
Abstract
Aims/hypothesis
Epidemiological data on the associations of circulating fatty acid levels with type 2 diabetes are inconsistent. We conducted a two-sample Mendelian randomisation study to explore the causal associations of plasma levels of ten fatty acids with type 2 diabetes and glycaemic traits.
Methods
Thirteen SNPs associated with circulating levels of ten individual fatty acids at the genome-wide significance level (p < 5 × 10−8) were selected as instrumental variables for the exposures. For the outcomes, summary-level data were obtained from the DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) consortium for type 2 diabetes (898,130 individuals) and from the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) for the glycaemic traits (up to 46,186 non-diabetic individuals). The inverse-variance weighted method was used for analyses.
Results
Genetic predisposition to higher plasma levels of eight of the ten fatty acids were statistically significantly associated with lower or higher odds of type 2 diabetes. The OR per one SD increment of each fatty acid was 0.93 (95% CI 0.90, 0.96; p = 2.21 × 10−5) for α-linolenic acid, 0.96 (95% CI 0.94, 0.98; p = 1.85 × 10−4) for linoleic acid, 0.86 (95% CI 0.81, 0.91; p = 6.68 × 10−7) for palmitoleic acid, 0.87 (95% CI 0.81, 0.93; p = 2.21 × 10−5) for oleic acid, 1.08 (95% CI 1.03, 1.12; p = 0.002) for eicosapentaenoic acid, 1.04 (95% CI 1.02, 1.07; p = 0.001) for docosapentaenoic acid, 1.03 (95% CI 1.02, 1.05; p = 2.51 × 10−5) for arachidonic acid and 1.09 (95% CI 1.03, 1.15; p = 0.003) for stearic acid. The same eight fatty acids were also associated with fasting glucose levels and HOMA-B. The associations, except that for palmitoleic acid, were driven by variants in FADS1/2.
Conclusions/interpretation
Genetic predisposition to higher circulating levels of eight out of ten fatty acids was associated with type 2 diabetes, fasting glucose and islet beta cell function. However, the associations, except that for palmitoleic acid, were driven by variants in FADS1/2, which encode enzymes with a key role in fatty acid metabolism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.

Introduction
Type 2 diabetes mellitus is a common chronic disease and its prevalence has quadrupled in the past three decades globally [1]. It is estimated that around 8–9% of adults had type 2 diabetes in 2015 [1, 2]. Affected individuals have high morbidity risk and mortality risk due to complications such as cardiovascular and kidney diseases, which impose a huge burden on medical systems and society [3].
Among the major driving factors of the global type 2 diabetes epidemic, dietary fatty acid intake has been shown to be a potentially modifiable risk factor for type 2 diabetes [4]. The concentration of different fatty acids in plasma and erythrocytes partly reflects dietary intake but is also determined by endogenous fatty acid production mediated by various fatty acid desaturases encoded by the FADS1, FADS2 and SCD genes as well as various fatty acid elongases encoded by different ELOVL genes (Fig. 1) [5].

The role of enzymes encoded by FADS, SCD and ELOVL in the metabolism of fatty acids. Desaturation enzymes are shown in red and elongases in blue
Available evidence from observational studies of the associations of circulating levels or intake of n-3 polyunsaturated fatty acids (PUFAs) [6, 7], n-6 PUFAs [8, 9] and saturated fatty acids (SFAs) [10, 11] with risk of type 2 diabetes are inconsistent. Data on monounsaturated fatty acids (MUFAs) in relation to type 2 diabetes are scarce. The inconsistent findings may partly be related to measurement errors in the assessment of dietary fatty acid intake, which could bias the true association between a categorical exposure and outcome in any direction [12]. The disagreements may also be due to residual confounding introduced by certain health behaviours or nutrients that are correlated with fatty acid levels or intake and also related to type 2 diabetes risk [4].
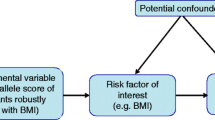
Mendelian randomisation (MR) studies can strengthen the causal inference on exposure–outcome relationships by using genetic variants as instrumental variables for an exposure [13]. This method diminishes confounding since genetic variants (alleles) are randomly assorted at meiosis, thereby having no connection to self-selected lifestyle factors and behaviours [13]. It also overcomes reverse causality, a potential drawback in observational studies, as allelic randomisation precedes the onset of disease [13]. There are three assumptions for an MR study design (Fig. 2): (1) the genetic variants selected as instrumental variables should be robustly associated with the exposure; (2) the used instrumental variables should not be associated with any potential confounders; (3) the genetic variants of an exposure should affect the risk of the outcome merely through the risk factor, not via other alternative pathways [14].

Schematic diagram of the MR assumptions underpinning an MR analysis of the association of plasma fatty acid levels with type 2 diabetes, fasting glucose, fasting insulin, HOMA-B and HOMA-IR. The dashed lines represent potential causal associations between variables that would represent violations of the MR assumption
We conducted a two-sample MR study to explore the causal associations of ten major fatty acid plasma levels and type 2 diabetes. As complementary analyses, we assessed whether the ten fatty acids were associated with fasting glucose and insulin levels, islet beta cell function and insulin resistance. HOMA-B and HOMA-IR were used as proxies of the islet beta cell function and insulin resistance, respectively.
Methods
Study design
An overview of the study design is displayed in Fig. 2. The present two-sample MR study is based on publicly available summary-level data on genetic associations with ten fatty acids, type 2 diabetes and glycaemic traits from genome-wide association studies (GWASs) (electronic supplementary material [ESM] Table 1). In all underlying studies included in the GWASs, the genetic association estimates were adjusted for age, sex and study-specific covariates. All these studies had been approved by a relevant institutional review board and participants had provided informed consents. The present MR study was approved by the Swedish Ethical Review Authority.
SNP selection
Summary-level data of fatty acid levels were obtained from the hitherto largest GWASs of plasma phospholipid fatty acids or total plasma fatty acids (ESM Table 1) [15,16,17]. The ten fatty acids included in this MR study were α-linolenic acid (ALA), eicosapentaenoic acid (EPA), docosapentaenoic acid (DPA), docosahexaenoic acid (DHA), linoleic acid (LA), arachidonic acid (AA), palmitoleic acid (POA), oleic acid (OA), palmitic acid (PA) and stearic acid (SA). Fifteen SNPs associated with one or more of the fatty acids were selected at the genome-wide significance level (p < 5 × 10−8) (Table 1). Two SNPs (rs780093 and rs780094) were located in the GCKR locus, which is a highly pleiotropic locus associated with a number of phenotypes, such as lipids, BMI, alcohol intake and serum calcium levels [18]. In particular, those two SNPs were more strongly associated with type 2 diabetes and glycaemic traits than with plasma DPA and POA levels (Table 1 and ESM Table 2) and were therefore excluded from all analyses. Detailed information on the SNPs is displayed in Table 1 and ESM Table 2. SNPs associated with n-3 and n-6 PUFAs were identified in GWASs of 8866 and 8631 individuals of European ancestry, respectively [16, 17]. SNPs associated with n-7 MUFAs, n-9 MUFAs and SFAs were identified from five prospective studies with 8961 individuals of European descent [15]. For each fatty acid, all selected SNPs were in different gene regions and linkage equilibrium (i.e. uncorrelated). However, SNPs located in or close to genes (FADS1, FADS2, SCD and ELOVL2) that encode enzymes with a central role in the metabolic pathway of fatty acids (Fig. 1) were associated with multiple fatty acids. Since the percentage of each fatty acid among the total plasma fatty acids varies largely, we used 1 SD as the unit of change of individual fatty acid level [19]. One SD change in the present study corresponded to 2.69, 1.96, 0.05, 0.30, 0.17, 0.89, 1.17, 0.18, 1.64 and 1.19 percentage units of total fatty acid for LA, AA, ALA, EPA, DPA, DHA, OA, POA, PA and SA, respectively [19].
Outcome sources
Summary-level data for the association of fatty-acid-associated SNPs with type 2 diabetes were obtained from the DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) consortium, a publicly available GWAS of 32 studies with a total of 898,130 individuals (74,124 with type 2 diabetes and 824,006 without) of European ancestry (ESM Table 1) [20]. We used the type 2 diabetes data without BMI adjustment in our primary analyses, and then performed a sensitivity analysis by using BMI-adjusted data.
Summary-level data for the fatty-acid-related SNPs with fasting glucose, fasting insulin, HOMA-B and HOMA-IR were obtained from the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC). Fasting glucose data was based on a meta-analysis of 21 GWASs including 46,186 non-diabetic individuals of European ancestry [21]. Summary-level data of fasting insulin, HOMA-B and HOMA-IR were acquired from meta-analysis of 20 GWASs with, respectively, 38,238, 36,466 and 37,037 non-diabetic individuals of European descent [21].
Statistical analyses
The fixed-effects inverse-variance weighted method was used to assess the associations of plasma phospholipid fatty acid levels with the outcomes. The OR of type 2 diabetes was calculated per 1 SD increment in genetically predicted plasma fatty acid levels. For fasting glucose and insulin, HOMA-B and HOMA-IR, β estimates were scaled per 1 SD increment in plasma fatty acid levels. Sensitivity analyses based on the random-effect inverse-variance weighted method and the weighted median method [22] were performed where more than three SNPs were available as instrumental variables. This was the case only for POA. All p values were two-sided. Associations with p values below 0.005 were deemed statistically significant after Bonferroni correcting for ten fatty acids. A p value between 0.001 and 0.05 was regarded as suggestive evidence of an association. The statistical analyses were performed in Stata/SE 15.0 (StataCorp, College Station, TX, USA) by using the mrrobust package [23].
Results
Fatty acids and type 2 diabetes
Genetic predisposition to higher levels of eight of the ten fatty acids was significantly associated with lower or higher odds of type 2 diabetes in analyses based on all SNPs (Figs 3, 4). The OR per 1 SD increase in plasma levels of each fatty acid was 0.93 (95% CI 0.90, 0.96; p = 2.21 × 10−5) for ALA, 0.96 (95% CI 0.94, 0.98; p = 1.85 × 10−4) for LA, 0.86 (95% CI 0.81, 0.91; p = 6.68 × 10−7) for POA, 0.87 (95% CI 0.81, 0.93; p = 2.21 × 10−5) for OA, 1.08 (95% CI 1.03, 1.12; p = 0.002) for EPA, 1.04 (95% CI 1.02, 1.07; p = 0.001) for DPA, 1.03 (95% CI 1.02, 1.05; p = 2.51 × 10−5) for AA and 1.09 (95% CI 1.03, 1.15; p = 0.003) for SA (Fig. 3). Only the association with POA remained after exclusion of SNPs (rs174547 and rs102275) in or close to FADS1/2, which generally explained the major proportion of variance in plasma fatty acid levels (Table 1). The minor allele (C) of both rs174547 and rs102275 was associated with higher plasma ALA, LA, POA and OA levels and lower plasma DPA, AA and SA levels as well as with lower odds of type 2 diabetes (Table 1). The SNPs in the FADS1/2 gene cluster had no significant pleiotropic associations with potential confounders (ESM Table 3). SNPs in ELOVL2 were associated with plasma levels of all three long-chain n-3 PUFAs (EPA, DPA and DHA) but were not associated with type 2 diabetes (Table 1) and had no significant pleiotropic associations with any other traits (ESM Table 3). The association between genetic predisposition to higher POA levels and lower odds of type 2 diabetes remained in analyses based on the random-effects inverse-variance weighted method and the weighted median method (ESM Fig. 1). No significant differences between the results based on type 2 diabetes data without and with adjustment for BMI were observed (ESM Fig. 2).

Associations between plasma fatty acid levels and type 2 diabetes from MR analyses. aNumber of SNPs included in the analysis of each fatty acid

MR associations of plasma fatty acid levels with type 2 diabetes, fasting glucose, fasting insulin, HOMA-B and HOMA-IR. FA, fatty acid; FG, fasting glucose; FI, fasting insulin; T2DM, type 2 diabetes
Fatty acids and glycaemic traits
Genetic predisposition to higher plasma ALA, LA, POA and OA levels were associated with lower fasting glucose levels and higher HOMA-B score, whereas genetic predisposition to higher plasma AA, EPA, DPA and SA levels were associated with higher fasting glucose levels and lower HOMA-B score (Fig. 4 and ESM Table 4). These associations seemed to be driven by SNPs in or close to FADS1/2 (ESM Table 2). None of the fatty acids was associated with fasting insulin levels or HOMA-IR (Fig. 4 and ESM Table 4).
Discussion
The present MR study found evidence that genetic predisposition to higher plasma levels of ALA, LA, POA and OA were associated with lower odds of type 2 diabetes, lower fasting glucose and higher HOMA-B score, whereas genetic predisposition to higher plasma levels of EPA, DPA, AA and SA were associated with higher odds of type 2 diabetes, higher fasting glucose and lower HOMA-B score. The associations except that for POA were driven by variants in FADS1/2, which explained the major proportion of variance in plasma fatty acid levels.
A major limitation of this MR study is that SNPs in the FADS1/2 gene cluster were associated with most fatty acids and only three or fewer SNPs were available as instrumental variables for individual fatty acids, except for POA. This limited the possibility for disentangling the association of individual fatty acids with type 2 diabetes, and performing sensitivity analyses to explore pleiotropy. The strong associations of FADS1/2 with fatty acid levels are not surprising because FADS1 and FADS2 encode Δ-5 desaturase and Δ-6 desaturase, respectively, which are key rate-limiting enzymes in the metabolism of fatty acids (Fig. 1). Strong correlations between circulating biomarkers is not only an issue in the present MR study but can also be an issue in for example MR studies on metabolomics where several metabolites can be highly correlated.
Our results are broadly in agreement with those of previous smaller studies of the associations of FADS1 and FADS2 genetic variants or Δ-5 and Δ-6 desaturase activities with type 2 diabetes. A Chinese case–control study, including 331 individuals with type 2 diabetes and 421 healthy control individuals, found that the minor allele (T) of rs174616 in the FADS1-FADS2 gene cluster was associated with decreased AA/LA ratio and lower odds of type 2 diabetes [24]. Results from a nested case-cohort study of 2653 German adults showed no overall association of rs174546, which is in complete linkage disequilibrium with rs174547, with type 2 diabetes [25]. However, that study revealed that a higher proportion of LA in erythrocytes and lower proportions of γ-linolenic acid and dihomo-γ-linolenic acid were associated with lower risk of type 2 diabetes. Furthermore, fatty acid ratios that reflect Δ-5 desaturase and Δ-6 desaturase activity were respectively inversely and positively associated with type 2 diabetes [25]. Similarly, results from a Finnish cohort study (407 overweight adults) [26] and an Iranian case–control study (95 individuals with type 2 diabetes and 95 without) [27] showed that Δ-5 desaturase activity was inversely associated with type 2 diabetes. Finally, a cross-sectional study of 576 Korean men showed that variants in the FADS gene cluster were associated with the proportions of LA, dihomo-γ-linolenic acid, and AA in serum phospholipids as well as with fasting insulin levels and HOMA-IR [28].
Available data on the associations of circulating POA levels with the risk of type 2 diabetes and glycaemic traits are inconsistent [29,30,31,32,33]. Several population-based observational studies found that increased plasma or erythrocyte membrane POA level was associated with higher blood glucose levels [29], insulin resistance [30] and incidence of diabetes [31]. However, in a prospective longitudinal study including 3630 Americans, circulating level of POA was not associated with the risk of diabetes and the positive association between POA level and insulin resistance was merely observed among men [30]. In addition, several studies concluded that increasing plasma POA level lowered fasting insulin, insulin resistance and the risk of type 2 diabetes [32]; this has been verified by cell experimental studies [33]. In the present study, genetic evidence, excluding the influences from FADS and GCKR, supports a protective effect of increasing POA levels on fasting glucose level and type 2 diabetes. Discrepancy between these findings may be attributable to residual confounding commonly affecting observational studies, such as type 2 diabetes-related nutrients, lifestyle determinants or endogenous sources introduced by dietary POA intake or de novo lipogenesis (e.g. key enzymes in fatty acid metabolism, inflammatory factors and metabolic traits encoded by POA synthesis-related genes).
As for potential biological mechanisms, the associations of plasma fatty acid levels and genetic variations in FADS1/2 with type 2 diabetes may be mediated by impaired glucose tolerance and beta cell dysfunction, as suggested by the present MR study. It has been postulated that POA may prevent beta cell apoptosis induced by glucose or SFAs [34]. Fatty acids are crucial structural components of cell membranes. The flexibility of a membrane, determined by the ratio of unsaturated to saturated fatty acyl chains of its phospholipids, influences the effectiveness of glucose transport via insulin-dependent glucose transporters [35]. A shift from unsaturated towards saturated fatty acyl chains of membrane phospholipids results in a decrease in glucose effectiveness and insulin sensitivity [36]. Fatty acids are further precursors of eicosanoids, which may affect type 2 diabetes risk through inflammation [36, 37]. An experimental study in mice showed that loss of Δ-5 desaturase activity promoted hepatic inflammation and implied that endogenously synthesised AA and EPA are key determinants of inflammatory disease progression [38]. Associations of fatty acids with type 2 diabetes might also be explained by the effects of fatty acids on the gastrointestinal tract [37], intestinal microbiome [39] and nervous system [37].
A further mechanism whereby fatty acids might affect the risk of type 2 diabetes is through blood cholesterol levels, which are inversely associated with risk of type 2 diabetes [40]. A meta-analysis of 60 trials revealed that SFAs increase total cholesterol and LDL-cholesterol levels, whereas MUFAs and PUFAs have the opposite effect, and that all types of fatty acid increase HDL-cholesterol levels [41]. The minor allele (C) of both rs174547 and rs102275 is associated with decreased levels of LDL-cholesterol, HDL-cholesterol and total cholesterol [42]. Thus, we would have expected a higher rather than lower odds of type 2 diabetes for the minor allele of rs174547 and rs102275 if the associations were mediated by cholesterol levels.
Conclusions
This MR study showed that genetic predisposition to higher plasma ALA, LA, POA and OA levels and lower EPA, DPA, AA and SA levels was associated with lower odds of type 2 diabetes, lower fasting glucose levels and higher HOMA-B score. However, the causality for any individual fatty acid, except POA, cannot be inferred from this study due to the high correlation between the fatty acids. Notably, the observed associations, except that for POA, appeared to be driven by genetic variants in or close to FADS1/2. Replication of our finding for POA in an independent MR study would provide strong evidence for a causal effect. Further research is needed to unravel the role of individual fatty acids in the prevention of type 2 diabetes.
Data availability
All data included in this study are available in the article and ESM.
Abbreviations
- AA:
-
Arachidonic acid
- ALA:
-
α-Linolenic acid
- DHA:
-
Docosahexaenoic acid
- DIAGRAM:
-
DIAbetes Genetics Replication And Meta-analysis
- DPA:
-
Docosapentaenoic acid
- EPA:
-
Eicosapentaenoic acid
- GWAS:
-
Genome-wide association study
- LA:
-
Linoleic acid
- MAGIC:
-
Meta-Analyses of Glucose and Insulin-related traits Consortium
- MR:
-
Mendelian randomisation
- MUFA:
-
Monounsaturated fatty acid
- OA:
-
Oleic acid
- PA:
-
Palmitic acid
- POA:
-
Palmitoleic acid
- PUFA:
-
Polyunsaturated fatty acid
- SA:
-
Stearic acid
- SFA:
-
Saturated fatty acid
References
Zheng Y, Ley SH, Hu FB (2018) Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol 14:88–98
Jaacks LM, Siegel KR, Gujral UP, Narayan KM (2016) Type 2 diabetes: a 21st century epidemic. Best Pract Res Clin Endocrinol Metab 30:331–343
Roglic G, Unwin N (2010) Mortality attributable to diabetes: estimates for the year 2010. Diabetes Res Clin Pract 87:15–19
Ley SH, Hamdy O, Mohan V, Hu FB (2014) Prevention and management of type 2 diabetes: dietary components and nutritional strategies. Lancet 383:1999–2007
Lattka E, Illig T, Koletzko B, Heinrich J (2010) Genetic variants of the fads1 fads2 gene cluster as related to essential fatty acid metabolism. Curr Opin Lipidol 21(1):64
Wallin A, Di Giuseppe D, Orsini N, Patel PS, Forouhi NG, Wolk A (2012) Fish consumption, dietary long-chain n-3 fatty acids, and risk of type 2 diabetes: systematic review and meta-analysis of prospective studies. Diabetes Care 35:918–929
Wu JH, Micha R, Imamura F et al (2012) Omega-3 fatty acids and incident type 2 diabetes: a systematic review and meta-analysis. Br J Nutr 107(Suppl 2):S214–S227
Wu JHY, Marklund M, Imamura F et al (2017) Omega-6 fatty acid biomarkers and incident type 2 diabetes: pooled analysis of individual-level data for 39 740 adults from 20 prospective cohort studies. Lancet Diabetes Endocrinol 5:965–974
Yary T, Voutilainen S, Tuomainen TP, Ruusunen A, Nurmi T, Virtanen JK (2016) Serum n-6 polyunsaturated fatty acids, Δ5- and Δ 6-desaturase activities, and risk of incident type 2 diabetes in men: the Kuopio Ischaemic heart disease risk factor study. Am J Clin Nutr 103:1337–1343
de Souza RJ, Mente A, Maroleanu A et al (2015) Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: systematic review and meta-analysis of observational studies. BMJ 351:h3978
Patel PS, Sharp SJ, Jansen E et al (2010) Fatty acids measured in plasma and erythrocyte-membrane phospholipids and derived by food-frequency questionnaire and the risk of new-onset type 2 diabetes: a pilot study in the European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk cohort. Am J Clin Nutr 92:1214–1222
Hutcheon JA, Chiolero A, Hanley JA (2010) Random measurement error and regression dilution bias. BMJ 340(7761):1402–1406
Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG (2015) Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol 30:543–552
Davies NM, Holmes MV, Davey Smith G (2018) Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 362:k601
Wu JH, Lemaitre RN, Manichaikul A et al (2013) Genome-wide association study identifies novel loci associated with concentrations of four plasma phospholipid fatty acids in the de novo lipogenesis pathway: results from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium. Circ Cardiovasc Genet 6:171–183
Lemaitre RN, Tanaka T, Tang W et al (2011) Genetic loci associated with plasma phospholipid n-3 fatty acids: a meta-analysis of genome-wide association studies from the CHARGE consortium. PLoS Genet 7:e1002193
Guan W, Steffen BT, Lemaitre RN et al (2014) Genome-wide association study of plasma N6 polyunsaturated fatty acids within the cohorts for heart and aging research in genomic epidemiology consortium. Circ Cardiovasc Genet 7:321–331
Larsson SC, Burgess S, Michaelsson K (2017) Association of genetic variants related to serum calcium levels with coronary artery disease and myocardial infarction. JAMA 318:371–380
Yamagishi K, Nettleton JA, Folsom AR (2008) Plasma fatty acid composition and incident heart failure in middle-aged adults: the atherosclerosis risk in communities (ARIC) study. Am Heart J 156:965–974
Mehta NN (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes mellitus. Circ Cardiovasc Genet 5:708–710
Dupuis J, Langenberg C, Prokopenko I et al (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42:105–116
Bowden J, Davey Smith G, Haycock PC, Burgess S (2016) Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 40:304–314
Spiller W, Davies NM, Palmer TM (2018) Software application profile: mrrobust—a tool for performing two-sample summary Mendelian randomization analyses. Int J Epidemiol 48(3):684–690
Yao M, Li J, Xie T et al (2015) Polymorphisms of rs174616 in the FADS1-FADS2 gene cluster is associated with a reduced risk of type 2 diabetes mellitus in northern Han Chinese people. Diabetes Res Clin Pract 109:206–212
Kroger J, Zietemann V, Enzenbach C et al (2011) Erythrocyte membrane phospholipid fatty acids, desaturase activity, and dietary fatty acids in relation to risk of type 2 diabetes in the European prospective investigation into Cancer and nutrition (EPIC)-Potsdam study. Am J Clin Nutr 93:127–142
Takkunen MJ, Schwab US, de Mello VD et al (2016) Longitudinal associations of serum fatty acid composition with type 2 diabetes risk and markers of insulin secretion and sensitivity in the Finnish diabetes prevention study. Eur J Nutr 55:967–979
Mazoochian L, Mohammad Sadeghi HM, Pourfarzam M (2018) The effect of FADS2 gene rs174583 polymorphism on desaturase activities, fatty acid profile, insulin resistance, biochemical indices, and incidence of type 2 diabetes. J Res Med Sci 23(47)
Kim OY, Lim HH, Yang LI, Chae JS, Lee JH (2011) Fatty acid desaturase (FADS) gene polymorphisms and insulin resistance in association with serum phospholipid polyunsaturated fatty acid composition in healthy Korean men: cross-sectional study. Nutr Metab (Lond) 8:24
Lankinen MA, Stancakova A, Uusitupa M et al (2015) Plasma fatty acids as predictors of glycaemia and type 2 diabetes. Diabetologia 58:2533–2544
Mozaffarian D, Cao H, King IB et al (2010) Circulating palmitoleic acid and risk of metabolic abnormalities and new-onset diabetes. Am J Clin Nutr 92:1350–1358
Wang L, Folsom AR, Zheng ZJ, Pankow JS, Eckfeldt JH (2003) Plasma fatty acid composition and incidence of diabetes in middle-aged adults: the atherosclerosis risk in communities (ARIC) study. Am J Clin Nutr 78:91–98
Mozaffarian D, de Oliveira Otto MC, Lemaitre RN et al (2013) trans-Palmitoleic acid, other dairy fat biomarkers, and incident diabetes: the Multi-Ethnic Study of Atherosclerosis (MESA). Am J Clin Nutr 97:854–861
Yang ZH, Miyahara H, Hatanaka A (2011) Chronic administration of palmitoleic acid reduces insulin resistance and hepatic lipid accumulation in KK-ay mice with genetic type 2 diabetes. Lipids Health Dis 10:120
Welters HJ, Tadayyon M, Scarpello JHB, Smith SA, Morgan NG (2004) Mono-unsaturated fatty acids protect against β-cell apoptosis induced by saturated fatty acids, serum withdrawal or cytokine exposure. FEBS Lett 560(1):103–108
Weijers RN (2012) Lipid composition of cell membranes and its relevance in type 2 diabetes mellitus. Curr Diabetes Rev 8:390–400
Ferrucci L, Cherubini A, Bandinelli S et al (2006) Relationship of plasma polyunsaturated fatty acids to circulating inflammatory markers. J Clin Endocrinol Metab 91:439–446
Kahn SE, Cooper ME, Del Prato S (2014) Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383:1068–1083
Gromovsky AD, Schugar RC, Brown AL et al (2018) Δ-5 fatty acid desaturase FADS1 impacts metabolic disease by balancing proinflammatory and proresolving lipid mediators. Arterioscler Thromb Vasc Biol 38:218–231
Menni C, Zierer J, Pallister T et al (2017) Omega-3 fatty acids correlate with gut microbiome diversity and production of N-carbamylglutamate in middle aged and elderly women. Sci Rep 7:11079
White J, Swerdlow DI, Preiss D et al (2016) Association of lipid fractions with risks for coronary artery disease and diabetes. JAMA Cardiol 1:692–699
Mensink RP, Zock PL, Kester AD, Katan MB (2003) Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: a meta-analysis of 60 controlled trials. Am J Clin Nutr 77:1146–1155
Willer CJ, Schmidt EM, Sengupta S et al (2013) Discovery and refinement of loci associated with lipid levels. Nat Genet 45:1274–1283
Acknowledgements
Open access funding provided by Karolinska Institute. Summary-level data for genetic associations with the type 2 diabetes has been contributed by the DIAGRAM consortium and were downloaded from http://diagram-consortium.org/downloads.html (accessed 24 May 2019). Summary-level data for genetic associations with fasting glucose, fasting insulin, HOMA-B and HOMA-IR have been contributed by the MAGIC consortium and were downloaded from https://www.magicinvestigators.org/ (accessed 24 May 2019). The authors thank all investigators for sharing these data.
Funding
This work was supported by the Swedish Research Council.
Author information
Authors and Affiliations
Contributions
SCL designed the study. SY and SCL analysed and interpreted the data. The manuscript was drafted by SY with contributions from SCL. Both authors provided final approval of the version to be published. SCL is the guarantor of this work.
Corresponding author
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM Tables
(XLSX 31 kb)
ESM Figures
(PDF 295 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yuan, S., Larsson, S.C. Association of genetic variants related to plasma fatty acids with type 2 diabetes mellitus and glycaemic traits: a Mendelian randomisation study. Diabetologia 63, 116–123 (2020). https://doi.org/10.1007/s00125-019-05019-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-05019-0