
Abstract
Aims/hypothesis
O-GlcNAcylation is implicated in modulating mitochondrial function, which is closely involved in regulating muscle metabolism. The presence of O-GlcNAcase (OGA), the enzyme involved in the removal of O-GlcNAc, in mitochondria was recently confirmed in rats. In the present study, we investigated the regulation of myogenesis and muscle insulin sensitivity to OGA in mice, with a focus on mitochondria.
Methods
C57BL/6J mice fed a high-fat diet for 4 months were used to observe mitochondrial density, activity and O-GlcNAcylation in muscle. Small interfering RNA and overexpression vectors were used to modulate protein content in vitro.
Results
High-fat feeding decreased the OGA level and largely increased mitochondrial O-GlcNAcylation in mouse skeletal muscle that was accompanied by decreased levels of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), decreased mitochondrial density and disrupted mitochondrial complex activities. Knockdown of OGA in C2C12 myoblasts promoted PGC-1α degradation, resulting in the suppression of mitochondrial biogenesis and myogenesis, whereas neither knockdown of O-GlcNAc transferase nor overexpression of OGA had significant effects on myogenesis. Mitochondrial dysfunction as evidenced by decreased ATP content and increased reactive oxygen species production, and increased lipid and protein oxidation was observed in both myoblasts and myotubes after OGA knockdown. Meanwhile, elevated O-GlcNAcylation through either OGA knockdown or treatment with the OGA inhibitor PUGNAc and the O-GlcNAc transferase substrate d-GlcNAc suppressed myotube insulin signalling transduction and glucose uptake. OGA overexpression had no significant effect on insulin sensitivity but sufficiently improved the insulin resistance induced by d-GlcNAc treatment.
Conclusions/interpretation
These data suggest that OGA can modulate mitochondrial density via PGC-1α and mitochondrial function via protein O-GlcNAcylation. In this manner, OGA appears to play a key role in myogenesis and the development of muscle insulin resistance.
Similar content being viewed by others
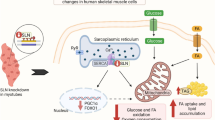


Introduction
Skeletal muscle, the largest endocrine organ in the body, plays a vital role in glucose clearance and utilisation [1]. Insulin resistance in skeletal muscle is considered an early hallmark in the development of type 2 diabetes [2]. A long-term diabetic condition usually results in skeletal muscle myopathy, which is a common source of disability in both type 1 and 2 diabetic populations and is increasing in prevalence [3–5]. Despite accumulating studies, the molecular mechanism underlying the skeletal muscle defects in the diabetic condition remains limited.
O-GlcNAcylation is a nutrient-sensitive, post-translational modification that targets thousands of proteins and modulates their physiological functions [6, 7]. Aberrant O-GlcNAc signalling has been observed in a study of several chronic diseases, including obesity, diabetes, neurodegeneration and cancer [8, 9]. In the study of diabetes, streptozotocin (STZ) has been used to induce a type 1 diabetic animal model by damaging pancreatic beta cells, resulting in hypoinsulinaemia and hyperglycaemia, while chronic high-caloric or high-fat diets (HFD) are usually used to induce type 2 diabetic animal models that are closely associated with obesity and insulin resistance; previous studies have revealed increased protein O-GlcNAcylation modification in both of these diabetic animal models [10–13], whereas exercise training has been shown to mitigate aberrant protein O-GlcNAcylation in diabetic mice [13].
As skeletal muscle is the major organ in exercise and glucose metabolism, skeletal muscle function, such as skeletal muscle contraction [14], myogenin protein content and myogenesis [15], has been suggested to be associated with O-GlcNAc signalling. Furthermore, higher levels of O-GlcNAcylation are associated with protein degradation and muscle atrophy [16]. These findings suggest that dysregulated O-GlcNAc cycling may be a critical component of muscle dysfunction and insulin resistance. The regulation of O-GlcNAcylation is highly dynamic but requires only two enzymes: O-GlcNAc transferase (OGT) [17], an enzyme that catalyses the addition of O-GlcNAc to target proteins; and O-GlcNAcase (OGA) [18], an enzyme that catalyses the removal of O-GlcNAc from target substrates. Disruption of the OGT/OGA balance results in aberrant protein O-GlcNAc modification that contributes to physiological changes. A number of studies have demonstrated that increased OGT protein can contribute to insulin resistance [9, 14, 19]. Although the role of OGA in O-GlcNAc modification is known, the involvement of OGA in other biological processes remains limited.
Mitochondria are major energy producers in living cells. Impaired content or activity of enzymes related to mitochondrial function has been reported in STZ- or HFD-induced diabetic animal models [20–23]. Following the observation of OGT localisation to the mitochondria by Love et al [24], the presence of OGA was recently reported in cardiac mitochondria in rats [25]. Despite a recently identified association between OGA-mediated O-GlcNAcylation and diabetes progression [25, 26], there have been no reports of the effects of OGA activity on mitochondrial regulation. In the current study in mice, we report that OGA deficiency promotes peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) degradation by increasing its O-GlcNAcylation, leading to suppressed mitochondrial biogenesis and myogenesis. In addition, we show that OGA deficiency increases mitochondrial protein O-GlcNAcylation, contributing to mitochondrial dysfunction and insulin resistance. Together, our data suggest that OGA activity is important for maintaining normal mitochondria and skeletal muscle function.
Methods
Chemicals
d-GlcNAc, FCCP (carbonyl cyanide 4-[trifluoromethoxy] phenylhydrazone), antimycin A and oligomycin were purchased from Sigma (St. Louis, MO, USA). Antibodies against complex I, II, III, IV and V were obtained from Invitrogen (Carlsbad, CA, USA). Antibodies against myosin heavy chain (MyHC), PGC-1α, GAPDH, OGA, OGT and O-GlcNAc (CTD110.6) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies against α-tubulin, AKT, p-AKT (S473), GSKα/β, and p-GSKα/β (S21/9) were obtained from Cell Signaling Technology (Danvers, MA, USA). Small interfering RNA (siRNA) oligos and PCR primers were synthesised by Genepharma (Shanghai, China). Insulin was purchased from Fitzgerald (Acton, MA, USA). The Oga plasmid was a generous gift from G. W. Hart (Johns Hopkins University School of Medicine, Baltimore, MD, USA); Oga is also known as Mgea5. See electronic supplementary material (ESM) Methods for further details.
Animals
Four-week-old male C57BL/6 mice were purchased from the SLAC Laboratory Animal Co. (Shanghai, China). After 1 week of acclimatisation, the mice were randomly divided into two feeding groups (n = 6 in each group): mice fed a normal diet (control, 12% energy fat content) and mice fed an HFD (45% energy fat content). After 16 weeks, the mice were fasted overnight and killed for tissue collection. All animals were housed in a temperature-controlled (25–28°C) and humidity-controlled (60%) animal room and maintained on a 12 h light/12 h dark cycle with food and water provided during the experiments. All the methods were performed in accordance with approved guidelines, and all efforts were made to minimise the suffering and the number of animals used in this study. Experimenters were not blind to group assignment or outcome assessment. See ESM Methods for the HFD formula.
C2C12 cell differentiation
Mouse mycoplasma free C2C12 myoblasts were purchased from the ATCC (Manassas, VA, USA). Cells were induced into myotubes after 6 days following previously reported method [27]. siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen), and DNA transfections were performed using X-tremeGENE HP DNA transfection reagent (Roche, Basel, Switzerland) as described in the supplier’s manuals. For MyHC immunocytochemistry analysis, cells were washed, fixed, stained and visualised by confocal microscopy (Zeiss, Jena, Germany). See ESM Methods for further details.
Mitochondrial complex activity assays
Mitochondria isolation from cultured mouse C2C12 myoblasts, myotubes and skeletal muscle tissues were performed as previously described [27, 28]. Mitochondrial complex activities were measured by spectrophotometer as described in previous reports [29, 30]. See ESM Methods for further details
Cell oxygen consumption rate measurement
Mitochondrial oxygen consumption rate of cultured mouse C2C12 myoblasts and myotubes were measured using an extracellular flux analysis (Seahorse Biosciences, N. Billerica, MA, USA). See ESM Methods for further details.
ATP measurement
Cellular ATP contents of cultured mouse C2C12 myoblasts and myotubes were performed using an ATP assay kit following the manufacturer’s protocol (FLAA, Sigma). See ESM Methods for further details.
Oxidative stress assays
Cellular oxidative stress of cultured mouse C2C12 myoblasts and myotubes was assessed by analysing intracellular reactive oxygen species (ROS) with the fluorescence probe 2ˊ,7ˊ-dichlorofluorescein (H2DCF-DA), carbonyl protein with the OxyBlot protein oxidation detection kit (Cell Biolabs, San Diego, CA, USA), 4-hydroxynonenal (4-HNE) with ELISA kit (RD Systems, Shanghai, China), and glutathione (GSH) with 2,3-naphthalenedicarboxyaldehyde (NDA) as previously described [23, 27]. See ESM Methods for further details.
Glucose uptake measurement
Medium glucose of cultured mouse C2C12 myotubes was detected using a commercial kit from Sigma (St. Louis, MO, USA). Glucose uptake was determined by calculating the fresh medium glucose minus the reduced medium glucose. See ESM Methods for further details.
Real-time PCR
Real-time quantitative PCR was used to determine the relative expression levels of mRNAs in cultured mouse C2C12 myoblasts and myotubes. See ESM Methods for further details.
Western blot
Protein samples from cells, skeletal muscles, mitochondria and co-immunoprecipitates were analysed by western blot. See ESM Methods for further details.
Statistical analysis
The results are presented as the means ± SEM. Statistical analyses were conducted using one-way ANOVA followed by least significant difference post hoc analyses or unpaired t tests. For all analyses, values of p < 0.05 were considered significant. No results were omitted or excluded in our study.
Results
Mitochondrial O-GlcNAcylation in skeletal muscle of diabetic mice
Obese and diabetic mice were developed by feeding a HFD for 4 months (ESM Fig. 1). Analysis of the skeletal muscle showed decreased OGA protein levels in both the total and mitochondrial portion of the HFD group, whereas no significant change was observed in the OGT levels (Fig. 1a). Compared with the overall O-GlcNAcylation, mitochondrial protein showed a more robust increase in O-GlcNAcylation (Fig. 1b). The protein level of PGC-1α was decreased in the HFD group and was accompanied by decreased mitochondrial complex subunits I and V (Fig. 1c, ESM Fig. 2). An analysis of mitochondrial electron transport chain complex activity showed decreased activities of complexes I and II as well as increased activities of complex III and V in the HFD group (Fig. 1d). These data suggest that OGA is more sensitive in diabetic muscle and that there may be a connection between dysregulated O-GlcNAcylation and mitochondrial dysfunction in diabetic muscle.

HFD-induced diabetes in mice is associated with increased O-GlcNAcylation and decreased mitochondrial function in skeletal muscle. Protein and mitochondria samples were collected from the skeletal muscle of C57BL6 mice fed an HFD for 16 weeks. (a) Total and mitochondrial portion of OGA and OGT levels. (b) Protein levels of PGC-1α and mitochondrial complexes. (c) Total and mitochondrial protein O-GlcNAcylation. (d) Mitochondrial complex activity. Grey, chow; black, HFD. The numbers to the right of each blot represent molecular mass in kDa. The values are the means ± SEM; n = 6; *p < 0.05; **p < 0.01
Mitochondrial O-GlcNAcylation in myogenesis
Mouse C2C12 myoblasts were induced to form myotubes after 6 days of differentiation. MyHC, the marker of myotubes, was increased in a time-dependent manner during C2C12 myogenesis (Fig. 2a, b). A robust increase in OGA protein content was observed at an early differentiation time (Fig. 2b), and the total protein O-GlcNAcylation decreased during myogenesis (Fig. 2b). Consistent with a previous report [31], myogenesis was accompanied with mitochondrial biogenesis as evidenced by increased fluorescence of MitoTracker green staining (Fig. 2a) and protein levels of mitochondrial complex subunits (Fig. 2c). In response to a recent report of localised OGA in mitochondria [25], we analysed the mitochondria and found that mitochondrial OGA levels sharply increased during myogenesis and were accompanied by decreased mitochondrial O-GlcNAcylation (Fig. 2d). The purity of the mitochondrial fraction was confirmed in both myoblasts and myotubes (ESM Fig. 3).

C2C12 cell differentiation is accompanied by mitochondrial biogenesis and decreased protein O-GlcNacylation. (a) Immunostaining of MyHC during myotube differentiation. (b) Protein levels of MyHC, OGA and O-GlcNAc. (c) Protein levels of mitochondrial complex subunits I, II, III, IV and V. (d) Protein levels of mitochondrial OGA and O-GlcNAc. Scale bar 200 μm. The numbers to the right of each blot represent molecular mass in kDa
OGA deficiency suppresses C2C12 myogenesis
C2C12 myoblasts were cultured with the OGA inhibitor PUGNAc during differentiation. The PUGNAc treatment decreased the MyHC protein content, indicating the suppression of myogenesis (Fig. 3a, ESM Fig. 2). To further confirm the dependency of myogenesis on OGA, we treated myoblasts with Oga siRNA and then subjected the cells to a 48 h differentiation period. The MyHC levels were significantly decreased by OGA knockdown (Fig. 3b). Similar decreases were also observed for the mRNA levels of the key regulators of myogenesis MyoD, myogenin and Mrf4 (also known as Mrf6) (Fig. 3c). OGA knockdown was accompanied by decreased OGT protein content; therefore, no significant change was observed in total protein O-GlcNAcylation (Fig. 3b). Interestingly, overexpression of OGA had no significant effect on MyHC (Fig. 3d), and MyoD, myogenin and Mrf4 mRNA levels (Fig. 3e), and further knockdown of OGT also showed no effects on MyHC protein content when subjected to 48 h differentiation (Fig. 3f); these findings indicate that OGA is required to maintain normal myogenesis activity. Myogenesis status was further confirmed by an immunofluorescence assay of MyHC (Fig. 3 g, h).

OGA deficiency suppresses C2C12 cell differentiation. (a) MyHC protein level of C2C12 myoblasts differentiated with or without 100 μmol/l PUGNAc. (b) Western blot and (c) real-time PCR assay of C2C12 myoblasts administered Oga siRNA for 24 h prior to 48 h of differentiation. Grey bars, control; black bars, Oga siRNA. (d) Western blot and (e) real-time PCR assay of C2C12 myoblasts administered Oga expression vector for 24 h prior to 48 h of differentiation. Grey bars, control; black bars, OGA overexpression. (f) Western blot assay of C2C12 myoblasts administered Ogt siRNA for 24 h prior to 48 h of differentiation. (g) Immunostaining of MyHC, and (h) myogenic index of C2C12 myoblasts with OGA knockdown or overexpression prior to 48 h of differentiation. Scale bar, 100 μm. The numbers to the right of each blot represent molecular mass in kDa. The values are the means ± SEM; n = 4; *p < 0.05; **p < 0.01. NC, negative control; siOga, Oga siRNA; siOgt, Ogt siRNA
PGC-1α is required for OGA-mediated C2C12 myogenesis
As a master regulator of mitochondrial biogenesis, PGC-1α was decreased by OGA knockdown, contributing to the suppression of mitochondrial biogenesis during myogenesis (Fig. 4a, ESM Fig. 2). Interestingly, OGA knockdown had no effect on Pgc-1α mRNA (Fig. 4b), and MG132, a proteasome inhibitor, inhibited the decrease in PGC-1α protein (Fig. 4c). Overexpression of OGA had no effect on PGC-1α and mitochondrial density (ESM Fig. 4). An analysis of PGC-1α immunoprecipitates showed increased O-GlcNAcylation and ubiquitination of PGC-1α in Oga siRNA-treated myoblasts (Fig. 4d), and a consistent increase was also observed in HFD-induced diabetic skeletal muscle (Fig. 4e). Additionally, the direct knockdown of PGC-1α significantly reduced the protein levels of MyHC (Fig. 4f) and the mRNA levels of MyoD, myogenin and Mrf4 (Fig. 4g). This suppressed myogenesis was further confirmed with MyHC immunofluorescence and a myogenic index analysis (Fig. 4h). The overexpression of PGC-1α could sufficiently prevent the OGA knockdown-induced decrease of MyHC and mitochondrial complex subunits (Fig. 4i). These data suggest that OGA could target PGC-1α to modulate mitochondrial biogenesis and C2C12 myogenesis.

PGC-1α is required for OGA-mediated C2C12 differentiation. (a) Protein levels of PGC-1α and mitochondrial complexes. (b) mRNA level of Pgc-1α in C2C12 myoblasts administered Oga siRNA for 24 h prior to 48 h of differentiation. (c) Protein level of PGC-1α in C2C12 myoblasts administered Oga siRNA for 24 h prior to 48 h of differentiation with or without 24 h of MG132 treatment. (d) C2C12 myoblasts administered Oga siRNA for 24 h prior to 48 h of differentiation. PGC-1α immunoprecipitates were then obtained to analyse the O-GlcNAc and ubiquitin levels. (e) PGC-1α immunoprecipitates obtained from skeletal muscle extracts were analysed by western blot for O-GlcNAc and ubiquitin levels. (f) Protein levels of PGC-1α and MyHC and (g) mRNA levels of MyoD, myogenin and Mrf4. (h) Immunofluorescence of MyHC, and myogenic index from C2C12 myoblasts administered PGC-1α siRNA for 24 h prior to 48 h of differentiation. (i) Protein levels of OGA, PGC-1α, mitochondrial complexes and MyHC from C2C12 myoblasts administered Oga siRNA and PGC-1α overexpression vector for 24 h prior to 48 h of differentiation. Grey bars, control; black bars, PGC-1α siRNA. Scale bar, 100 μm. The numbers to the right of each blot represent molecular mass in kDa. The values are the means ± SEM; n = 4; *p < 0.05; **p < 0.01. IB, immunoblotting; IP, immunoprecipitation; NC, negative control; α-UB, ubiquitin; siOga, Oga siRNA
OGA deficiency impairs mitochondrial function during C2C12 differentiation
Abnormal mitochondria could contribute to an abnormality in myogenesis [32]. Although no significant change in total protein O-GlcNAcylation was observed after OGA knockdown (Fig. 3), we were surprised to observe a robust increase in O-GlcNAcylation of the mitochondrial fraction (Fig. 5a). An oxygen consumption analysis showed that basal respiration, maximal respiration and ATP potential during differentiation were all increased by OGA knockdown (Fig. 5b). However, the ATP level (Fig. 5c) dramatically decreased. As determined using flow cytometry and immunofluorescence, the production of ROS increased (Fig. 5d, ESM Fig. 4). Accordingly, protein oxidation, as indicated by carbonyl protein analysis (Fig. 5e), and lipid oxidation, as indicated by 4-HNE analysis (Fig. 5f), were both increased by OGA knockdown during myogenesis, suggesting that OGA knockdown could impair mitochondrial function and induce oxidative stress during myogenesis. As one of the most abundant antioxidants in the cells, GSH was decreased by OGA knockdown, and treatment with buthionine sulphoximine (BSO), a specific inhibitor of γ-glutamylcysteine synthetase that is responsible for GSH production, also induced a similar decrease in GSH as OGA knockdown (Fig. 5g). However, H2DCF-DA staining showed that OGA knockdown induced a more significant increase in ROS than BSO treatment in normal cells, although BSO treatment alone in normal cells increased the ROS content (Fig. 5h, i). These data further support that ROS production in OGA knockdown cells is mainly attributed to mitochondrial dysfunction.

OGA deficiency impairs mitochondrial function during C2C12 differentiation. C2C12 myoblasts administered Oga siRNA for 24 h prior to 48 h of differentiation. (a) Protein levels of mitochondrial OGA, OGT and O-GlcNAc. (b) Extracellular flux (XF) analysis of mitochondrial oxygen consumption. (c) Relative ATP level. (d) Cellular ROS level. (e) Carbonyl protein level. (f) Relative 4-HNE level. C2C12 myoblasts were administered Oga siRNA for 48 h followed by BSO treatment for 24 h. (g) Cellular GSH content. (h) Fluorescence of H2DCF-DA in myoblasts. (i) Relative ROS content analysed by flow cytometry. Grey bars, control; black bars, Oga siRNA. Scale bar, 200 μm. The numbers to the right of each blot represent molecular mass in kDa. The values are the means ± SEM; n = 4; *p < 0.05; **p < 0.01. NC, negative control; siOga, Oga siRNA
OGA deficiency impairs mitochondrial function in myotubes
In fully differentiated myotubes, the knockdown of OGA in myotubes resulted in increased mitochondrial OGT levels and O-GlcNAcylation (Fig. 6a). The mitochondrial oxygen consumption capacity, including basal respiration, maximal respiration and ATP potential, were all elevated after OGA knockdown (Fig. 6b). Consistent with our observations during myogenesis, cellular ATP (Fig. 6c) decreased, whereas the cellular ROS level was robustly increased as revealed by flow cytometry (Fig. 6d) and immunofluorescence analysis (ESM Fig. 4). As a result of oxidative stress, the carbonyl protein (Fig. 6e) and cellular 4-HNE levels (Fig. 6f) were also significantly increased after OGA knockdown. The overexpression of OGA in myotubes had no significant effect on mitochondrial oxygen consumption (ESM Fig. 4). These data suggest that mitochondria are more sensitive to an OGA deficiency-associated increase in protein O-GlcNAcylation.

OGA deficiency impairs mitochondrial function in myotubes. C2C12 myoblasts were induced into myotubes for 6 days followed by Oga siRNA administration for another 72 h. (a) Protein levels of mitochondrial OGA, OGT and O-GlcNAc. (b) Extracellular flux (XF) analysis of mitochondrial oxygen consumption. (c) Relative ATP level. (d) Cellular ROS level. (e) Carbonyl protein level.(f) Relative 4-HNE level. Grey bars or points/lines, control; black bars or points/lines, Oga siRNA. The numbers to the right of each blot represent molecular mass in kDa. The values are means ± SEM; n = 4; *p < 0.05; **p < 0.01. siOga, Oga siRNA
OGA deficiency impairs insulin sensitivity in myotubes
In fully differentiated myotubes, the addition of the OGA inhibitor PUGNAc and the OGT substrate d-GlcNAc decreased p-Akt and p-GSK levels under insulin stimuli (Fig. 7a, ESM Fig. 2), which was accompanied by decreased glucose uptake (Fig. 7b), indicating impaired insulin sensitivity. OGA knockdown also impaired insulin signalling (Fig. 7c, ESM Fig. 2) and decreased glucose uptake (Fig. 7d) in myotubes. However, the overexpression of OGA did not increase or decrease insulin sensitivity (Fig. 7e); instead, the overexpression of OGA sufficiently eliminated d-GlcNAc-induced insulin resistance (Fig. 7f). Taken together, these data suggest the importance of OGA activity in maintaining myotube insulin sensitivity.

OGA deficiency impairs insulin sensitivity in myotubes. (a) Fully differentiated myotubes were treated with 100 μmol/l PUGNAc and 1 mol/l d-GlcNAc for 48 h with or without insulin challenge for another 10 min, followed by detection of p-Akt/Akt and p-GSK-3α/β/GSK-3α/β. (b) Glucose uptake was detected without insulin challenge. (c) Myotubes were administered Oga siRNA for 72 h with or without insulin challenge for another 10 min, followed by detection of p-Akt/Akt and p-GSK-3α/β/GSK-3α/β. (d) Glucose uptake was detected without insulin challenge. Grey bar, control; black bar, Oga siRNA. (e) Myotubes were transfected with pcDNA3.1-Oga for 72 h with or without insulin challenge for another 10 min, followed by detection of p-Akt/Akt and p-GSK-3α/β/GSK-3α/β. (f) Myotubes were transfected with pcDNA3.1-Oga for 24 h followed by 48 h of d-GlcNAc treatment (1 mol/l) with or without insulin challenge for another 10 min, followed by detection of p-Akt/Akt and p-GSK-3α/β/GSK-3α/β. The numbers to the right of each blot represent molecular mass in kDa. The values are the means ± SEM; n = 4; *p < 0.05; **p < 0.01. siOga, Oga siRNA
Discussion
Skeletal muscle insulin resistance is the primary type of dysregulation in type 2 diabetes [33]. A strong association between aberrant O-GlcNAc cycling and development of insulin resistance and diabetes has been proposed in recent years. Enhanced global O-GlcNAcylation has been found in the liver tissue of db/db mice and HFD-induced diabetic mouse models [33], and in the cardiac muscle and coronary endothelial cells of STZ-induced diabetic mouse models [13, 34], which is consistent with our observations in skeletal muscle. Meanwhile, the cytoprotective potential of increased O-GlcNAc signalling in the cardiovascular system has also been proposed in several studies [35, 36], suggesting that O-GlcNAc signalling has multiple roles in regulating physiological activities. More efforts are required to elucidate the details of the involvement of O-GlcNAc signalling in diabetes progression.
Although only two enzymes regulate O-GlcNAc cycling, the regulation of their activities and the regulation between the two enzymes remain unclear. In addition to adding O-GlcNAc to a target protein, OGT is also auto-GlcNAcylated within its catalytic domain, and this autoglycosylation may regulate its activity [37]. In our study, in the mitochondrial fraction of the HFD-induced diabetic skeletal muscle, neither OGT protein expression nor OGT O-GlcNAc levels were affected, whereas the OGA level showed a significant decrease leading to elevated O-GlcNAcylation (ESM Fig. 5). This result indicates that OGA is quite susceptible to insulin resistance in diabetic skeletal muscle, and a loss of OGA activity did not promote OGT autoglycosylation. Different from our in vivo observation, we found that OGA knockdown in C2C12 cells could decrease OGT protein content, which is consistent with observations in the intestines of OGA knockdown mice by Yang et al [38]. However, OGA knockdown in cancer cells had no effect on the OGT level [39], suggesting a complex relationship between OGA and OGT, and how OGA activity regulates OGT in different contexts remains to be elucidated.
Impaired mitochondrial O-GlcNAcylation has been associated with mitochondrial dysfunction in neurons [40] and cardiomyocytes [25, 41]. Thus, we hypothesised that elevated mitochondrial O-GlcNAcylation may contribute to mitochondrial dysfunction in diabetic skeletal muscle. OGA significantly decreased in diabetic muscle; therefore, we focused on OGA in in vitro studies. By use of C2C12 myoblasts as a myogenesis model, we showed that OGA levels increased with decreased cellular O-GlcNAcylation during myogenesis, which was consistent with a previous finding [42]. In our study, mitochondrial biogenesis was possibly induced in response to the higher energy demand required for myogenesis, whereas mitochondrial O-GlcNAcylation was dramatically reduced because of increased mitochondrial OGA levels. This finding is supported by the recent discovery of OGA mitochondrial localisation [25]. In addition to the mitochondrial density affected by OGA deficiency, we also showed that mitochondrial protein O-GlcNAcylation was altered in response to OGA knockdown during myogenesis and in fully differentiated myotubes, and was accompanied by impaired mitochondrial function, including significantly lowered ATP content, increased ROS content and protein and lipid oxidation. By employing the OGA inhibitor PUGNAc or siRNA, we showed that OGA deficiency could decrease myotube fusion and inhibit myogenesis. The overexpression of OGA had no significant effect on mitochondrial activity and myogenesis, with the exception of a dramatic decrease in O-GlcNAcylation. These data suggest that in addition to being closely involved in chronic diseases such as obesity, diabetes, neurodegeneration and cancer [8, 9], OGA is also required to maintain normal mitochondrial function during myogenesis. Further studies are called for to explore the specific regulators involved in myogenesis that are modulated by OGA.
As the master regulator of mitochondrial biogenesis, PGC-1α has been suggested to contribute to myogenesis [43], which was confirmed in the current study. We showed that the knockdown of OGA decreased PGC-1α protein content and suppressed mitochondrial biogenesis during myogenesis. In addition, the overexpression of PGC-1α sufficiently prevented OGA knockdown-induced mitochondrial loss and myogenesis decrease. These observations indicate that PGC-1α is a key myogenesis regulator that can be modulated by OGA. A recent report by Ruan et al showed that host cell factor C1 could recruit OGT to O-GlcNacylate PGC-1α and protect it from degradation [44]. Interestingly, we found that in both Oga siRNA-treated cells and diabetic skeletal muscle, both O-GlcNAcylation and ubiquitination of PGC-1α were increased and were accompanied by decreased PGC-1α protein content, suggesting a more complicated regulation of PGC-1α stability by O-GlcNAcylation, which requires further exploration.
Previous reports have shown that the loss of OGA in mice leads to defects in metabolic homeostasis, culminating in obesity and insulin resistance [26], and that the overexpression of OGA in db/db mice significantly improves the lipid profile in liver [45]. Consistently, we found impaired insulin signalling and glucose uptake capacity in myotubes after OGA knockdown or treatment with PUGNAc or d-GlcNAc, with concurrent increased O-GlcNAcylation and mitochondrial dysfunction. Additionally, OGA overexpression successfully improved the insulin sensitivity damaged by the addition of d-GlcNAc. Taken together, our data further indicate that OGA activity is important for maintaining regular myogenesis and sensing insulin signals in skeletal muscle, and modulating mitochondrial density and function may account for its benefits.
Abbreviations
- BSO:
-
Buthionine sulphoximine
- FCCP:
-
Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- GSH:
-
Glutathione
- H2DCF-DA:
-
2´,7´-Dichlorofluorescein
- HFD:
-
High-fat diet
- MyHC:
-
Myosin heavy chain
- OGA:
-
O-GlcNAcase
- OGT:
-
O-GlcNAc transferase
- PGC-1α:
-
Peroxisome proliferator-activated receptor γ coactivator 1α
- ROS:
-
Reactive oxygen species
- siRNA:
-
Small interfering RNA
- STZ:
-
Streptozotocin
- 4-HNE:
-
4-Hydroxynonenal
References
Timmons JA, Norrbom J, Scheele C, Thonberg H, Wahlestedt C, Tesch P (2006) Expression profiling following local muscle inactivity in humans provides new perspective on diabetes-related genes. Genomics 87:165–172
Schrauwen-Hinderling VB, Kooi ME, Schrauwen P (2016) Mitochondrial function and diabetes: consequences for skeletal and cardiac muscle metabolism. Antioxid Redox Signal 24:39–51
Ramchurn N, Mashamba C, Leitch E et al (2009) Upper limb musculoskeletal abnormalities and poor metabolic control in diabetes. Eur J Intern Med 20:718–721
Park SW, Goodpaster BH, Lee JS et al (2009) Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 32:1993–1997
Kim S, Valdez R (2015) Metabolic risk factors in U.S. youth with low relative muscle mass. Obes Res Clin Pract 9:125–132
Hanover JA, Krause MW, Love DC (2012) Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol 13:312–321
Butkinaree C, Park K, Hart GW (2010) O-linked beta-N-acetylglucosamine (O-GlcNAc): extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta 1800:96–106
Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O (2011) Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 80:825–858
Akimoto Y, Kreppel LK, Hirano H, Hart GW (2000) Increased O-GlcNAc transferase in pancreas of rats with streptozotocin-induced diabetes. Diabetologia 43:1239–1247
Marsh SA, Powell PC, Dell'italia LJ, Chatham JC (2013) Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci 92:648–656
Medford HM, Chatham JC, Marsh SA (2012) Chronic ingestion of a Western diet increases O-linked-beta-N-acetylglucosamine (O-GlcNAc) protein modification in the rat heart. Life Sci 90:883–888
Li SY, Liu Y, Sigmon VK, McCort A, Ren J (2005) High-fat diet enhances visceral advanced glycation end products, nuclear O-Glc-Nac modification, p38 mitogen-activated protein kinase activation and apoptosis. Diabetes Obes Metab 7:448–454
Bennett CE, Johnsen VL, Shearer J, Belke DD (2013) Exercise training mitigates aberrant cardiac protein O-GlcNAcylation in streptozotocin-induced diabetic mice. Life Sci 92:657–663
Yang X, Ongusaha PP, Miles PD et al (2008) Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 451:964–969
Ogawa M, Sakakibara Y, Kamemura K (2013) Requirement of decreased O-GlcNAc glycosylation of Mef2D for its recruitment to the myogenin promoter. Biochem Biophys Res Commun 433:558–562
Cieniewski-Bernard C, Mounier Y, Michalski JC, Bastide B (2006) O-GlcNAc level variations are associated with the development of skeletal muscle atrophy. J Appl Physiol 100:1499–1505
Kreppel LK, Blomberg MA, Hart GW (1997) Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem 272:9308–9315
Dong DL, Hart GW (1994) Purification and characterization of an O-GlcNAc selective N-acetyl-beta-d-glucosaminidase from rat spleen cytosol. J Biol Chem 269:19321–19330
McClain DA, Lubas WA, Cooksey RC et al (2002) Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc Natl Acad Sci U S A 99:10695–10699
Chao TT, Ianuzzo CD, Armstrong RB, Albright JT, Anapolle SE (1976) Ultrastructural alterations in skeletal muscle fibers of streptozotocin-diabetic rats. Cell Tissue Res 168:239–246
Chowdhury SK, Zherebitskaya E, Smith DR et al (2010) Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes 59:1082–1091
Zou X, Yan C, Shi Y et al (2014) Mitochondrial dysfunction in obesity-associated nonalcoholic fatty liver disease: the protective effects of pomegranate with its active component punicalagin. Antioxid Redox Signal 21:1557–1570
Cao K, Xu J, Zou X et al (2014) Hydroxytyrosol prevents diet-induced metabolic syndrome and attenuates mitochondrial abnormalities in obese mice. Free Radic Biol Med 67:396–407
Love DC, Kochan J, Cathey RL, Shin SH, Hanover JA (2003) Mitochondrial and nucleocytoplasmic targeting of O-linked GlcNAc transferase. J Cell Sci 116:647–654
Banerjee PS, Ma J, Hart GW (2015) Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc Natl Acad Sci U S A 112:6050–6055
Keembiyehetty C, Love DC, Harwood KR, Gavrilova O, Comly ME, Hanover JA (2015) Conditional knock-out reveals a requirement for O-linked N-acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J Biol Chem 290:7097–7113
Wang X, Li H, Zheng A et al (2014) Mitochondrial dysfunction-associated OPA1 cleavage contributes to muscle degeneration: preventative effect of hydroxytyrosol acetate. Cell Death Dis 5, e1521
Feng Z, Bai L, Yan J et al (2011) Mitochondrial dynamic remodeling in strenuous exercise-induced muscle and mitochondrial dysfunction: regulatory effects of hydroxytyrosol. Free Radic Biol Med 50:1437–1446
Li X, Liu Z, Luo C et al (2008) Lipoamide protects retinal pigment epithelial cells from oxidative stress and mitochondrial dysfunction. Free Radic Biol Med 44:1465–1474
Sun L, Luo C, Long J, Wei D, Liu J (2006) Acrolein is a mitochondrial toxin: effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Mitochondrion 6:136–142
Moyes CD, Mathieu-Costello OA, Tsuchiya N, Filburn C, Hansford RG (1997) Mitochondrial biogenesis during cellular differentiation. Am J Physiol 272:C1345–C1351
Kim B, Kim JS, Yoon Y, Santiago MC, Brown MD, Park JY (2013) Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. Am J Physiol Regul Integr Comp Physiol 305:R927–R938
DeFronzo RA, Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32(Suppl 2):S157–S163
Makino A, Dai A, Han Y et al (2015) O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am J Physiol Cell Physiol 309:C593–C599
Ngoh GA, Watson LJ, Facundo HT, Jones SP (2011) Augmented O-GlcNAc signaling attenuates oxidative stress and calcium overload in cardiomyocytes. Amino Acids 40:895–911
Zafir A, Readnower R, Long BW et al (2013) Protein O-GlcNAcylation is a novel cytoprotective signal in cardiac stem cells. Stem Cells 31:765–775
Tai HC, Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC (2004) Parallel identification of O-GlcNAc-modified proteins from cell lysates. J Am Chem Soc 126:10500–10501
Yang YR, Jang HJ, Yoon S et al (2014) OGA heterozygosity suppresses intestinal tumorigenesis in Apc(min/+) mice. Oncogenesis 3, e109
Yehezkel G, Cohen L, Kliger A, Manor E, Khalaila I (2012) O-linked beta-N-acetylglucosaminylation (O-GlcNAcylation) in primary and metastatic colorectal cancer clones and effect of N-acetyl-beta-d-glucosaminidase silencing on cell phenotype and transcriptome. J Biol Chem 287:28755–28769
Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL (2014) Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 158:54–68
Hu Y, Suarez J, Fricovsky E et al (2009) Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem 284:547–555
Ogawa M, Mizofuchi H, Kobayashi Y et al (2012) Terminal differentiation program of skeletal myogenesis is negatively regulated by O-GlcNAc glycosylation. Biochim Biophys Acta 1820:24–32
Lin J, Wu H, Tarr PT et al (2002) Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418:797–801
Ruan HB, Han X, Li MD et al (2012) O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab 16:226–237
Guinez C, Filhoulaud G, Rayah-Benhamed F et al (2011) O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 60:1399–1413
Acknowledgements
The authors thank G. W. Hart (Johns Hopkins University School of Medicine, Baltimore, MD, USA) and the NHLBI P01HL107153 Core C4 for providing Oga plasmids.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This study was supported by the National Basic Research Program (Nos. 2015CB553602 and 2014CB548200), the National Natural Science Foundation of China (81571050 and 31570777), Tianjin Science and Technology Planning Major Project (12JCZDJC34400), Tianjin Education Committee Sci-Tech Development Major Project (20112D05) and Tianjin Key Labs and Tech-Platform Project (10SYSYJC28400).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author contributions
YZ and JL designed the study. All authors were involved with the acquisition, analysis and interpretation of data. XW wrote the manuscript, with critical revision from ZF, XQW, LY, SH, KC, JX, LZ, YZ and JL. All authors approved the final version of the paper. ZF and JL are the guarantors of this work.
Additional information
Xun Wang and Zhihui Feng contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 120 kb)
ESM Fig. 1
(PDF 244 kb)
ESM Fig. 2
(PDF 356 kb)
ESM Fig. 3
(PDF 153 kb)
ESM Fig. 4
(PDF 1821 kb)
ESM Fig. 5
(PDF 128 kb)
Rights and permissions
About this article

Cite this article
Wang, X., Feng, Z., Wang, X. et al. O-GlcNAcase deficiency suppresses skeletal myogenesis and insulin sensitivity in mice through the modulation of mitochondrial homeostasis. Diabetologia 59, 1287–1296 (2016). https://doi.org/10.1007/s00125-016-3919-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3919-2