
Abstract
Aims/hypothesis
Aggregation of islet amyloid polypeptide (IAPP) to form amyloid contributes to beta cell dysfunction in type 2 diabetes. Human but not non-amyloidogenic rodent IAPP induces islet macrophage proIL-1β synthesis. We evaluated the effect of IL-1 receptor antagonist (IL-1Ra) on islet inflammation and dysfunction in a mouse model of type 2 diabetes with amyloid formation.
Methods
Lean and obese male mice (A/a or A vy /A at the agouti locus, respectively) with or without beta cell human IAPP expression (hIAPP Tg/0) were treated with PBS or IL-1Ra (50 mg kg−1 day−1) from 16 weeks of age. Intraperitoneal glucose and insulin tolerance tests were performed after 8 weeks. Pancreases were harvested for histology and gene expression analysis.
Results
Aggregation of human IAPP was associated with marked upregulation of proinflammatory gene expression in islets of obese hIAPP Tg/0 mice, together with amyloid deposition and fasting hyperglycaemia. IL-1Ra improved glucose tolerance and reduced plasma proinsulin:insulin in both lean and obese hIAPP Tg/0 mice with no effect on insulin sensitivity. The severity and prevalence of islet amyloid was reduced by IL-1Ra in lean hIAPP Tg/0 mice, suggesting a feed-forward mechanism by which islet inflammation promotes islet amyloid at the early stages of disease. IL-1Ra limited Il1a, Il1b, Tnf and Ccl2 expression in islets from obese hIAPP Tg/0 mice, suggesting an altered islet inflammatory milieu.
Conclusions/interpretation
These data provide the first in vivo evidence—using a transgenic mouse model with amyloid deposits resembling those found in human islets—that IAPP-induced beta cell dysfunction in type 2 diabetes may be mediated by IL-1. Anti-IL-1 therapies may limit islet inflammation and dysfunction associated with amyloid formation.
Similar content being viewed by others
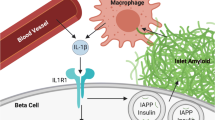


Introduction
Islet amyloid deposition [1], macrophage infiltration [2, 3], and upregulation of proinflammatory cytokines [4, 5] are common pathological features of pancreatic islets from patients with type 2 diabetes. Amyloid deposits are composed primarily of islet amyloid polypeptide (IAPP), a 37-amino-acid peptide that is co-secreted with insulin by beta cells. Recent evidence suggests that IAPP aggregates trigger islet secretion of IL-1, which acts alone or in combination with other proinflammatory cytokines to impair beta cell insulin secretion [6, 7]. Human but not non-amyloidogenic rodent IAPP induces IL-1β synthesis in bone marrow-derived and intra-islet macrophages [8, 9]. IAPP also activates the NACHT, LRR and PYD domain-containing protein 3 (NLRP3) inflammasome, required for processing of proIL-1β into its mature form [10]. Blockade of IL-1 signalling with IL-1 receptor (IL-1R) antagonist (IL-1Ra) limits IAPP-induced secretion of proinflammatory cytokines by cultured macrophages and islets [9], suggesting that this cytokine is a central mediator of islet inflammation in the setting of amyloid formation.
Increased expression of IL-1-related genes is a characteristic of islets from patients with type 2 diabetes [11]. Moreover, multiple clinical studies in patients with type 2 diabetes have demonstrated improved insulin secretion in response to anti-IL-1 therapy with no detectable effects on insulin resistance [12]. These data suggest that the islet may be particularly susceptible to IL-1-induced inflammation, perhaps due in part to an islet-localised stimulus for IL-1 such as IAPP aggregation. Here, we assessed the effect of IL-1Ra on amyloid formation, islet inflammation and beta cell function in a transgenic mouse model of islet amyloid formation.
Methods
Mice
C57BL/6 and FVB/NJ mice were from the Jackson Laboratory (Bar Harbor, ME, USA). Hemizygous FVB/N-Tg(Ins2-IAPP)RHFSoel/J mice were bred with wild-type FVB mice to produce hemizygous human IAPP transgenic mice (hIAPP Tg/0). To generate hIAPP Tg/0 mice and wild-type littermates with a viable yellow allele at the agouti locus, female hIAPP Tg/0 mice (A/A at the agouti locus) were crossed with male B6.C3-A vy/J mice (A vy /a) as described previously [13]. Agouti allele status was determined based on coat colour [14]. Homozygous hIAPP Tg/Tg Sprague–Dawley rats from transgenic Line 9 generated by Butler et al [15] were obtained from Pfizer (New York, NY, USA). Transgenic rats were bred with wild-type Sprague–Dawley rats (University of British Columbia, Vancouver, BC, Canada) to generate hemizygous hIAPP Tg/0 rats. Rodents were maintained in compliance with Canadian Council on Animal Care guidelines. Studies were approved by the University of British Columbia Committee on Animal Care. All rodents were maintained on a 13 kJ% fat diet (PicoLab Rodent Diet 20-5053, Lab Diet, St Louis, MO, USA).
Metabolic testing
Sixteen-week-old male A vy-hIAPP Tg/0 or a-hIAPP Tg/0 mice and wild-type littermates were treated s.c. with 50 mg/kg/day IL-1Ra (anakinra; Sobi, Stockholm, Sweden). Plasma insulin and proinsulin, glucose tolerance and insulin sensitivity were assessed after 7–8 weeks according to previously published methods [8]. Tail vein glucose was measured by a glucometer (OneTouch, Burnaby, BC, Canada). Glucose-stimulated insulin secretion in isolated islets was assessed as described previously [16]. Plasma hIAPP was measured by ELISA (Millipore, Etobicoke, ON, Canada).
Gene expression analysis
Islets were isolated by ductal collagenase injection and filtration [17]. Islet culture, RNA isolation, cDNA synthesis and reverse transcription-quantitative PCR (RT-qPCR) were performed as described previously [8], with primer sequences from PrimerBank [18]. Gene expression in islets isolated from Avy-hIAPP Tg/0 and control mice was assessed with a mouse Inflammatory Response and Autoimmunity Array and analysed with RT2 Profiler PCR Array Data Analysis Version 3.5 (Qiagen, Mississauga, ON, Canada).
Immunohistochemistry
Formalin-fixed, paraffin-embedded sections (5 μm) from two to three areas of pancreas were deparaffinised and rehydrated. Antigen retrieval was performed in Target Retrieval Solution (Dako, Carpinteria, CA, USA) for 20 min. Sections were blocked with 0.25% casein (Dako) for 30 min. Primary antibodies were diluted in 0.1% BSA in PBS and applied overnight at 4°C as follows: guinea pig anti-insulin (Dako), 1:200; rat anti-F4/80 (Cedarlane, Burlington, ON, Canada), 1:75; rabbit anti-PDX1 (Abcam, Toronto, ON, Canada), 1:200; rabbit anti-MAFA (Bethyl Laboratories, Montgomery, TX, USA), 1:200. For nuclear staining, tissue was permeabilised with 0.25% Triton-X100 (Sigma-Aldrich, Oakville, ON, Canada) for 15 min prior to blocking. Alexa 594 goat anti-guinea pig and Alexa 488 goat anti-rat secondary antibodies (1:100; Life Technologies, Burlingon, ON, Canada) were applied for 1 h at room temperature. Sections were incubated with 0.1% thioflavin S for 2 min followed by an ethanol wash. Slides were mounted using Vectashield mounting medium with DAPI (Vector Laboratories, Burlington, ON, Canada) and imaged on a BX61 microscope (Olympus, Center Valley, PA, USA). Quantification was performed using Image-Pro software (MediaCybernetics, Bethesda, MD, USA).
Statistical analyses
Data were analysed with GraphPad Prism (GraphPad Software, La Jolla, CA, USA) and are expressed as means ± SEM of the indicated number of trials. Differences between two groups were evaluated by two-tailed t test. Differences among three or more groups were evaluated by one-way or two-way ANOVA and Bonferroni post tests.
Results
Amyloid accumulation in obese Avy-hIAPPTg/0 mice is associated with upregulation of islet proinflammatory cytokines and decreased beta cell gene expression
We generated obese, insulin-resistant hIAPP Tg/0 mice expressing the agouti viable yellow allele (A vy) as described previously [13]. We compared the phenotypes of A vy-expressing mice (yellow, obese and insulin-resistant; A vy-hIAPP Tg/0 and A vy-WT) to their brown, leaner littermates (a-hIAPP Tg/0 and a-WT). hIAPP transgene expression in beta cells of obese A vy-expressing mice caused islet amyloid formation, with small flecks of amyloid in ~15% of islets at 12 weeks of age and larger pericapillary deposits affecting ~60% of islets and comprising ~20% of islet area by 24 weeks of age (Fig. 1a–c). At 12 weeks, hIAPP transgene expression significantly impaired glucose tolerance in obese A vy-expressing mice, with a similar trend in a-hIAPP Tg/0 vs a-WT mice (Fig. 1d, e). The hIAPP-induced glucose intolerance could not be attributed to insulin resistance, as A vy-hIAPP Tg/0 mice were more insulin-sensitive than their wild-type littermates (Fig. 1f, g). A vy expression caused increased body weight, with no difference between hIAPP Tg/0 and wild-type mice (Fig. 1h, i). Amyloid deposition was associated with hyperglycaemia in A vy-hIAPP Tg/0 but not a-hIAPP Tg/0 mice at both 12 and 24 weeks of age (Fig. 1h, i).

Beta cell expression of hIAPP in obese A vy-expressing mice promotes amyloid accumulation between 12 and 24 weeks. Pancreases were isolated from 12- and 24-week-old male hIAPP Tg/0 or wild-type (WT) mice with or without agouti viable yellow expression (A vy and a, respectively). (a) Formalin-fixed, paraffin-embedded pancreas sections from hIAPP Tg/0 mice were stained for insulin (red), amyloid (green, thioflavin S) and nuclei (DAPI, blue). Note that the overlap between thioflavin S and DAPI is due to the broad emission spectrum of thioflavin S. Scale bars, 50 μm. (b) Amyloid prevalence (proportion of islets containing amyloid) and (c) amyloid severity (proportion of islet area occupied by amyloid) were assessed at each time point. a-hIAPP Tg/0, white bars; A vy-hIAPP Tg/0, black bars. (d) Glucose tolerance at 12 weeks was assessed by i.p. injection of 0.75 g/kg glucose and (e) evaluation of area under the glycaemia curve up to 120 min. a-WT, white circles; a-hIAPP Tg/0, black circles; A vy-WT, white squares; A vy-hIAPP Tg/0, black squares. (f) Insulin sensitivity at 12 weeks was assessed by i.p. injection of 1 U/kg insulin and (g) evaluation of area under baseline (normalised to 1) and above the glycaemia curve up to 60 min. Body weight and fasting glucose were assessed at (h) 12 and (i) 24 weeks. WT, white bars; hIAPP Tg/0, black bars. Data represent means ± SEM of 7–11 mice per group. *p < 0.05, **p < 0.01, ***p < 0.001
To determine whether amyloid deposition is associated with changes in proinflammatory gene expression, we used an RT-qPCR array to analyse gene expression in islets from 24-week-old a-hIAPP Tg/0 and A vy -hIAPP Tg/0 mice and their wild-type littermates. Of 84 inflammation-related genes evaluated, 66 could be detected in islets (electronic supplementary material [ESM] Table 1, Fig. 2a–c). In lean a-hIAPP Tg/0 mice, only Itgb2 (which together with Itgam encodes the αMβ2 integrin) was significantly upregulated compared with a-WT littermates. Indeed, other proinflammatory cytokines, as well as the anti-inflammatory cytokines Il1rn and Il10, were downregulated, consistent with our previous observations in a model of early amyloid formation with high-fat feeding [8]. The obese, insulin-resistant state caused by A vy expression in wild-type mice on its own also had minimal effects on islet proinflammatory gene expression (Fig. 2). However, islets from obese A vy -hIAPP Tg/0 mice with extensive amyloid deposition showed upregulation of 33 inflammation-related genes (ESM Table 1; Fig. 2), including anti-inflammatory cytokines (such as Il1rn, encoding IL-1Ra) that are often induced in parallel, pointing to hIAPP aggregation as an inciting mechanism for islet inflammation.

hIAPP-induced islet dysfunction and amyloid deposition in obese A vy-expressing mice is associated with upregulation of islet proinflammatory gene expression. Islets were isolated from 24-week-old male hIAPP Tg/0 or wild-type (WT) mice with or without agouti viable yellow expression (A vy and a, respectively). Islets were lysed for RNA purification 4 h following isolation. Gene expression was evaluated by RT-qPCR array. Volcano plots for the indicated comparisons (a–c) show the log2 fold change in gene expression (x-axis, threshold = 2) relative to the log10 p value (y-axis, threshold = 0.05). Expression was normalised to a panel of housekeeping genes (Actb, Gapdh, Gusb, Hsp90ab1). Data represent the average fold change of four mice per group
Because both Il1b and Il1rn were among the amyloid-associated upregulated cytokines, we next assessed expression of other IL-1-related genes. Islets from A vy-hIAPP Tg/0 mice expressed elevated Il1a, Il1b, Il1rn, Casp1 and Nlrp3 compared with islets from A vy-WT littermates, while expression of Il18, Il1r1, Il1rap and Pycard were unchanged (Fig. 3a). In wild-type mice, A vy expression was associated with decreased expression of the beta cell transcription factors Pdx1 and Mafa as well as the prohormone processing enzymes Pcsk1 and Pcsk2, with no effect on Ins1 or Ins2 (Fig. 3b). Expression of hIAPP in lean mice also caused downregulation of Pdx1, Mafa and Pcsk1, while islets from A vy-hIAPP Tg/0 vs A vy-WT mice displayed significantly reduced expression of all beta cell genes analysed (Fig. 3b). There was no change in expression of IL-1-related genes in livers from the same mice, suggesting an islet-specific proinflammatory response (Fig. 3c). Thus, A vy-hIAPP Tg/0 mice show evidence of islet-localised inflammation associated with amyloid deposition, reduced expression of key genes required for beta cell function and hyperglycaemia relative to their A vy-WT littermates.

Amyloid-containing islets have elevated expression of IL-1 and decreased expression of genes associated with beta cell function. Islets and livers were isolated from 24-week-old male hIAPP Tg/0 or wild-type (WT) mice with or without agouti viable yellow expression (A vy and a, respectively). (a) Islets were lysed for RNA purification 4 h following isolation. Expression of IL-1 family genes and (b) beta cell genes was assessed by RT-qPCR. (c) Livers were lysed immediately following isolation. Expression of IL-1 family genes was assessed by RT-qPCR and normalised to the housekeeping gene Rplp0. a-WT, white bars; a-hIAPP Tg/0, black bars; A vy-WT, light grey bars; A vy-hIAPP Tg/0, dark grey bars. Data represent means ± SEM of four to six mice per group. *p < 0.05, **p < 0.01, ***p < 0.001 compared with WT islets with the same agouti phenotype. †† p < 0.01, ††† p < 0.001 comparison between A vy-WT and a-WT mice
IL-1Ra improves glucose tolerance in both lean a-hIAPPTg/0 and obese Avy-hIAPPTg/0 mice
We next evaluated the effects of IL-1R blockade on glucose homeostasis and islet inflammation in hIAPP Tg/0 mice. For these studies, we treated 16-week-old lean mice (a-hIAPP Tg/0 and a-WT; Fig. 4a–d) and obese mice (A vy -hIAPP Tg/0 and A vy-WT; Fig. 4e–h) with IL-1Ra (50 mg/kg/day s.c.) or PBS for 8 weeks. We chose mice 16 weeks of age to begin IL-1Ra treatment because by that age small amounts of islet amyloid associated with impaired glucose tolerance are detectable in both lean and obese hIAPP transgenic mice. Thus, at the same age, lean and obese littermates provide a model of early (lean) and later (obese) stages in development of diabetes. IL-1Ra had no effect on body weight or fasting glycaemia (Fig. 4a, b). IL-1Ra improved glucose tolerance in hIAPP Tg/0 but not wild-type mice, with no detectable effect on insulin sensitivity (Fig. 4d, h). In a-hIAPP Tg/0 mice, there was a trend towards reduced fasting plasma insulin compared with wild-type mice, which was unaffected by IL-1Ra (Fig. 5b). However, hIAPP expression increased proinsulin:insulin ratios (Fig. 5c), which were significantly improved with IL-1Ra. There was no effect of IL-1Ra on plasma hIAPP levels in the non-fasting state (Fig. 5d). In A vy-hIAPP Tg/0 mice, there was no significant effect of IL-1Ra on fasting (Fig. 5e) or non-fasting (Fig. 5f) plasma insulin. As in the lean mice, hIAPP expression in obese animals was associated with an increased proinsulin:insulin ratio, which was reduced with IL-1Ra (Fig. 5g). Again, IL-1Ra had no effect on plasma hIAPP (Fig. 5h). We were unable to detect changes in plasma insulin levels at the 15 and 30 min time points of the glucose tolerance test (data not shown). Therefore, to determine the effect of IL-1Ra on amyloid-induced beta cell dysfunction, we cultured a-hIAPP Tg/0 islets for 7 days at 16.7 mmol/l glucose, a model known to result in extensive amyloid formation and loss of insulin secretion. We chose this model of in vitro amyloid formation rather than studying isolated islets following amyloid formation in vivo, because of the difficulty in isolating amyloid-laden islets. IL-1Ra restored the impaired glucose-stimulated insulin secretion in hIAPP Tg/0 islets (Fig. 5i), with no significant effect on insulin content (Fig. 5j). We also measured KCl-stimulated insulin secretion, which—in parallel with content—was decreased by approximately 50% in hIAPP Tg/0 islets (2.1 ± 0.8 vs 1.1 ± 0.2 ng/ml), consistent with a defect downstream of glucose metabolism such as exocytosis or insulin content of the releasable granule pool. Interestingly, in contrast to the impaired glucose-stimulated insulin secretion induced by hIAPP, the defective KCl-stimulated insulin release was not corrected by IL-1Ra treatment (1.1 ± 0.2 vs 0.8 ± 0.2 ng/islet), suggesting an additional impact of hIAPP independent of IL-1-mediated inflammation (e.g. beta cell loss due to direct toxicity of hIAPP). In addition, IL-1Ra had no noticeable effects on islet morphology in vivo (ESM Fig. 1a). hIAPP transgene expression was associated with decreased insulin-positive islet area in both lean and obese mice, with no significant effect of IL-1Ra (ESM Fig. 1b). Thus, IL-1Ra improves hIAPP-induced glucose intolerance and beta cell dysfunction in the absence of significant changes in islet morphology or insulin content.

IL-1Ra improves glucose tolerance in both lean and obese hIAPP transgenic mice. Sixteen-week-old male hIAPP Tg/0 or wild-type (WT) mice with a (a–d; lean) or A vy (e–h; obese) agouti allele expression were treated s.c. with 50 mg/kg/day IL-1Ra for 8 weeks. (a, e) Body weight was assessed at 24 weeks of age. PBS, white bars; IL-1Ra, black bars. (b, f) Glucose tolerance was assessed by i.p. injection of 1 g/kg glucose and (c, g) calculation of total area under the glycaemia curve. PBS-treated WT, white circles; IL-1Ra-treated WT, black circles; PBS-treated hIAPP Tg/0, white squares; IL-1Ra-treated hIAPP Tg/0, black squares. (d, h) Insulin sensitivity was evaluated following i.p. injection of 1 U/kg insulin. Data represent means ± SEM of four–six mice per group. *p < 0.05, ***p < 0.01

IL-1Ra improves islet function in hIAPP transgenic islets. Sixteen-week-old male hIAPP Tg/0 or wild-type (WT) mice with a (a–d; lean) or A vy (e–h; obese) agouti allele expression were treated s.c. with 50 mg/kg/day IL-1Ra for 8 weeks. (a, e) Fasting plasma insulin, (b, f) non-fasting insulin, (c, g) proinsulin:insulin and (d, h) non-fasting hIAPP were determined by ELISA. PBS, white bars; IL-1Ra, black bars. (i) Islets were isolated from 12–20-week-old a-hIAPP Tg/0 mice or a-WT littermate controls. Islets were cultured for 7 days at 16.7 mmol/l glucose to promote amyloid formation, in the presence or absence of IL-1Ra (2 μg/ml). Glucose-stimulated insulin secretion and (j) insulin content were determined by ELISA. PBS-treated WT islets, open bars; IL-1Ra-treated WT islets, black bars; PBS-treated a-hIAPP Tg/0 islets, light grey bars; IL-1Ra-treated a-hIAPP Tg/0 islets, dark grey bars. Data represent means ± SEM of four to six mice per group. *p < 0.05, **p < 0.01
IL-1Ra limits hIAPP-induced islet inflammation
To determine the effect of IL-1Ra on hIAPP-induced islet inflammation, we evaluated cytokine expression in islets isolated from PBS- and IL-1Ra-treated mice. Of note, islet yield was reduced by an average of ~50% in mice with significant amyloid formation, which may bias the population of isolated islets towards those with less amyloid. Beta cell hIAPP expression caused modest upregulation of both Il1a and Il1b (Fig. 6a, b) but not Tnf or Ccl2 (Fig. 6c, d) in islets of a-hIAPP Tg/0 mice, and significant upregulation of all four cytokines in association with amyloid deposition in islets of A vy-hIAPP Tg/0 mice, an effect that was blocked with IL-1Ra. Expression of the macrophage markers Itgam (CD11b) and Emr1 (F4/80) was upregulated in both a-hIAPP Tg/0 and A vy-hIAPP Tg/0 mice (Fig. 6e, f), consistent with our previous observation of increased macrophage glycoprotein expression by macrophages in hIAPP-expressing islets [8]. IL-1Ra did not prevent this upregulation, suggesting that changes in proinflammatory gene expression are not due to significant changes in the size of the islet macrophage population. hIAPP expression also significantly decreased expression of the beta cell genes Ins2, Pdx1 and Mafa in PBS, but not in IL-1Ra-treated mice (ESM Fig. 2a). A previous study in db/db mice showed that IL-1Ra prevents cytoplasmic translocation of the transcription factor pancreatic and duodenal homeobox factor 1 (PDX1) [19], while others have observed loss of nuclear v-Maf musculoaponeurotic fibrosarcoma oncogene homologue A (MAFA) in db/db mouse beta cells with no change in PDX1 [20]. We did not detect a significant decrease in nuclear PDX1 staining intensity with hIAPP transgene expression. However, nuclear MAFA intensity was markedly decreased in islet sections from both a-hIAPP Tg/0 and A vy -hIAPP Tg/0 mice relative to wild-type littermates (ESM Fig. 2b, c). IL-1Ra limited the hIAPP-induced loss of nuclear MAFA in obese A vy-expressing mice, consistent with increased beta cell gene expression and improved beta cell function.

IL-1Ra limits islet inflammation in hIAPP transgenic mice. Sixteen-week-old male hIAPP Tg/0 or wild-type (WT) mice with or without expression of the agouti viable yellow allele (A vy and a, respectively) were treated s.c. with 50 mg/kg/day IL-1Ra. Islets were isolated after 8 weeks and lysed for RNA isolation after overnight recovery. Expression of cytokines (a–d) and macrophage markers (e, f) was assessed by RT-qPCR and normalised to the housekeeping gene Rplp0. PBS, white bars; IL-1Ra, black bars. Data represent means ± SEM of four to six mice per group. *p < 0.05, **p < 0.01, ***p < 0.001
IL-1Ra limits islet amyloid formation in lean a-hIAPPTg/0 but not obese Avy-hIAPPTg/0 mice
To determine whether IL-1Ra treatment affected islet amyloid deposition, we stained pancreas sections from IL-1Ra- and PBS-treated hIAPP Tg/0 mice with thioflavin S (Fig. 7a). As expected, no thioflavin-S-positive amyloid deposits were observed in wild-type mice. IL-1Ra reduced amyloid prevalence (proportion of islets containing amyloid; Fig. 7b) and severity (area occupied by amyloid; Fig. 7c) in a-hIAPP Tg/0 but not obese A vy-hIAPP Tg/0 mice. These data suggest that—at least early in disease progression—IL-1Ra may limit IL-1-induced beta cell dysfunction leading to amyloid formation. However, in mice with more extensive amyloid deposits, IL-1Ra improved glucose homeostasis and limited islet inflammation without affecting amyloid deposition. To rule out possible peripheral effects of IL-1Ra in the context of amyloid formation, we also evaluated the effects of IL-1Ra on amyloid formation and insulin secretion in cultured hIAPP Tg/0 rat islets, which develop islet amyloid after 7 days of culture with 22 mmol/l glucose [21]. IL-1Ra reduced amyloid severity by 50% (ESM Fig. 3a, b) and improved glucose-stimulated insulin secretion in hIAPP Tg/0 but not wild-type islets, with no effect on insulin content in this model (ESM Fig. 3c). Thus, IL-1Ra can act directly on the islet to limit amyloid formation.

IL-1Ra limits early islet amyloid formation in hIAPP transgenic mice. Sixteen-week-old male hIAPP Tg/0 or wild-type (WT) mice with or without expression of the agouti viable yellow allele (A vy and a, respectively) were treated s.c. with 50 mg/kg/d IL-1Ra. (a) Formalin-fixed, paraffin-embedded pancreas sections from hIAPP Tg/0 mice were stained for insulin (red), amyloid (thioflavin S, green) and nuclei (DAPI, blue) to assess (b) amyloid prevalence and (c) amyloid severity. White arrow: small amyloid deposit. Scale bars, 50 μm. PBS, white bars; IL-1Ra, black bars. Data represent means ± SEM of four to six mice per group. **p < 0.01
Discussion
Chronic exposure to elevated IL-1 causes beta cell death and dysfunction due to altered gene transcription, changes in protein activity and induction of oxidative stress [22]. Data from clinical studies suggest that anti-IL-1 agents can improve beta cell function without affecting insulin sensitivity [12]. Recent work by us [8, 9] and others [10] demonstrated that IAPP—the principal component of islet amyloid—induces IL-1 synthesis and secretion by cultured macrophages. Moreover, islet macrophages are the major source of hIAPP-induced IL-1β in islets, and islet macrophage depletion can improve hIAPP-induced islet dysfunction (7). Given that IAPP can deliver the two signals required for IL-1β secretion [9, 10, 23] and that anti-IL-1 therapy can improve beta cell function in patients with type 2 diabetes [12], here we asked whether IL-1 mediates hIAPP-induced islet dysfunction in vivo. We found that IL-1Ra markedly improved glycaemia and islet inflammation in a mouse model of islet amyloid formation and type 2 diabetes.
Most strains of hIAPP Tg/0 mice develop extracellular islet amyloid resembling that found in humans under conditions that promote beta cell dysfunction, including ex vivo culture [9, 24, 25], high-fat diet [26] or obesity due to expression of the A vy allele [13]. A previous study showed that A vy-hIAPP Tg/0 mice develop slow-onset diabetes associated with extracellular amyloid deposits, decreased plasma insulin and pancreatic insulin content, and decreased beta cell mass by 41–52 weeks of age [13]. We found that impaired glucose tolerance is present in A vy-hIAPP Tg/0 mice at 12 weeks of age in association with small extracellular flecks of amyloid and becomes more pronounced by 24 weeks of age, together with increased amyloid severity. Although we did not detect changes in plasma insulin levels associated with impaired glucose tolerance at the 15 and 30 min time points sampled during the IPGTT, we did observe impaired glucose-stimulated insulin secretion in islets isolated from a-hIAPP Tg/0 mice compared with those from their non-transgenic littermates, suggesting the presence of beta cell dysfunction associated with amyloid formation. Extensive amyloid deposition and hyperglycaemia in A vy-hIAPP Tg/0 mice was associated with increased islet expression of a number of proinflammatory cytokines and chemokines that were not upregulated in a-hIAPP Tg/0 mice with low amyloid severity and normoglycaemia, in keeping with a critical role for hIAPP aggregation in islet inflammation. As the islet yield from mice with more severe amyloid deposition was limited, the actual differences in gene expression between amyloid-free and amyloid-laden islets may be greater than those measured. These data are consistent with a recent study demonstrating increased expression of proinflammatory cytokines, inflammasome components and macrophage markers in the islets of hIAPP Tg/0 mice fed a high-fat diet [27]. Lean hIAPP-expressing mice with few detectable thioflavin-S-positive amyloid deposits may have insufficient fibrillar IAPP to induce maximal NLRP3 activation and IL-1β secretion by islet macrophages, thus limiting autocrine/paracrine induction of other proinflammatory mediators in the early stages of disease. Nonetheless, a-hIAPP Tg/0 mice still had elevated proinsulin/insulin ratios compared with lean non-transgenic mice, and this impairment in proinsulin processing was corrected by IL-1Ra treatment, supporting the notion that pre-fibrillar hIAPP aggregates are sufficient to induce an IL-1-dependent defect in beta cell function.
While it is possible that hyperglycaemia rather than islet amyloid is the major driver of islet proinflammatory gene expression in this model, several lines of evidence point to a specific and more important role for hIAPP-induced islet inflammation. First, hIAPP transgene expression was not associated with the same proinflammatory response in liver, a tissue with a high content of macrophages that respond to systemic stimuli. Second, both synthetic and endogenous hIAPP induce Il1b expression in isolated islets [8] under the same culture conditions as rodent IAPP-treated or wild-type islets. Third, IL-1Ra improved the function of hIAPP Tg/0 but not wild-type rodent islets in high-glucose culture. Finally, A vy expression alone (i.e. in A vy-WT mice) was not associated with elevated expression of IL-1-related genes in islets or liver despite impaired glucose tolerance relative to a-WT mice. However, we cannot exclude the possibility that amyloid-associated proinflammatory gene expression is in part due to liberation of danger signals produced by beta cells damaged by hIAPP.
It may be difficult to uncouple the toxic and proinflammatory effects of hIAPP given that pre-fibrillar oligomers are critical participants in both hIAPP-induced beta cell death and proinflammatory cytokine synthesis. Whether fibrillar species are entirely inert is controversial [28], and studies of other amyloidogenic peptides suggest that amyloid fibrils activate NLRP3 [29, 30]. Moreover, we have now found protective effects of IL-1Ra in multiple models in which it does not affect insulin-positive beta cell area, including: NOD/SCID (non-obese diabetic/severe combined immunodeficiency) recipients of hIAPP-expressing islet transplants [9]; both the a-hIAPP Tg/0 and A vy-hIAPP Tg/0 mice in the present study; and cultured hIAPP Tg/0 rodent islets. Changes in the ratio of proinsulin:insulin provide further evidence for hIAPP-induced islet dysfunction independent of beta cell death. The marked decrease in nuclear MAFA localisation, which was partially rescued by IL-1Ra, suggests that this transcription factor—which is a critical regulator of genes important for normal beta cell function [31]—is a sensitive indicator of beta cell dysfunction in the setting of amyloid formation. Beta cell expression of MAFA is downregulated in response to IL-1 [32], but may also be downregulated in response to effects of hIAPP that are not IL-1-dependent, such as oxidative stress. We also cannot rule out an effect of IL-1Ra on the expression and localisation of other beta cell transcription factors. Further studies are required to understand the key transcriptional mediators of hIAPP-induced beta cell dysfunction.
We propose two mechanisms by which IL-1Ra may improve hIAPP-induced islet dysfunction. First, IL-1Ra may suppress IL-1-induced amplification of the inflammatory response to hIAPP as well as the direct deleterious effect of IL-1 on beta cell function [6, 33–35]. Indeed, treatment with IL-1Ra suppressed upregulation of hIAPP-induced islet Il1a, Il1b and Tnf, suggesting that IL-1Ra indeed acts on macrophages, the major source of these hIAPP-induced cytokines [8]. Signalling via the IL-1R can be induced not only by IL-1β but also by IL-1α and IL-18 [36]. Although we did not observe upregulation of IL-18 expression in A vy-hIAPP Tg/0 islets, the relative contributions of IL-1α and IL-1β to IL-1R-mediated islet inflammation requires further study. Given current clinical trials under way using anti-IL-1α and anti-IL-1β monoclonal antibodies, this distinction may be clinically relevant [37]. In any case, the dramatic effects of hIAPP aggregation on the islet inflammatory milieu suggest that the more commonly used rodent models of obesity and type 2 diabetes that do not express hIAPP may lack a central trigger for islet inflammation that contributes to beta cell dysfunction. These models may therefore be inadequate for the evaluation of anti-inflammatory therapies, and arguably for any type 2 diabetes therapy.
Improving beta cell function by blocking beta cell IL-1R (or reducing the amount of IL-1 present) may also reduce the formation of proinflammatory IAPP aggregates. Consistent with this hypothesis, IL-1Ra limited amyloid formation in a-hIAPP Tg/0 mice and cultured hIAPP Tg/0 rat islets. The effect of IL-1Ra to decrease amyloid formation could not be attributed to an inhibition of hIAPP production, since although plasma hIAPP levels were higher in obese compared with lean hIAPP Tg/0 mice, IL-1Ra administration had no effect on plasma hIAPP levels in either lean or obese mice. Since impaired proIAPP processing may promote islet amyloid formation [38], it is possible that IL-1-induced beta cell dysfunction alters the processing of proIAPP leading to increased amyloid. Indeed, IL-1β and TNF-α impair proinsulin processing [39], which occurs via a similar pathway as for proIAPP [40–42]. No effect of IL-1Ra on amyloid deposition in A vy-hIAPP Tg/0 mice was detected, despite improved glucose tolerance, suggesting either a reduction in pre-fibrillar hIAPP aggregates not detected with thioflavin S or protective effects on the beta cell despite the persistence of the initiating stimulus. In this more advanced stage of hIAPP deposition, IL-1Ra may be unable to prevent additional amyloid formation given the presence of extensive deposits that act as seeds for further aggregation. In less severe disease, for example in lean a-hIAPP Tg/0 mice or in A vy-hIAPP Tg/0 mice younger than 16 weeks of age, improved beta cell function in response to IL-1Ra may limit amyloid formation. This interpretation of our data would support a model in which cytokine production begets amyloid formation (which, in turn, leads to further inflammation) in a feed-forward cycle. This model further points to the importance of amyloid and islet inflammation as therapeutic targets in type 2 diabetes.
While in vitro evidence suggests that interaction of hIAPP with macrophages induces release of cytokines such as IL-1β that at high local concentrations are known to cause beta cell dysfunction [10, 23], it is likely that IL-1 also plays an important physiological role in regulating islet homeostasis. For example, low concentrations of IL-1β promote beta cell proliferation, and IL-1β knockout mice have impaired glucose tolerance and decreased islet Pdx1 expression [43]. That IL-1Ra did not alter glucose tolerance in wild-type mice suggests that blockade of this pathway under physiological conditions provides no benefit but also does not cause harm. Moreover, insulin sensitivity was not significantly improved by IL-1Ra, pointing to a specific role for IL-1 in islet pathology. Somewhat surprisingly, insulin sensitivity was already enhanced in obese hIAPP Tg/0 mice compared with their non-transgenic counterparts. This finding suggests that hIAPP overexpression in obese mice might directly impact insulin sensitivity by an unknown mechanism, although earlier studies have suggested that supraphysiological levels of IAPP decrease, rather than increase, insulin sensitivity [44]. It should also be noted that IL-1Ra treatment caused a trend towards reduced plasma insulin levels in lean wild-type mice. Although previous studies have demonstrated an effect of IL-1Ra on insulin sensitivity in other rodent models [35, 45], there was no effect of IL-1Ra on insulin sensitivity in this model as determined by an insulin tolerance test. The lower fasting insulin levels and increased proinsulin:insulin ratios observed in IL-1Ra-treated wild-type mice may have resulted from inhibition of beneficial effects of IL-1 on the beta cell, since physiological levels of IL-1 are thought to be important for normal beta cell function [43].
Collectively, these findings provide a mechanistic link between two important islet pathologies in type 2 diabetes, amyloid and inflammation. They provide the first in vivo evidence of a hIAPP-induced, IL-1-mediated islet autoinflammatory process in a mouse model of type 2 diabetes and suggest that hIAPP-induced beta cell dysfunction can be improved even in the presence of widespread amyloid deposition. While no current therapies are available that specifically target islet amyloid formation, anti-NLRP3, caspase-1 or IL-1 agents may provide an alternative approach for protecting the beta cell against hIAPP-induced dysfunction.
Abbreviations
- hIAPP:
-
Human islet amyloid polypeptide
- hIAPP Tg/0 :
-
Hemizygous human islet amyloid polypeptide transgenic
- IAPP:
-
Islet amyloid polypeptide
- IL-1R:
-
Interleukin-1 receptor
- IL-1Ra:
-
Interleukin-1 receptor antagonist
- MAFA:
-
v-Maf musculoaponeurotic fibrosarcoma oncogene homologue A
- NLRP3:
-
NACHT, LRR and PYD domain-containing protein 3
- PDX1:
-
Pancreatic and duodenal homeobox factor 1
References
Hull RL, Westermark GT, Westermark P, Kahn SE (2004) Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab 89:3629–3643
Ehses JA, Perren A, Eppler E et al (2007) Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56:2356–2370
Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG (2009) Islet-associated macrophages in type 2 diabetes. Diabetologia 52:1686–1688
Donath MY, Böni-Schnetzler M, Ellingsgaard H, Halban PA, Ehses JA (2010) Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol Metab 21:261–267
Westwell-Roper C, Nackiewicz D, Dan M, Ehses JA (2014) Toll-like receptors and NLRP3 as central regulators of pancreatic islet inflammation in type 2 diabetes. Immunol Cell Biol 92:314–323
Bendtzen K, Mandrup-Poulsen T, Nerup J, Nielsen JH, Dinarello CA, Svenson M (1986) Cytotoxicity of human pI 7 interleukin-1 for pancreatic islets of Langerhans. Science 232:1545–1547
Corbett JA, Wang JL, Sweetland MA, Lancaster JR, McDaniel ML (1992) Interleukin 1 beta induces the formation of nitric oxide by beta-cells purified from rodent islets of Langerhans. Evidence for the beta-cell as a source and site of action of nitric oxide. J Clin Invest 90:2384–2391
Westwell-Roper CY, Ehses JA, Verchere CB (2013) Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1beta production and beta cell dysfunction. Diabetes 63:1698–1711
Westwell-Roper C, Dai DL, Soukhatcheva G et al (2011) IL-1 blockade attenuates islet amyloid polypeptide-induced proinflammatory cytokine release and pancreatic islet graft dysfunction. J Immunol 187:2755–2765
Masters SL, Dunne A, Subramanian SL et al (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol 11:897–904
Mahdi T, Hanzelmann S, Salehi A et al (2012) Secreted frizzled-related protein 4 reduces insulin secretion and is overexpressed in type 2 diabetes. Cell Metab 16:625–633
Donath MY, Dalmas E, Sauter NS, Böni-Schnetzler M (2013) Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell Metab 17:860–872
Soeller WC, Janson J, Hart SE et al (1998) Islet amyloid-associated diabetes in obese A(vy)/a mice expressing human islet amyloid polypeptide. Diabetes 47:743–750
Waterland RA, Jirtle RL (2003) Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23:5293–5300
Butler AE, Jang J, Gurlo T, Carty MD, Soeller WC, Butler PC (2004) Diabetes due to a progressive defect in beta-cell mass in rats transgenic for human islet amyloid polypeptide (HIP Rat): a new model for type 2 diabetes. Diabetes 53:1509–1516
Hutton M, Soukhatcheva G, Johnson JD, Verchere CB (2010) Role of the TLR signaling molecule TRIF in β-cell function and glucose homeostasis. Islets 2:104–111
Salvalaggio PRO, Deng S, Ariyan CE et al (2002) Islet filtration: a simple and rapid new purification procedure that avoids ficoll and improves islet mass and function. Transplantation 74:877–879
Spandidos A, Wang X, Wang H, Seed B (2010) PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res 38:D792–D799
Ardestani A, Sauter NS, Paroni F et al (2011) Neutralizing interleukin-1beta (IL-1beta) induces beta-cell survival by maintaining PDX1 protein nuclear localization. J Biol Chem 286:17144–17155
Harmon JS, Bogdani M, Parazzoli SD et al (2009) Beta-cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 150:4855–4862
Montane J, Klimek-Abercrombie A, Potter KJ, Westwell-Roper C, Verchere CB (2012) Metabolic stress, IAPP and islet amyloid. Diabetes Obes Metab 14(Suppl 3):68–77
Novotny GW, Lundh M, Backe MB et al (2012) Transcriptional and translational regulation of cytokine signaling in inflammatory beta-cell dysfunction and apoptosis. Arch Biochem Biophys 528:171–184
Westwell-Roper C, Dunne A, Kim ML, Verchere CB, Masters SL (2013) Activating the NLRP3 inflammasome using the amyloidogenic peptide IAPP. Methods Mol Biol 1040:9–18
Zraika S, Hull RL, Udayasankar J et al (2007) Glucose- and time-dependence of islet amyloid formation in vitro. Biochem Biophys Res Commun 354:234–239
MacArthur DL, de Koning EJ, Verbeek JS, Morris JF, Clark A (1999) Amyloid fibril formation is progressive and correlates with beta-cell secretion in transgenic mouse isolated islets. Diabetologia 42:1219–1227
Hull RL, Andrikopoulos S, Verchere CB et al (2003) Increased dietary fat promotes islet amyloid formation and beta-cell secretory dysfunction in a transgenic mouse model of islet amyloid. Diabetes 52:372–379
Meier DT, Morcos M, Samarasekera T, Zraika S, Hull RL, Kahn SE (2014) Islet amyloid formation is an important determinant for inducing islet inflammation in high-fat-fed human IAPP transgenic mice. Diabetologia 57:1884–1888
Zraika S, Hull RL, Verchere CB et al (2010) Toxic oligomers and islet beta cell death: guilty by association or convicted by circumstantial evidence? Diabetologia 53:1046–1056
Halle A, Hornung V, Petzold GC et al (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9:857–865
Gustot A, Raussens V, Dehousse M et al (2013) Activation of innate immunity by lysozyme fibrils is critically dependent on cross-beta sheet structure. Cell Mol Life Sci 70:2999–3012
Matsuoka TA, Kaneto H, Stein R et al (2007) MafA regulates expression of genes important to islet beta-cell function. Mol Endocrinol 21:2764–2774
Oetjen E, Blume R, Cierny I et al (2007) Inhibition of MafA transcriptional activity and human insulin gene transcription by interleukin-1beta and mitogen-activated protein kinase kinase kinase in pancreatic islet beta cells. Diabetologia 50:1678–1687
Arnush M, Heitmeier MR, Scarim AL, Marino MH, Manning PT, Corbett JA (1998) IL-1 produced and released endogenously within human islets inhibits beta cell function. J Clin Invest 102:516–526
Maedler K, Sergeev P, Ris F et al (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110:851–860
Ehses JA, Lacraz G, Giroix M-H et al (2009) IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proc Natl Acad Sci U S A 106:13998–14003
Arend WP, Palmer G, Gabay C (2008) IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 223:20–38
Dinarello CA, Simon A, van der Meer JW (2012) Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 11:633–652
Marzban L, Rhodes CJ, Steiner DF, Haataja L, Halban PA, Verchere CB (2006) Impaired NH2-terminal processing of human proislet amyloid polypeptide by the prohormone convertase PC2 leads to amyloid formation and cell death. Diabetes 55:2192–2201
Hostens K, Pavlovic D, Zambre Y et al (1999) Exposure of human islets to cytokines can result in disproportionately elevated proinsulin release. J Clin Invest 104:67–72
Wang J, Xu J, Finnerty J, Furuta M, Steiner DF, Verchere CB (2001) The prohormone convertase enzyme 2 (PC2) is essential for processing pro-islet amyloid polypeptide at the NH2-terminal cleavage site. Diabetes 50:534–539
Marzban L, Trigo-Gonzalez G, Zhu X et al (2004) Role of beta-cell prohormone convertase (PC)1/3 in processing of pro-islet amyloid polypeptide. Diabetes 53:141–148
Marzban L, Soukhatcheva G, Verchere CB (2005) Role of carboxypeptidase E in processing of pro-islet amyloid polypeptide in beta-cells. Endocrinology 146:1808–1817
Maedler K, Schumann DM, Sauter N et al (2006) Low concentration of interleukin-1beta induces FLICE-inhibitory protein-mediated beta-cell proliferation in human pancreatic islets. Diabetes 55:2713–2722
Leighton B, Cooper GJ (1988) Pancreatic amylin and calcitonin gene-related peptide cause resistance to insulin in skeletal muscle in vitro. Nature 335:632–635
Sauter NS, Schulthess FT, Galasso R, Castellani LW, Maedler K (2008) The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology 149:2208–2218
Acknowledgements
We thank D. L. Dai and G. Soukhatcheva (University of British Columbia, Vancouver, Canada) for assistance with islet isolation. We thank J. C. Cheng (University of British Columbia, Vancouver, Canada) for assistance with rat islet histology.
Funding
This work was supported by grants from the Canadian Institutes of Health Research (CIHR; MOP-123338 to CBV) and the Canadian Diabetes Association (CDA; OG-3-11-3413-CV) and by core support to the Child & Family Research Institute (CFRI) from the BC Children’s Hospital Foundation and Canucks for Kids Fund Childhood Diabetes Laboratories. CYW-R is supported by a Vanier Canada Graduate Studentship. HCD is supported by postdoctoral fellowships from the JDRF and the CDA. JAE is supported by a scholar award from the CDA and an investigator award from the CFRI. CBV is supported by the Irving K. Barber Chair in Diabetes Research at the University of British Columbia and an investigator award from the CFRI.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
CYW-R, JAE and CBV contributed to the conception and design of the experiments. CYW-R, CAC, HCD and JAC conducted the experiments and analysed the data. CYW-R drafted the manuscript. All authors critically revised the manuscript and approved the final version to be published. CBV is the guarantor of this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 2514 kb)
ESM Fig. 2
(PDF 1319 kb)
ESM Fig. 3
(PDF 958 kb)
ESM Table 1
(PDF 112 kb)
Rights and permissions
About this article

Cite this article
Westwell-Roper, C.Y., Chehroudi, C.A., Denroche, H.C. et al. IL-1 mediates amyloid-associated islet dysfunction and inflammation in human islet amyloid polypeptide transgenic mice. Diabetologia 58, 575–585 (2015). https://doi.org/10.1007/s00125-014-3447-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3447-x