Int J Mol Sci 21(15):5291. https://doi.org/10.3390/ijms21155291
Miron VE, Boyd A, Zhao JW et al (2013) M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16(9):1211–1218. https://doi.org/10.1038/nn.3469
McMurran CE, Jones CA, Fitzgerald DC, Franklin RJ (2016) CNS Remyelination and the innate immune system. Front Cell Dev Biol 4:38. https://doi.org/10.3389/fcell.2016.00038
Wynn TA, Vannella KM (2016) Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44(3):450–462. https://doi.org/10.1016/j.immuni.2016.02.015
Denroche HC, Miard S, Salle-Lefort S, Picard F, Verchere CB (2021) T cells accumulate in non-diabetic islets during ageing. Immun Ageing 18(1):8. https://doi.org/10.1186/s12979-021-00221-4
Carrero JA, Calderon B, Towfic F, Artyomov MN, Unanue ER (2013) Defining the transcriptional and cellular landscape of type 1 diabetes in the NOD mouse. PLoS One 8(3):e59701. https://doi.org/10.1371/journal.pone.0059701
Vomund AN, Zinselmeyer BH, Hughes J et al (2015) Beta cells transfer vesicles containing insulin to phagocytes for presentation to T cells. Proc Natl Acad Sci U S A 112(40):E5496–E5502. https://doi.org/10.1073/pnas.1515954112
Ferris ST, Zakharov PN, Wan X et al (2017) The islet-resident macrophage is in an inflammatory state and senses microbial products in blood. J Exp Med 214(8):2369–2385. https://doi.org/10.1084/jem.20170074
Westwell-Roper CY, Ehses JA, Verchere CB (2014) Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1β production and β-cell dysfunction. Diabetes 63(5):1698–1711. https://doi.org/10.2337/db13-0863
Westwell-Roper CY, Chehroudi CA, Denroche HC, Courtade JA, Ehses JA, Verchere CB (2015) IL-1 mediates amyloid-associated islet dysfunction and inflammation in human islet amyloid polypeptide transgenic mice. Diabetologia 58(3):575–585. https://doi.org/10.1007/s00125-014-3447-x
Larsen CM, Faulenbach M, Vaag A et al (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356(15):1517–1526. https://doi.org/10.1056/NEJMoa065213
Moran A, Bundy B, Becker DJ et al (2013) Interleukin-1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double-blind, placebo-controlled trials. Lancet 381(9881):1905–1915. https://doi.org/10.1016/S0140-6736(13)60023-9
Quattrin T, Haller MJ, Steck AK et al (2020) Golimumab and Beta-cell function in youth with new-onset type 1 diabetes. N Engl J Med 383(21):2007–2017. https://doi.org/10.1056/NEJMoa2006136
Xiao X, Gittes GK (2015) Concise review: new insights into the role of macrophages in β-cell proliferation. Stem Cells Transl Med 4(6):655–658. https://doi.org/10.5966/sctm.2014-0248
Böni-Schnetzler M, Häuselmann SP, Dalmas E et al (2018) β cell-specific deletion of the IL-1 receptor antagonist impairs β cell proliferation and insulin secretion. Cell Rep 22(7):1774–1786. https://doi.org/10.1016/j.celrep.2018.01.063
Hajmrle C, Smith N, Spigelman AF et al (2016) Interleukin-1 signaling contributes to acute islet compensation. JCI Insight 1(4):e86055. https://doi.org/10.1172/jci.insight.86055
Saunders DC, Aamodt KI, Richardson TM et al (2021) Coordinated interactions between endothelial cells and macrophages in the islet microenvironment promote beta cell regeneration. NPJ Regen Med 6(1):22. https://doi.org/10.1038/s41536-021-00129-z
Cao X, Han Z-B, Zhao H, Liu Q (2014) Transplantation of mesenchymal stem cells recruits trophic macrophages to induce pancreatic beta cell regeneration in diabetic mice. Int J Biochem Cell Biol 53:372–379. https://doi.org/10.1016/j.biocel.2014.06.003
Criscimanna A, Coudriet GM, Gittes GK, Piganelli JD, Esni F (2014) Activated macrophages create lineage-specific microenvironments for pancreatic acinar- and β-cell regeneration in mice. Gastroenterology 147(5):1106–1118.e1111. https://doi.org/10.1053/j.gastro.2014.08.008
Rulifson IC, Karnik SK, Heiser PW et al (2007) Wnt signaling regulates pancreatic beta cell proliferation. Proc Natl Acad Sci U S A 104(15):6247–6252. https://doi.org/10.1073/pnas.0701509104
Ying W, Lee YS, Dong Y et al (2019) Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab 29(2):457–474.e455. https://doi.org/10.1016/j.cmet.2018.12.003
Sahay P, Bava EP, Iyer S, Dudeja V (2020) Modulation of macrophage polarity for treatment of acute pancreatitis: are we there yet? EBioMedicine 60:103002. https://doi.org/10.1016/j.ebiom.2020.103002
Wofford KL, Cullen DK, Spiller KL (2019) Modulation of macrophage phenotype via phagocytosis of drug-loaded microparticles. J Biomed Mater Res A 107(6):1213–1224. https://doi.org/10.1002/jbm.a.36617
Vaithilingam V, Evans MDM, Lewy DM, Bean PA, Bal S, Tuch BE (2017) Co-encapsulation and co-transplantation of mesenchymal stem cells reduces pericapsular fibrosis and improves encapsulated islet survival and function when allografted. Sci Rep 7(1):10059. https://doi.org/10.1038/s41598-017-10359-1
Vasandan AB, Jahnavi S, Shashank C, Prasad P, Kumar A, Prasanna SJ (2016) Human mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2-dependent mechanism. Sci Rep 6:38308. https://doi.org/10.1038/srep38308
Josefowicz SZ, Lu LF, Rudensky AY (2012) Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30:531–564. https://doi.org/10.1146/annurev.immunol.25.022106.141623
Spence A, Purtha W, Tam J et al (2018) Revealing the specificity of regulatory T cells in murine autoimmune diabetes. Proc Natl Acad Sci U S A 115(20):5265–5270. https://doi.org/10.1073/pnas.1715590115
Spence A, Tang Q (2016) Restoring regulatory T cells in type 1 diabetes. Curr Diab Rep 16(11):110. https://doi.org/10.1007/s11892-016-0807-6
Cepika A-M, Sato Y, Liu JM-H, Uyeda MJ, Bacchetta R, Roncarolo MG (2018) Tregopathies: monogenic diseases resulting in regulatory T-cell deficiency. J Allergy Clin Immunol 142(6):1679–1695. https://doi.org/10.1016/j.jaci.2018.10.026
Bettini M, Bettini ML (2021) Function, failure, and the future potential of Tregs in type 1 diabetes. Diabetes. https://doi.org/10.2337/dbi18-0058
Bluestone JA, Buckner JH, Fitch M et al (2015) Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 7(315):315ra189–315ra189. https://doi.org/10.1126/scitranslmed.aad4134
Raffin C, Vo LT, Bluestone JA (2020) Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol 20(3):158–172. https://doi.org/10.1038/s41577-019-0232-6
Nosbaum A, Prevel N, Truong HA et al (2016) Cutting edge: regulatory T cells facilitate cutaneous wound healing. J Immunol 196(5):2010–2014. https://doi.org/10.4049/jimmunol.1502139
Ali N, Rosenblum MD (2017) Regulatory T cells in skin. Immunology 152(3):372–381. https://doi.org/10.1111/imm.12791
Burzyn D, Kuswanto W, Kolodin D et al (2013) A special population of regulatory T cells potentiates muscle repair. Cell 155(6):1282–1295. https://doi.org/10.1016/j.cell.2013.10.054
Delacher M, Simon M, Sanderink L et al (2021) Single-cell chromatin accessibility landscape identifies tissue repair program in human regulatory T cells. Immunity. https://doi.org/10.1016/j.immuni.2021.03.007
Munoz-Rojas AR, Mathis D (2021) Tissue regulatory T cells: regulatory chameleons. Nat Rev Immunol. https://doi.org/10.1038/s41577-021-00519-w
Lam AJ, MacDonald KN, Pesenacker AM et al (2019) Innate control of tissue-reparative human regulatory T cells. J Immunol 202(8):2195–2209. https://doi.org/10.4049/jimmunol.1801330
Zaiss DM, Minutti CM, Knipper JA (2019) Immune- and non-immune-mediated roles of regulatory T-cells during wound healing. Immunology 157(3):190–197. https://doi.org/10.1111/imm.13057
Arpaia N, Green JA, Moltedo B et al (2015) A distinct function of regulatory T cells in tissue protection. Cell 162(5):1078–1089. https://doi.org/10.1016/j.cell.2015.08.021
Wan YY, Flavell RA (2007) ‘Yin–Yang’ functions of transforming growth factor-β and T regulatory cells in immune regulation. Immunol Rev 220(1):199–213. https://doi.org/10.1111/j.1600-065x.2007.00565.x
Cook L, Stahl M, Han X et al (2019) Suppressive and gut-reparative functions of human type 1 T regulatory cells. Gastroenterology 157(6):1584–1598. https://doi.org/10.1053/j.gastro.2019.09.002
Lužnik Z, Anchouche S, Dana R, Yin J (2020) Regulatory T cells in angiogenesis. J Immunol 205(10):2557–2565. https://doi.org/10.4049/jimmunol.2000574
Leung OM, Li J, Li X et al (2018) Regulatory T cells promote Apelin-mediated sprouting angiogenesis in type 2 diabetes. Cell Rep 24(6):1610–1626. https://doi.org/10.1016/j.celrep.2018.07.019
Kikuchi K (2020) New function of zebrafish regulatory T cells in organ regeneration. Curr Opin Immunol 63:7–13. https://doi.org/10.1016/j.coi.2019.10.001
Zhou L, He X, Cai P et al (2021) Induced regulatory T cells suppress Tc1 cells through TGF-β signaling to ameliorate STZ-induced type 1 diabetes mellitus. Cell Mol Immunol 18(3):698–710. https://doi.org/10.1038/s41423-020-00623-2
Shikhagaie MM, Germar K, Bal SM, Ros XR, Spits H (2017) Innate lymphoid cells in autoimmunity: emerging regulators in rheumatic diseases. Nat Rev Rheumatol 13(3):164–173. https://doi.org/10.1038/nrrheum.2016.218
Crome SQ, Nguyen LT, Lopez-Verges S et al (2017) A distinct innate lymphoid cell population regulates tumor-associated T cells. Nat Med 23(3):368–375. https://doi.org/10.1038/nm.4278
Neo SY, Yang Y, Record J et al (2020) CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J Clin Invest 130(3):1185–1198. https://doi.org/10.1172/JCI128895
Ramirez-Ramirez D, Padilla-Castaneda S, Galan-Enriquez CS et al (2019) CRTAM(+) NK cells endowed with suppressor properties arise in leukemic bone marrow. J Leukoc Biol 105(5):999–1013. https://doi.org/10.1002/JLB.MA0618-231R
Wang S, Xia P, Chen Y et al (2017) Regulatory innate lymphoid cells control innate intestinal inflammation. Cell 171(1):201–216.e218. https://doi.org/10.1016/j.cell.2017.07.027
Morita H, Kubo T, Rückert B et al (2019) Induction of human regulatory innate lymphoid cells from group 2 innate lymphoid cells by retinoic acid. J Allergy Clin Immunol 143(6):2190–2201.e2199. https://doi.org/10.1016/j.jaci.2018.12.1018
Seehus CR, Kadavallore A, Torre BDL et al (2017) Alternative activation generates IL-10 producing type 2 innate lymphoid cells. Nat Commun 8(1):1–13. https://doi.org/10.1038/s41467-017-02023-z
Bruce DW, Stefanski HE, Vincent BG et al (2017) Type 2 innate lymphoid cells treat and prevent acute gastrointestinal graft-versus-host disease. J Clin Invest 127(5):1813–1825. https://doi.org/10.1172/JCI91816
Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DMW, Artis D (2015) IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A 112(34):10762–10767. https://doi.org/10.1073/pnas.1509070112
Monticelli LA, Sonnenberg GF, Abt MC et al (2011) Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12(11):1045–1054. https://doi.org/10.1031/ni.2131
Rak GD, Osborne LC, Siracusa MC et al (2016) IL-33-dependent group 2 innate lymphoid cells promote cutaneous wound healing. J Invest Dermatol 136(2):487–496. https://doi.org/10.1038/JID.2015.406
Cao Q, Wang Y, Niu Z et al (2018) Potentiating tissue-resident type 2 innate lymphoid cells by IL-33 to prevent renal ischemia-reperfusion injury. J Am Soc Nephrol 29(3):961–976. https://doi.org/10.1681/ASN.2017070774
Von Moltke J, Ji M, Liang H-E, Locksley RM (2016) Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature 529(7585):221–225. https://doi.org/10.1038/nature16161
Cella M, Fuchs A, Vermi W et al (2009) A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457(7230):722–725. https://doi.org/10.1038/nature07537
Kudira R, Malinka T, Kohler A et al (2016) P2X1-regulated IL-22 secretion by innate lymphoid cells is required for efficient liver regeneration. Hepatology 63(6):2004–2017. https://doi.org/10.1002/hep.28492
Hanash AM, Dudakov JA, Hua G et al (2012) Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity 37(2):339–350. https://doi.org/10.1016/j.immuni.2012.05.028
Petri RM, Hackel A, Hahnel K et al (2017) Activated tissue-resident mesenchymal stromal cells regulate natural killer cell immune and tissue-regenerative function. Stem Cell Rep 9(3):985–998. https://doi.org/10.1016/j.stemcr.2017.06.020
Miani M, Le Naour J, Waeckel-Enée E et al (2018) Gut microbiota-stimulated innate lymphoid cells support β-Defensin 14 expression in pancreatic endocrine cells, preventing autoimmune diabetes. Cell Metab 28(4):557–572.e556. https://doi.org/10.1016/j.cmet.2018.06.012
Zakharov PN, Hu H, Wan X, Unanue ER (2020) Single-cell RNA sequencing of murine islets shows high cellular complexity at all stages of autoimmune diabetes. J Exp Med 217(6). https://doi.org/10.1084/jem.20192362
Marca V, Gianchecchi E, Fierabracci A (2018) Type 1 Diabetes and Its Multi-Factorial Pathogenesis: The Putative Role of NK Cells. Int J Mol Sci 19(3). https://doi.org/10.3390/ijms19030794
Fasolino M, Schwartz GW, Golson ML et al (2021) Multiomics single-cell analysis of human pancreatic islets reveals novel cellular states in health and type 1 diabetes. In. Cold Spring Harbor Laboratory
Damond N, Engler S, Zanotelli VRT et al (2019) A map of human type 1 diabetes progression by imaging mass cytometry. Cell Metab 29(3):755–768.e755. https://doi.org/10.1016/j.cmet.2018.11.014
Fraker C, Bayer AL (2016) The expanding role of natural killer cells in type 1 diabetes and immunotherapy. Curr Diab Rep 16(11):109. https://doi.org/10.1007/s11892-016-0806-7
Molofsky AB, Nussbaum JC, Liang HE et al (2013) Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med 210(3):535–549. https://doi.org/10.1084/jem.20121964
Hasnain SZ, Borg DJ, Harcourt BE et al (2014) Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med 20(12):1417–1426. https://doi.org/10.1038/nm.3705
Borg DJ, Wang R, Murray L et al (2017) The effect of interleukin-22 treatment on autoimmune diabetes in the NOD mouse. Diabetologia 60(11):2256–2261. https://doi.org/10.1007/s00125-017-4392-2
Graves CL, Li J, LaPato M et al (2017) Intestinal epithelial cell regulation of adaptive immune dysfunction in human type 1 diabetes. Front Immunol 7(JAN). https://doi.org/10.3389/fimmu.2016.00679
Beilke JN, Kuhl NR, Van Kaer L, Gill RG (2005) NK cells promote islet allograft tolerance via a perforin-dependent mechanism. Nat Med 11(10):1059–1065. https://doi.org/10.1038/nm1296
Huang Q, Ma X, Wang Y et al (2020) IL −10 producing type 2 innate lymphoid cells prolong islet allograft survival. EMBO Mol Med 12(11). https://doi.org/10.15252/emmm.202012305
Tosti L, Hang Y, Debnath O et al (2020) Single-nucleus and in situ RNA–sequencing studies reveal cell topographies in the human pancreas. Gastroenterology. https://doi.org/10.1053/j.gastro.2020.11.010
Bakhti M, Scheibner K, Tritschler S et al (2019) Establishment of a high-resolution 3D modeling system for studying pancreatic epithelial cell biology in vitro. Mol Metab 30:16–29. https://doi.org/10.1016/j.molmet.2019.09.005
Atkinson MA, Campbell-Thompson M, Kusmartseva I, Kaestner KH (2020) Organisation of the human pancreas in health and in diabetes. Diabetologia 63(10):1966–1973. https://doi.org/10.1007/s00125-020-05203-7
Qadir MMF, Álvarez-Cubela S, Weitz J et al (2020) Long-term culture of human pancreatic slices as a model to study real-time islet regeneration. Nat Commun 11(1). https://doi.org/10.1038/s41467-020-17040-8
Panzer JK, Hiller H, Cohrs CM et al (2020) Pancreas tissue slices from organ donors enable in situ analysis of type 1 diabetes pathogenesis. JCI Insight 5(8). https://doi.org/10.1172/jci.insight.134525
Zhang B, Kumar RB, Dai H, Feldman BJ (2014) A plasmonic chip for biomarker discovery and diagnosis of type 1 diabetes. Nat Med 20(8):948–953. https://doi.org/10.1038/nm.3619
Rogal J, Zbinden A, Schenke-Layland K, Loskill P (2019) Stem-cell based organ-on-a-chip models for diabetes research. Adv Drug Deliv Rev 140:101–128. https://doi.org/10.1016/j.addr.2018.10.010
Kolb H, Von Herrath M (2017) Immunotherapy for type 1 diabetes: why do current protocols not halt the underlying disease process? Cell Metab 25(2):233–241. https://doi.org/10.1016/j.cmet.2016.10.009
Herold KC, Vignali DA, Cooke A, Bluestone JA (2013) Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol 13(4):243–256. https://doi.org/10.1038/nri3422
Boni-Schnetzler M, Meier DT (2019) Islet inflammation in type 2 diabetes. Semin Immunopathol 41(4):501–513. https://doi.org/10.1007/s00281-019-00745-4
Tsai S, Clemente-Casares X, Zhou AC et al (2018) Insulin receptor-mediated stimulation boosts T cell immunity during inflammation and infection. Cell Metab 28(6):922–934 e924. https://doi.org/10.1016/j.cmet.2018.08.003
Wu D, Wong CK, Han JM et al (2020) T reg-specific insulin receptor deletion prevents diet-induced and age-associated metabolic syndrome. J Exp Med 217(8). https://doi.org/10.1084/jem.20191542
Corliss BA, Azimi MS, Munson JM, Peirce SM, Murfee WL (2016) Macrophages: an inflammatory link between angiogenesis and Lymphangiogenesis. Microcirculation 23(2):95–121. https://doi.org/10.1111/micc.12259
Staels W, Heremans Y, Heimberg H, De Leu N (2019) VEGF-A and blood vessels: a beta cell perspective. Diabetologia 62(11):1961–1968. https://doi.org/10.1007/s00125-019-4969-z
Acknowledgements
Graphical abstract created with BioRender.com.
Authors’ relationships and activities
CBV is a member of the editorial board for Diabetologia and declares no competing interests in the publication of this manuscript, nor any involvement in the review and assessment process for its publication merit. The authors declare that there are no other relationships or activities that might bias, or be perceived to bias, this work.
Funding
The authors’ own work in this area is supported by the Canadian Institutes of Health Research (HH3-168005; PJT-165943) and JDRF Canadian Clinical Trials Network (4-SRA-2020-953-A-N).
Author information
Authors and Affiliations
Contributions
All authors were responsible for drafting, reviewing, editing and critically evaluating the intellectual content of the article. All authors have approved this version of the article as submitted.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Matthew A. Budd, Mahdis Monajemi and Sarah J. Colpitts are joint first authors. Sarah Q. Crome, C. Bruce Verchere and Megan K. Levings are joint senior authors.
Supplementary Information
Figure slide
(PPTX 495 kb)
Rights and permissions
About this article

Cite this article
Budd, M.A., Monajemi, M., Colpitts, S.J. et al. Interactions between islets and regulatory immune cells in health and type 1 diabetes. Diabetologia 64, 2378–2388 (2021). https://doi.org/10.1007/s00125-021-05565-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-021-05565-6