
Abstract
Mitochondria and the endoplasmic reticulum (ER) have a synergistic relationship and are key regulatory hubs in maintaining cell homeostasis. Communication between these organelles is mediated by mitochondria ER contact sites (MERCS), allowing the exchange of material and information, modulating calcium homeostasis, redox signalling, lipid transfer and the regulation of mitochondrial dynamics. MERCS are dynamic structures that allow cells to respond to changes in the intracellular environment under normal homeostatic conditions, while their assembly/disassembly are affected by pathophysiological conditions such as ageing and disease. Disruption of protein folding in the ER lumen can activate the Unfolded Protein Response (UPR), promoting the remodelling of ER membranes and MERCS formation. The UPR stress receptor kinases PERK and IRE1, are located at or close to MERCS. UPR signalling can be adaptive or maladaptive, depending on whether the disruption in protein folding or ER stress is transient or sustained. Adaptive UPR signalling via MERCS can increase mitochondrial calcium import, metabolism and dynamics, while maladaptive UPR signalling can result in excessive calcium import and activation of apoptotic pathways. Targeting UPR signalling and the assembly of MERCS is an attractive therapeutic approach for a range of age-related conditions such as neurodegeneration and sarcopenia. This review highlights the emerging evidence related to the role of redox mediated UPR activation in orchestrating inter-organelle communication between the ER and mitochondria, and ultimately the determination of cell function and fate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Disruption of organelle communication plays a pivotal role in the altered cellular homeostasis in older organisms and during disease progression. The cellular response to perturbations within the intracellular environment can be an adaptive and ultimately beneficial response, or a hormesis effect, where low levels of stress renders cells resistant to a subsequent challenge [1]. The beneficial hormesis effect is often preceded by an acute change in the cellular environment, such as in skeletal muscle during exercise where there is a site-specific increase in ROS that activates specific signalling pathways, such as Nrf2 activation [2, 3]. Chronic changes in the intracellular redox environment, result in maladaptive responses that can be detrimental and often described in pathological conditions and age-related diseases [4]. Cellular homeostasis is maintained by a constant flow of information from the external environment but also critically by inter-organelle communication, facilitating the exchange of material and information in response to biological perturbations. The endoplasmic reticulum (ER) and mitochondria are key regulatory hubs in maintaining cell homeostasis and they have a synergistic relationship that can determine their function and response to the cellular environment. Mitochondrial-ER contact sites (MERCS) mediate the exchange of information between these organelles and help determine how the cell responds to disruption in the cellular environment. The regulation of the assembly and disassembly of MERCS is an active area of research, in particular in the context of how MERCS change during development, age and disease and with subsequent effects on the function of both the ER and mitochondria.
Endoplasmic reticulum stress and the unfolded protein response
The endoplasmic reticulum (ER) is the largest of the cell's membrane-bound organelles (~ 10% cell volume), it is composed of a continuous network of tubules and sacs surrounded by membranes or cisternae [5]. The ER contributes to proteostasis by regulating protein synthesis, folding and transport [6]. It is the main intracellular store of calcium (Ca2+), the ER releases Ca2+ into the cytosol in response to cellular signals, initiating a signalling cascade that can modulate a wide range of cellular functions [7]. The rough ER is composed of sacs with a high density of ribosomes attached to the cytosolic domain and involved in protein biosynthesis, while the smooth ER contains tubules that specialise in lipid synthesis [5, 8].
Protein folding is a key regulatory step in proteostasis and disruption can result in the accumulation of misfolded proteins. The ER has a unique environment that facilitates protein folding, its oxidising nature favouring the formation of disulphide bonds [6]. ER homeostasis can be altered by physiological and pathological conditions, leading to an accumulation of misfolded proteins in the ER lumen, referred to as ER stress and results in the activation of the unfolded protein response (UPR) [9]. A variety of cellular stress conditions can alter ER proteostasis, including disruption of Ca2+ homeostasis, protein glycosylation, redox imbalance and an accumulation of misfolded proteins [10]. The adaptive UPRER aims to restore proteostasis and alleviate ER stress by reducing protein translation, increasing the chaperone capacity of the ER and stimulating the degradation of misfolded proteins [6, 9].
UPR activation
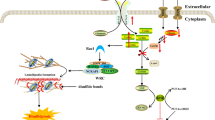
The UPRER comprises three branches: inositol-requiring enzyme 1α (IRE1α), protein kinase RNA-like ER kinase (PERK) and activating transcription factor 6 (ATF6) [9]. These ER signalling proteins have a similar structure, consisting of ER luminal and cytosolic domains. The ER luminal domains are formed by a single pass through the membrane [9], while cytosolic domains are the mediators of the UPRER [9, 11]. Under physiological conditions, the chaperone BiP/glucose-regulated protein 78 (GRP78), binds to the luminal domains of the mediators of the UPRER, repressing their activation [12, 13]. Upon accumulation of excessive unfolded or misfolded proteins in the ER lumen, BiP binds to misfolded proteins on the substrate-binding site and the ATPase domain dissociates from the transmembrane receptors, allowing allosteric activation of the UPRER regulators by oligomerisation [14, 15] (Fig. 1a).

The UPRER. A Adaptive UPRER. Following ER stress, BiP binds to misfolded proteins on the substrate-binding site and the ATPase domain dissociates from the transmembrane receptors, allowing allosteric activation of the UPRER regulators by oligomerisation and phosphorylation [14, 15]. (1) IRE1α RNase activity mediates unconventional splicing of XBP1 [16,17,18], XBP1s translocates to the nucleus to promote expression of genes related to quality control [9, 19]. IRE1α also mediates the cleavage and degradation of mRNAs and microRNAs; regulated IRE1α-dependent decay (RIDD), decreasing the protein load in the ER lumen [20]. (2) PERK phosphorylates eIF2α [21], promoting rapid attenuation of global mRNA translation [22, 23]. Phosphorylated eIF2α also regulates the translation of the transcription factor ATF4 [24]. ATF4 regulates the feedback loop responsible for the restoration of protein synthesis. ATF4 induction of CHOP, upregulates the expression of GADD34 which forms a complex with PP1 to dephosphorylate eIF2α [25, 26]. (3) ATF6α translocates to the Golgi apparatus, where it is cleaved to generate ATF6f, which acts as a transcription factor that promotes the expression of ER chaperones [27, 28]. ATF6α promotes the expression of Xbp1 mRNA, enhancing the substrate load for IRE1α splicing [29]. B Maladaptive UPRER. Following prolonged ER stress the homeostatic capacity of the UPRER becomes saturated that can activate pro-apoptotic signalling. (1) IRE1α interacts with TRAF2 to promote a kinase signalling cascade that activates JNK [30, 31]. JNK promotes the oligomerisation of BAX and BAK on the mitochondrial membrane and the assembly of the apoptosome [32, 33]. RIDD can promote apoptosis by degrading essential cell-survival mRNAs such as the negative regulators of TXNIP, promoting the assembly of the inflammasome leading to apoptosis [34, 35]. (2) PERK-eIF2α induces the translation of ATF4, activation of CHOP and GADD34 [25, 26]. CHOP promotes the expression of PUMA, NOXA, BIM and BID, which induce the mitochondrial BCL-2 pro-apoptotic proteins. CHOP can also activate the translation of ERO1α, promoting the oxidation of the ER environment [36, 37]. PERK-ATF4-CHOP arm regulates IP3R-mediated Ca2+ leakage from the ER [38, 39]. Sustained and excessive Ca2+ transport from the ER to the mitochondria impairs mitochondrial metabolism and lead to opening of the mPTP and pro-apoptotic signalling [40, 41]
UPRER signalling
IRE1α is the most conserved signalling branch of the UPRER, it is a type I transmembrane protein with Ser/Thr protein kinase and endoribonuclease activities [42]. Upon accumulation of misfolded proteins, BiP dissociates from IRE1α, inducing its oligomerisation and autophosphorylation [43, 44]. Phosphorylated IRE1α RNase activity mediates the unconventional splicing of an intronic region of XBP1 in the cytoplasm independently from the spliceosome, generating the active form, spliced XBP1 (XBP1s) [16,17,18] (Fig. 1a). XBP1s contains a basic leucine zipper domain (bZIP), it can translocate to the nucleus to induce expression of ER stress-response elements (ERSE), related to quality control (protein folding, translocation, and degradation) [9, 19]. IRE1α also mediates the cleavage and degradation of mRNAs and microRNAs; in a process known as regulated IRE1α-dependent decay (RIDD), decreasing the abundance of some mRNAs and reducing the protein load in the ER lumen [20]. IRE1α regulation of mRNAs and microRNAs depends on the presence of an IRE1α cleavage site formed by a stem-loop containing the sequence “CUGCAG” [45]. IRE1α has been demonstrated to degrade miR-17, -34a, -96, and -125b, these microRNAs target mRNA encoding the pro-apoptotic protein caspase-2, increasing the levels of this protein and initiating activation of apoptosis [46]. Furthermore, the cytosolic domain of IRE1α can interact with adapter proteins to establish crosstalk with other stress-mediator pathways [47]. The interaction of IRE1α with TRAF2 (tumour necrosis factor receptor (TNFR)-associated factor-2) promotes the activation of ASK1/JNK [30], ERK and p38 [48], protein kinases involved in autophagy, apoptosis and NF-κB inflammatory pathways [49].
PERK is a type I protein kinase that dissociates from BiP under ER stress, it is activated by dimerization and autophosphorylation [12]. Active PERK phosphorylates eIF2α at serine 51 [21], promoting a rapid attenuation of global mRNA translation, reducing the protein load for folding in the ER [22, 23]. Phosphorylated eIF2α also controls the selective translation of the transcription factor ATF4 [24] (Fig. 1a). ATF4 promotes the translation of ER stress genes related to the restoration of cellular homeostasis: protein synthesis, amino acid metabolism, redox homeostasis, apoptosis and autophagy [9]. ATF4 orchestrates the restoration of protein synthesis when the ER stress levels have been reestablished by regulating a feedback loop responsible for eIF2α dephosphorylation. The feedback loop is mediated by the induction of C/EBP homologous protein (CHOP) by ATF4, upregulation of GADD34 (growth arrest and DNA damage 34), which forms a complex with PP1 (a serine/threonine-protein phosphatase) to dephosphorylate eIF2α [25, 26].
ATF6α is a type II transmembrane protein that possesses a cytosolic N-terminus containing a bZIP motif [50]. ATF6α is located on the ER membrane with BiP bound to its Golgi localisation sequences. Under ER stress BiP is released from ATF6α, allowing translocation to the Golgi apparatus [51]. In the Golgi apparatus ATF6α is cleaved by Site-1 and 2 proteases (S1P and S2P), generating the N-terminal cytoplasmic fragment (ATF6f) containing the bZIP motif [27, 28] (Fig. 1a). ATF6f, following translocation to the nucleus, promotes the expression of ERSE and the ER chaperones (BiP and GRP94), affecting protein folding, maturation, translocation, and degradation [27, 28]. ATF6f and IRE1α constitute a regulatory hub of signalling pathways that are normally activated simultaneously for the regulation of XBP1s [29]. ATF6α promotes the expression of XBP1 mRNA, enhancing the substrate load for IRE1α splicing [29]. ATF6α also heterodimerises with XBP1s for the transcription of genes required for ER associated degradation (ERAD). Finally, XBP1s and ATF6f promote cellular secretory capacity by inducing the expansion of the ER and Golgi apparatus [52,53,54].
ERAD is activated alongside the UPRER [10, 55]. ERAD involves the recognition of misfolded proteins in the ER, their retrotranslocation to the cytoplasm, ubiquitination and subsequent degradation by the proteasome [55]. The induction of ERAD is regulated by the UPRER, although there is crosstalk between these two mechanisms as ERAD can coordinate the expression of IRE1α [56].
Adaptive UPRER signalling
The regulation and activation of the UPRER is dose-dependant, a low dose of an ER stressor can activate adaptive UPRER, while in response to higher doses or chronic ER stress, maladaptive UPRER is induced [57] (Fig. 1). Adaptive UPRER activation (Fig. 1a) can promote an increase in the translation of chaperones, Ca2+ binding proteins and activation of antiapoptotic and antioxidant signalling pathways [58,59,60]. Ageing is associated with an alteration of ER morphology and the expression levels of ER chaperones and transducers, resulting in an impairment of the adaptive UPRER [59]. Subsequently cells are more susceptible to alterations in proteostasis and the ability to adapt to disrupted homeostasis [61].
Adaptive UPRER has been linked to a signalling network that improves the ageing phenotype. The stage of life of the organism, whether during development or maturity, can determine the hormesis effect of activation of the UPRER which is related to the decline in the inducibility of these pathways with age [62]. In C. elegans it was demonstrated that the inducibility of the UPRER peaks in the early developmental stages and declines in adulthood [62]. Exposure of C. elegans during larval development to low doses of tunicamycin (0.125 µg/ml) for 24 h resulted in increased lifespan and animals that had a delayed age-associated reduction in inducible UPRER activation [63]. Activation of IRE1-XBP1 arm can improve organismal development, stress resistance, and longevity [63,64,65,66]. During dietary restriction in C. elegans, the IRE1-XBP1 arm activates ERAD and results in increased longevity [63]. Similarly in C. elegans, it was demonstrated that expression of XBP1s in neurons, led to extended lifespan by triggering an adaptive UPRER in distant non-neuronal cells [65]. Activation of the ATF4 signalling pathway has also been demonstrated to extend lifespan in C. elegans [67, 68] and Saccharomyces cerevisiae [69].
Maladaptive UPRER signalling
Following prolonged ER stress, the homeostatic capacity of the UPRER becomes saturated and results in pro-apoptotic signalling, regulated by IRE1α and PERK, with increased Ca2+ release from the ER (Fig. 1b). Under prolonged ER stress phosphorylated IRE1α interacts with TRAF2 to promote a kinase signalling cascade that ultimately activates JNK (Jun amino-terminal kinase) [30, 31]. JNK can promote apoptosis through activation of the mitochondrial BCL-2 pro-apoptotic proteins, BAX and BAK [32]. Oligomerisation of BAX and BAK promotes the assembly of the apoptosome [33]. Activation of the RIDD pathway by IRE1α can promote apoptosis by degrading essential cell-survival mRNAs such as chaperone BiP [70]. RIDD can degrade microRNAs that negatively target the expression of caspase 2, mediating BAX/BAK dependant apoptosis [46]. Finally, RIDD is involved in the degradation of negative regulators of thioredoxin-interacting protein (TXNIP), promoting the assembly of the inflammasome leading to apoptosis [34, 35] (Fig. 1b).
The PERK-eIF2α branch of the UPRER induces the translation of ATF4, activation of CHOP and GADD34 [25, 26]. CHOP regulates mitochondrial BCL-2 pro-apoptotic proteins, BAX and BAK through upstream regulators such as BH1-3 pro-apoptotic proteins; PUMA, NOXA [71], BIM [72] and BID [73, 74]. Activation of GADD34 by CHOP can restore protein translation in homeostatic conditions, however when proteostasis is not recovered, they can disrupt oxidative folding and result in altered ROS generation in the ER lumen [36, 75]. In addition, CHOP can activate the translation of ERO1α, involved in the formation of disulphide bonds in nascent proteins, but during ER stress promotes oxidation of the ER environment [36, 37]. The disruption of the ER redox state promotes leakage of H2O2 to the cytoplasm that can further induce apoptotic signalling [36] (Fig. 1b).
Redox regulation of the ER
The intracellular redox environment is closely linked to the initiation of ER stress and UPRER activation. For example the disulphide reducing agent, dithiothreitol, is commonly used as an inducer of ER stress, as it can interfere with the redox dependent protein folding mechanisms within the ER [76]. In response to both endogenous and external stressors, the ER increases its protein folding capacity and activates defence mechanisms, such as autophagy and the antioxidant response [77, 78]. In the ER there is a constitutive production of H2O2 as a biproduct of oxidative protein folding, that promotes the formation of covalent disulphide bonds on nascent polypeptide chains [76]. The ER has a more oxidising environment compared to the cytosol that facilitates thiol disulphide exchange for correct protein folding and the ratio of GSH/GSSG is much lower compared to other organelles [79]. Oxidative folding is catalysed by ER-resident protein disulphide isomerases (PDIs), endoplasmic reticulum protein 72 (Erp72) and endoplasmic reticulum 57 (Erp57) [80]. The Cys residues located in the active site of PDI’s are reduced upon oxidation of the polypeptide, promoting the formation of disulphide bonds and subsequently re-oxidised by ER oxidoreductase 1 (ERO1) [81, 82]. ERO1 can transfer electrons to molecular oxygen (O2) and as a result generate H2O2, constituting a basal source of ROS in the ER [83]. Correct oxidative folding of proteins is essential for maintaining ER homeostasis, as impairment can induce the accumulation of both unfolded proteins and ROS in the lumen of the ER [76]. In C. elegans it was demonstrated that during ageing there is a shift in the redox state of the ER to more reducing conditions compared to the cytosol, which becomes more oxidised with age [84]. As a result of reduced folding capacity within the ER, cells are more sensitive to maladaptive UPR signalling or ER stress response failure, as described in metabolic disease and ageing [85]. Organelle specific changes in the redox environment reflect the distinct functions of these organelles and how redox homeostasis within different compartments needs to be regulated. Table 1 contains ER and MERCS localised proteins identified with redox specific post transcriptional modifications.
The UPRER response can also be activated by alterations in the redox state of the ER. Activation of the UPRER in response to oxidative stress is mediated by two members of the PDIs, PDIA5 and PDIA6, which facilitate thiol–disulphide exchange on Cys residues of the luminal domains of IRE1, PERK and ATF6 [86, 87]. ER generated ROS can induce ATF6 signalling, PDIA5 cleaves disulphide bonds in ATF6, promoting oligomer dissociation and translocation from the ER to Golgi and expression of ATF6 target genes [87]. ROS can also activate IRE1α and PERK signalling, when PDIA6 binds to the luminal domain of both UPRER sensors and promotes thiol disulphide exchange, similar to ATF6 activation [86, 88, 89]. Following initial ER stress PERK Cys216 can be reversibly oxidised allowing formation of covalent interactions with ERO1α, resulting in a tightening of MERCS formation and increased Ca2+ flux into mitochondria and regulating mitochondrial bioenergetics [90]. IRE1α also provides a metabolic link between UPRER, redox signalling and mitochondrial function. Sulfenylation of a conserved Cys residue located in the IRE1α kinase loop can inhibit its kinase activity and promote p38 activation of the Nrf2/SKN-1 dependent antioxidant response, regulating cytoplasmic ROS and inhibiting the UPR [91]. IRE1α therefore lies at a metabolic hub that dictates cell fate via activation of the UPR, initiation of the antioxidant response (via Nrf2 activation) or activation of apoptotic cell death via enhanced Ca2+ entry into mitochondria.
Calcium signalling in the ER
The ER is the main Ca2+ store in metazoan cells, regulating Ca2+ homeostasis which is vital for cellular function. In the lumen of the ER, chaperones including calreticulin, calnexin, BiP, GRP94 and PDI, maintain Ca2+ levels within a physiological range [92]. Many of these chaperones are implicated in ER stress and ROS sensing, connecting these responses with Ca2+ homeostasis [76]. Ca2+ flux within the ER is mediated by sarco/endoplasmic reticulum Ca2+ transport ATPase (SERCA) family, which regulates the pumping of Ca2+ inside the ER in an ATP-dependent process [93]. Thapsigargin, an inhibitor of SERCA and commonly used to promote the induction of ER stress. The release of Ca2+ to the cytosol is controlled by the inositol 1,4,5-trisphosphate receptor (IP3R) and the ryanodine receptor (RyR) families [94, 95].
Under ER stress conditions a decrease in ER Ca2+ levels has been associated with the inhibition of SERCA activity [96,97,98] and passive leak of Ca2+ from the ER due to altered IP3R activity [99]. An increase in ROS within the ER has been demonstrated to promote the release of Ca2+ from the ER, linked with oxidation of specific Cys residues of Ca2+ regulators including SERCA [100], IP3R [101] and RyR [102]. Perturbations in Ca2+ homeostasis within the ER will inhibit the function of Ca2+-dependent ER chaperones potentially resulting in ER stress [103]. Ca2+ flux between the ER, cytoplasm and mitochondria can also determine apoptotic signalling during prolonged ER stress [104]. The PERK-ATF4-CHOP arm regulates Ca2+ flux by CHOP induction of ERO1α, that subsequently induces IP3R-mediated Ca2+ leakage from the ER [38, 39]. Sustained ER stress and excessive Ca2+ transport from the ER to the mitochondria can impair mitochondrial metabolism and lead to opening of the mitochondrial membrane permeability transition pore (mPTP) and pro-apoptotic signalling [40, 41] (Fig. 1b). Ca2+ release into the cytoplasm also activates Calpain proteases, which cleaves and activates caspase 12, triggering the induction of apoptosis [105, 106].
Mitochondria
Mitochondria are essential organelles with multi-faceted functions including energy generation via oxidative phosphorylation, iron metabolism, ion and phospholipid homeostasis. Mitochondria are also involved in the generation of ROS and subsequent redox signalling, Ca2+ homeostasis, apoptosis and autophagy. Disruption of mitochondrial function has been implicated in almost all age-related diseases including sarcopenia, neurodegeneration and cancer [107]. Mitochondria are in constant dynamic flux determined by the balance between biogenesis, mitochondrial fusion and fission along with selective degradation via mitophagy [107]. Mitochondrial morphology has been linked to substrate use, with fragmented mitochondria demonstrating increased fatty acid oxidation, linking mitochondrial dynamics and cellular fuel preference [108]. Indeed mitochondrial morphology can change rapidly in response to metabolic demand during exercise [109] or in proliferating cells such as in stem cells or cancerous cells, where mitochondrial fission predominates over fusion and is characterised by a fragmented mitochondrial network [110].
Mitochondria and ER are linked at MERCS facilitating the dynamic flow of information between the organelles, allowing changes in ER homeostasis to regulate mitochondrial function [111, 112]. Early adaptive ER stress promotes the formation of contact sites and facilitates Ca2+ transfer to mitochondria that increases mitochondrial metabolism [113], increasing energetics to alleviate ER stress [114].
Mitochondrial dynamics
Mitochondrial biogenesis is a complex process requiring the integration of mitochondrial DNA, lipids and proteins, responding to stimuli such as hypoxia and metabolic demand [115]. Mitochondrial division stimulates the recruitment of proteins and components to existing mitochondrial compartments and complexes, ensuring that biogenesis is closely coupled to mitochondrial fusion and fission [116]. The regulation of mitochondrial degradation via mitophagy is controlled by a number of pathways including: Ubiquitin dependent degradation via the Pink/Parkin pathway, receptor mediated mitophagy via BNIP3, BNIP3L/NIX and FUNDC1, that facilitate direct interaction with the autophagosome [117, 118]. AMPK mediated mitophagy has also been described in conditions of high metabolic demand, with AMPK interacting antagonistically with mTORC1 to promote mitophagy [119]. The precise mechanisms underlying basal levels of mitochondrial degradation or in response to acute and chronic stress are still to be defined, although it is increasingly recognised that MERCS play a key role in determining mitochondrial dynamics [112, 120, 121]. Key regulators of mitochondrial biogenesis and turnover such as PGC1α, DRP1, MFN2 and OPA1 have been demonstrated to be regulated by the redox environment [122, 123].
Mitochondrial stress sensing
Mitochondrial DNA (mtDNA) contains 37 genes, of which 13 encode structural polypeptides of components of electron transport chain (ETC) complexes [124]. Most proteins that constitute the mitochondrial proteome are synthesised in the cytoplasm, targeted and imported into mitochondria, where they bind to mitochondrial-localised chaperones to help their translocalisation and assembly [125]. Trafficking of proteins into the mitochondrial matrix via the TOM/TIM complex (translocase of the outer membrane/translocase of the inner membrane) [126] needs to be carefully controlled since disruption could impair mitochondrial proteostasis and overwhelm the chaperone capacity within mitochondria, inducing mitochondrial stress [127] (Fig. 2). Any perturbation of mitochondrial proteostasis that induces mitochondrial stress, activates pathways related to the integrated stress response (ISR) [128]. The ISR is activated to restore homeostasis in response to various types of stress conditions and ultimately results in the phosphorylation of eIF2α Ser51 [129]. Phosphorylated eIF2α activates ATF4, inducing the attenuation of protein translation and promoting the expression of mRNAs encoding CHOP and ATF4, which promotes expression of ATF5 [130, 131].

The UPRmt. Most proteins that constitute the mitochondrial proteome are synthesised in the cytoplasm, targeted and imported into mitochondria [125] via the TOM/TIM complex [126], perturbation of this trafficking can impair mitochondrial proteostasis and induce mitochondrial stress [127]. A The canonical axis of the UPRmt is controlled by the expression of ATF5, ATF4 and CHOP [132]. CHOP alleviates proteotoxic stress by inducing the expression of the mitochondrial chaperones HSP10 and HSP60 [134]. ATF5 is normally imported into mitochondria via TOM and TIM, where it is degraded by proteases [138]. Mitochondrial proteotoxic stress will perturb mitochondrial import efficiency, resulting in the activation of ATF5 by p-eIF2α and its translocation to the nucleus [139]. ATF5 promotes the transcription of genes related to chaperones, proteases and antioxidant proteins [137]. B The sirtuin axis of the UPRmt activates SIRT3, which deacetylates FOXO3A, promoting its translocation to the nucleus and transcription of SOD2 and catalase [142, 143]. C AKT mediates the ROS-dependant phosphorylation of ERα, which activates NRF1 and the IMS protease HTRA2 [144]. NRF1 stimulates mitochondrial respiration, proteasome activity and the IMS protease OMI. D Mitochondrial proteotoxic stress promotes epigenetic changes in the cellular DNA regulated by HSF1, it translocates to the nucleus where it interacts with SSBP1 to bind to the chromatin and boost the expression of mitochondrial chaperones [145, 146]
Mitochondrial UPR
The canonical axis of the UPRmt is controlled by the expression of ATF4, ATF5 and CHOP, three bZIP transcription factors central to the ISR [132]. ATF4 promotes the expression of genes related to the UPRmt, however it mainly acts as a regulator of both ATF5 and CHOP expression [133]. CHOP alleviates proteotoxic stress by inducing the expression of the mitochondrial chaperones HSP10 and HSP60 [134]. CHOP has been also proposed as a regulator of the protease complex ClpXP, which plays a key role in sensing and maintaining proteostasis (through the ClpP proteolytic subunit) inside the mitochondrial matrix [135]. ClpXP has been reported to activate UPRmt under conditions of mitochondrial proteotoxic stress [136]. ATF5 possesses a mitochondrial-targeting sequence (MTS) and a nuclear localisation sequence (NLS) [137]. Under homeostatic conditions, ATF5 is imported into healthy mitochondria via TOM and TIM, where it is degraded by proteases, thus acting as a sensor of mitochondrial import efficiency [138]. However, under overload of misfolded proteins, protein aggregation and perturbed mitochondrial import efficiency, ATF5 is activated by p-eIF2α and translocated to the nucleus, where it increases folding capacity via retrograde signalling [139] (Fig. 2a). ATF5 promotes the transcription of genes that aid in the recovery of normal proteostasis, for example by upregulating chaperonins, chaperones, proteases and antioxidant proteins [137]. Impaired mitochondrial protein import efficiency results in the accumulation of mistargeted mitochondrial proteins in the cytosol, that will activate the UPRam (UPR activated by mistargeted proteins), which enhances the assembly of the proteasome in order to degrade potentially toxic mislocalised proteins [140, 141].
The sirtuin axis of the UPRmt boosts the antioxidant capacity of the cell in response to disrupted proteostasis, driven by the increase in mitochondrial ROS derived from mitochondrial dysfunction and activation of the canonical UPRmt [131]. During mitochondrial proteotoxic stress, activation of SIRT3 results in deacetylation of FOXO3A, promoting its translocation to the nucleus and transcription of SOD2 and catalase [142, 143] (Fig. 2b). Under proteotoxic stress in the IMS, AKT mediates the ROS-dependant phosphorylation of ERα, which increases the expression of nuclear respiratory factor 1 (NRF1) and the IMS protease HTRA2 transcripts [144]. NRF1 mediates the activation of protein quality control by stimulating mitochondrial respiration [147], proteasome activity and the expression of the IMS protease OMI [144] (Fig. 2c). Mitochondrial proteotoxic stress also promotes epigenetic changes, through the induction of chromatin remodelling factors that facilitate the induction of mitochondrial chaperones [145]. These changes are regulated by HSF1, which also plays a key role in the heat-shock response and forms a complex with mitochondrial single-stranded DNA binding protein 1 (SSBP1) [145, 146]. HSF1 translocates to the nucleus where it binds to the chromatin remodelling factor BRG1 and completes the formation of the chromatin remodelling complex, which will ultimately increase the expression of chaperones to protect mitochondrial function [146] (Fig. 2d).
Acute mitochondrial stress activates the translation axis of the UPRmt, leading to a decrease in pre-RNA processed product and decreased mitochondrial translation, reducing the folding load in mitochondria [148]. This axis of the UPRmt works as a first defence mechanism against proteotoxic stress, it is activated in stressed mitochondria before the activation of the canonical UPRmt [148] (Fig. 2d). mtDNA is transcribed into long pre-RNAs, processed by the RNase P complex (formed by MRPP1, 2 and 3). Activation of the translation axis of the UPRmt, reduces MRPP3 levels, as a result some of the mitochondrial long pre-RNAs are not translated with a subsequent reduction in mitochondrial protein biosynthesis [149, 150].
Redox regulation of mitochondria
Mitochondrial respiration generates ATP but can also result in ROS generation, both superoxide and H2O2 as a result of electron leak from redox donors in the ETC, reducing molecular oxygen to superoxide and its subsequent conversion to H2O2 [151]. Mitochondrial ROS generation has been described at various sites along the ETC in particular at complex I and III by both forward and reverse electron transport as well as in conditions of hypoxia, indicating mitochondria are key regulatory hubs for redox signalling in cellular homeostasis and pathologies [151, 152]. ROS generation during reverse electron transport has been identified as a major cause of oxidative damage in conditions such as ischaemia where there is an accumulation of succinate [153]. Succinate is a substrate for the TCA enzyme succinate dehydrogenase at complex II, a FAD-dependent enzyme from the IMM that participates in the reduction of ubiquinone [152]. During conditions of mitochondrial hyperpolarisation, reverse electron transport results in electrons flowing back to complex I, generating NADH and superoxide [154]. Under controlled conditions where complex I and III are blocked and levels of succinate reduced, complex II has the capacity to generate significant levels of ROS both in forward (accepts electrons from succinate) and reverse (accepts electrons from ubiquinol) modes [155]. Cys39 of ND3 subunit of complex I has been identified as a critical redox switch in determining its catalytic active state, this Cys residue becomes accessible to alkylating agents in the inactive D-state [156]. Temporal reversible oxidation of Cys39 of ND3 has become a therapeutic target in ischaemia as when reversibly oxidised, complex I remains in an inactive state preventing reverse electron transport and subsequent superoxide generation [153]. In acute hypoxia, complex I acidifies the mitochondrial matrix which can solubilise Ca2+ and activate the Ca2+/Na+ antiporter, causing a decrease in IMM fluidity, this can result in a reduction in the diffusion rate of ubiquinol from complex II to complex III, promoting ROS generation [157]. ROS generation within mitochondria particularly the IMS has the capacity to result in redox modifications of sensitive proteins affecting their function and overall mitochondrial capacity. Redox modifications of proteins imported into the IMS can also affect mitochondrial activity as a result of disrupted assembly of complexes within the ETC due to the altered redox environment [158]. Table 1 contains mitochondrial localised proteins identified with redox specific post transcriptional modifications.
The mitochondrial redox environment also regulates mitochondrial dynamics, sites of mitochondrial fission have distinct ROS signatures, fission at the periphery or tip results in mitochondrial fragments destined for degradation while midzone fission is preferential for dynamics [159]. Disrupted mitophagy can result in an accumulation of dysfunctional mitochondria and has been associated with a range of age-related diseases particularly in tissues with high metabolic demand such as neurons and skeletal muscle [160, 161]. Chronic mitochondrial dysfunction leads to the accumulation of mitochondrial generated ROS, which can promote the unfolding/misfolding and aggregation of proteins inside the organelle and propagate mitochondrial dysfunction [162]. An increase in mitochondrial dysfunction can induce activation of the UPRmt, in particular ATF5 activation, in order to resolve proteotoxic and oxidative stress [137, 163]. In C. elegans it was demonstrated that the orthologue of ATF5, ATFS-1 has a dual action to protect cells from mitochondrial dysfunction, as it can upregulate genes involved in mitochondrial proteostasis (such as chaperones to restore protein homeostasis or glycolysis to boost ATP production) and bind promoters of NADH ubiquinone oxidoreductase assembly factors to maintain the function of the ETC complexes in order to optimise respiratory capacity during mitochondrial stress [164].
Low levels of mitochondrial stress can result in a mitohormesis response, the initial activation of stress signalling pathways that ultimately result in adaptive responses to improve stress resistance. A link between ROS production and mitohormesis has been repeatedly demonstrated in C. elegans, for example glucose deprivation resulted in enhanced respiration, increased ROS generation and extended the lifespan of the nematodes [165]. Inhibition of mitochondrial complex I with low doses of rotenone has also been demonstrated to promote lifespan extension in C. elegans [166]. The amount and duration of ROS generated by the ETC can influence lifespan and behaviour in model organisms [167, 168]. Similarly a recent study using Drosophila and mice pre-treated with N-acetyl-L-tyrosine, induced the production of ROS and promoted stress resistance related to mitohormesis [169].
Mitochondrial metabolism is modulated by Ca2+-dependent mechanisms linked to the ER stress response, through the stimulation of CHOP expression and phosphorylation of eIF2 and JNK [170]. The exchange of information via metabolites, ions and lipids between the ER and mitochondria can alter ATP production and promote reorganisation of the mitochondrial network [113]. Induction of an adaptive UPRER has been demonstrated to increase mitochondrial biogenesis, through the PERK-Nrf2 pathway [171]. ER stress can promote changes in the morphology of mitochondrial by promoting UPRER induced mitochondrial hyperfusion, in a process dependent on the phosphorylation of eIF2α by PERK [172]. A study in Drosophila demonstrated mitochondrial ETC disruption specifically activated PERK, while the other branches of the UPRER were not responsive [173]. This was attributed to PERK localisation at mitochondria-associated ER membranes (MAMs), making it more sensitive to respond to local stress signals [173].
In C. elegans, ATFS-1 regulates mitochondrial biogenesis and network expansion during normal development [174]. High levels of mitochondrial protein synthesis are needed during development, this results in a reduction in the levels of ATFS-1 imported into mitochondria. Subsequently ATFS-1 is translocated to the nucleus and results in the activation of the UPRmt, promoting the expansion of the mitochondrial network [174]. Mild mitochondrial stress can initiate a hormesis response that increases lifespan in C. elegans, this effect can activate the UPRmt leading to descendants with higher levels of mtDNA that exhibit longer lifespans; increased resistance to infection, heat shock, and oxidative stress; although with slower development and lower fertility compared to those with normal mtDNA and UPRmt levels [175]. ATFS-1 regulates the accumulation of transcripts of OXPHOS from both the nuclear and mitochondrial genomes in order that biogenesis of the ETC complex aligns with the ability of the stressed organelles to fold proteins and assemble ETC complexes [164].
Mitochondrial ER contact sites
Organelle contacts are essential for the maintenance of cellular homeostasis and establish a link that allows inter-organelle signalling and transfer of metabolites [114, 176]. Contact sites refer to areas where two membranes are near each other, but do not merge as the individual organelles maintain their distinct identities. MERCS are dynamic structures that remodel in response to intra and extra cellular signals, affecting the function of both mitochondria and ER [5, 176]. MERCS are relatively stable structures that require the formation of molecular bridges established by interacting proteins anchored in the smooth ER and the mitochondrial outer membrane [5]. MERCS contain a defined subset of proteins involved in tethering membranes, Ca2+ homeostasis, lipid transfer, redox balance and mitochondrial homeostasis [5, 40] (Fig. 3). The contacts between ER and mitochondria can be classified as narrow (8–10 nm) and wide (40–50 nm), resulting in different responses against stress and metabolic changes [121].

Mitochondria-ER contact sites molecular components and cellular functions. MERCS are relatively stable structures that require the formation of molecular bridges established by interacting proteins anchored in the smooth ER and the OMM [5]. Tethering complexes are essential, structural and reversible bonds that stabilise MERCS [177]. A MERCS tethering complexes occur between ER MFN2 and mitochondrial MFN2 or ER MFN2 and MFN1 [178]. The MFN tethering complex is dependent on the interaction of MFN2 and PERK at the ER membrane, essential for the establishment of the contact sites [90, 179]. Other complexes reported as regulating the tethering of MERCS include the ER VAPB and the OMM PTPIP51 [180]. B MERCS regulate Ca2+ flux between the ER and the mitochondria by the complex that forms between IP3R from the ER and VDAC from the OMM [5, 177]. Ca2+ passes through the MCU to reach the mitochondrial matrix [185, 186]. DJ-1 [187] and GRP75 [188] regulate the connection between IP3R and VDAC [189]. Some components of the TCA cycle require the binding of Ca2+ for their function, the interaction of mitochondria and ER via MERCS supply Ca2+ to mitochondria for stimulating the TCA cycle, resulting in an increase in ATP production [190]. C MERCS control the processes of mitochondrial fusion, fission and mitophagy [111, 191]. The ER promotes the polymerisation of actin filaments and establishment of close contacts between the two organelles [192]. ER tubules will release Ca2+ ions into the mitochondria, triggering the inner mitochondrial membrane to divide [192, 193]. DRP1 assembles around mitochondria at the fission site, a DRP1 ring constricts with the aid of actin–myosin filaments, resulting in the formation of two daughter mitochondria. ER tubules guide the position and timing of mitochondria fusion through the tethering with mitochondria [191, 194]. During mitochondrial fusion the contact sites between the tubules and the mitochondria need to be maintained to avoid the disruption of these MERCS, the Ca2+ sensitive motorprotein Miro cease all transportation movements of the mitochondria involved [195]. In the mitochondria PINK1 phosphorylates MFN2, recruits Parkin at the MERCS, allowing Parkin dependent ubiquitination of ER MFN2, promoting the separation of the two organelles and the initiation of mitophagy [196]
Tethering of MERCS
The tethering complexes are essential, structural and reversible bonds that stabilise MERCS [177]. The most recognised MERCS tethering complexes occur between ER mitofusin-2 (MFN2) and mitochondrial MFN2 or ER MFN2 and mitochondrial mitofusin-1 (MFN1) [178]. The MFN tethering complex is dependent on the interaction of MFN2 and PERK on the ER membrane, suggesting a potential role of PERK (and ultimately the UPRER) as a key mediator of MERCS assembly [90]. The interaction of PERK with MFN2 is essential for the establishment of contact sites, inhibition of these components lead to a reduction in the number of MERCS [90, 179] (Fig. 3a). Ablation of MFN2 leads to an abnormal upregulation of the PERK-ATF4-CHOP pathway, resulting in an increase in ROS, abnormal mitochondrial Ca2+ transients and altered mitochondrial morphology [179]. Knockdown of PERK in this condition can restore these alterations, demonstrating that PERK is a key regulator of the mitochondrial antioxidant response [179]. Other members of the complexes reported as regulating the tethering of MERCS include the ER vesicle‐associated membrane protein B (VAPB) and the OMM tyrosine phosphatase‐interacting protein‐51 (PTPIP51) [180]. Disruption of these components lead to a delay in Ca2+ flux into mitochondria and mitochondrial aggregation [181, 182]. The ER membrane chaperone B-cell receptor-associated protein-31 (BAP-31) can also form a physical and regulatory tether with different mitochondrial proteins [177], such as the mitochondrial fission protein-1 (FIS1), which contributes to the physical tethering and can promote the transmission of apoptotic signals from the ER to mitochondria [183]. Similarly, the interaction of BAP-31 with TOMM40 establish a physical tether that allows BAP-31 to control the transmission of apoptotic signals and regulate mitochondrial homeostasis [184].
Calcium flux between the ER and the mitochondria
An important function of MERCS is regulation of Ca2+ flux between the ER and the mitochondria by the complex that forms between IP3R from the ER and VDAC from the OMM [5, 177]. Ca2+ passes through the MCU to reach the mitochondrial matrix [185, 186]. DJ-1 [187] and GRP75 [188] regulate the connection between IP3R and VDAC stabilising MERCS integrity allowing entry of Ca2+ into mitochondria [189] (Fig. 3b). It has been recently demonstrated that IRE1a is also involved in regulating ER-mitochondria Ca2+ transfer by interacting with IP3R, stimulating mitochondrial respiration and ATP production to maintain energy homeostasis [197]. Ca2+ entry into the mitochondrial matrix provides Ca2+ to mitochondrial membrane proteins, however in cases of chronic stress it promotes swelling of the mitochondria and the opening of the mPTP that can initiate apoptosis [5, 41]. Some components of the TCA cycle (isocitrate dehydrogenase, oxoglutarate dehydrogenase and pyruvate dehydrogenase) require the binding of Ca2+ for their function. The ER poses a much higher concentration of Ca2+ (100–500 μM) compared to the cytosol (~ 100 nM), the interaction of mitochondria and ER via MERCS can supply enough Ca2+ to mitochondria for stimulating the TCA cycle, resulting in an increase in ATP production [190] (Fig. 3). Excess Ca2+ transfer into mitochondria via IP3R can induce the opening of the mPTP, release of Cytochrome c and activation of the caspase signalling cascade and pro-apoptotic pathways [198] (Fig. 4).

MERCS regulation of cellular signalling in ageing and disease. Disruption of MERCS assembly and disassembly plays a key role in pathophysiological conditions particularly in ageing and age-related diseases. Disrupted Ca2+ flow from the ER to mitochondria can result in mitochondrial dysfunction with loss of mitochondrial membrane potential and mitochondrial ROS generation, that result in activation of apoptotic pathways or senescence [40]. Excess Ca2+ transfer into mitochondria via IP3R can induce the opening of the mPTP, release of cytochrome c and activation of the caspase signalling cascade and pro-apoptotic pathways [198]. On mitochondria PINK1 phosphorylates MFN2, recruits Parkin at the MERCS, allowing Parkin dependent ubiquitination of ER MFN2, promoting the separation of the two organelles and the initiation of mitophagy [196]. Release of mtDNA through channels such as VDAC (located in or close to MERCS) has emerged as a potential regulator for the inflammatory response [201]
Regulation of mitochondrial homeostasis
Mitochondrial fusion, fission and mitophagy and the organisation of the mitochondrial network regulate mitochondrial function and fuel utilisation [199]. The ER can coordinate these processes by establishing contact sites between ER tubules and mitochondria [111, 191]. The ER inverted formin-2 (INF2) interacts with the OMM actin nucleator Spire1c to polymerise actin filaments and establish close contacts between the two organelles [192]. Actin polymerisation around mitochondria stimulates ER tubules to release Ca2+ ions into mitochondria through the VDAC1 channel, triggering the inner mitochondrial membrane to divide [192, 193]. The inner membrane scission is followed by constriction of the outer membrane, which occurs when the cytosolic GTPase DRP1 assemble around mitochondria at the fission site, guided by the OMM receptors FIS1 and MFF [193, 200]. This DRP1 ring constricts with the aid of actin–myosin filaments, resulting in the formation of two daughter mitochondria [191, 193] (Fig. 3). During mitochondrial fission, the original mitochondrion needs to transfer a copy of mtDNA to daughter mitochondria, MERCS mediate the replication and distribution of the mtDNA along the mitochondrial network, in a process that depends on DRP1 [201, 202]. Disruption of mitochondrial dynamics and subsequently mtDNA replication, may result in the release of mtDNA into the cytoplasm and in the generation of an inflammatory response [203, 204]. Considering that the release of mtDNA is thought to occur through channels such VDAC (located in or close to MERCS), and as MFN2 mediates the tethering of ER with mitochondria, contact sites between these two organelles emerge as a potential regulator of the inflammatory response [201] (Fig. 4). If mitophagy is activated, the pre-autophagosome markers ATG14L and ATG5 [205] and the mitophagy regulator PINK1 and Parkin localise to MERCS [206]. In the mitochondria PINK1 phosphorylates MFN2, recruits Parkin at MERCS, allowing Parkin dependent ubiquitination of ER MFN2, promoting the separation of the two organelles and the initiation of mitophagy [196] (Figs. 3 and 5).

PERK regulation of mitochondrial capacity. PERK is a key regulator of both the UPRER and the UPRmt, that localises at MERCS [90]. The adaptive ER stress response promotes mitochondrial elongation and network establishment [172]. The modulation of mitochondrial metabolism by PERK results in improved cristae formation, assembly of the ETC and oxidative phosphorylation efficiency [220]. 1 PERK regulates the expression of the mitochondrial contact site and cristae-organizing system (MICOS) [221]. 2 The activation of ATF4 by PERK promotes the expression of SCAF1 helps mediate assembly of the ETC [218, 222]. 3 The adaptive UPRER also promotes one-carbon metabolism [223]. 4 PERK can promote cell survival by increasing antioxidant capacity through the activation of Nrf2 [224]. 5 During the adaptive UPRER response, there is an upregulation of TFEB [225], which can induce the ISR via activation of ATF4 and CHOP, activate mitophagy machinery and boost mitochondrial biogenesis by promoting expression of PGC1α, TFAM and NRF1 [219]. 6 The formation of PERK-ERO1⍺ complex can restore mitochondrial homeostasis and promote the formation of MERCS [188, 226]. 7 PERK is essential for the activation of UPRmt transcription factor ATF5 [139] and can reduce mitochondrial protein import by promoting the degradation of mitochondrial translocase TIM17A by phosphorylation of eIF2α [227]
It has been proposed that ER tubules guide the position and timing of mitochondria fusion through the tethering with mitochondria [191, 194]. Fusion of the OMM is mediated by MFN1 and MFN2 homodimers [207, 208], while the IMM fusion is regulated by OPA1 [209]. During mitochondrial fusion the contact sites between the tubules and the mitochondria need to be maintained to avoid the disruption of MERCS and decrease mitochondrial motility [210]. In yeast during mitochondrial fusion, the Ca2+ sensitive motorprotein Miro, is involved in both actin filament and microtubule transport, that ceases all transportation movements of the mitochondria involved [195] (Fig. 3).
Redox Regulation of MERCS
The connection established by MERCS between the ER and mitochondria implies that disruption of redox homeostasis in one organelle will affect the other, generating a regulatory hub. It has been reported that ROS production in mitochondria leads to an exacerbation of ER stress, suggesting the existence of a feed-back loop that generates ROS in both organelles [211]. Within MERCS there is a constant production of ROS, generated from the oxidative protein folding activity of the ER chaperone ERO1α and the ER NADPH oxidase activity of NOX4 [212]. The presence of ROS within MERCS generates redox nanodomains between the two organelles, in a Ca2+-dependent process, allowing for effective redox crosstalk [213]. Targeting a H2O2-specific fluorescent probe to MAMs, it was reported that these redox nanodomains promoted IP3R-mediated Ca2+ release via MERCS,resulting in the swelling of the mitochondrial matrix, reduction of the cristae and release of H2O2 [213].
PERK is a key regulator of both the UPRER and the UPRmt and localises at MERCS [90, 214]. Mouse embryonic fibroblasts with PERK knocked out, have a disrupted MERCS network, altered ER morphology, disrupted redox signalling and impaired Ca2+ transport [215, 216]. PERK is a regulatory signalling hub that monitors stress in both organelles and its Cys216 can be reversibly oxidised allowing formation of covalent interactions with ERO1α and tightening of MERCS [90, 217]. UPRER and UPRmt establish a crosstalk in response to proteotoxic stress through PERK activation, regulating the coactivation of CHOP and ATF4 and increasing the expression of ATF5, promoting the translation of ER and mitochondrial chaperones to alleviate proteotoxic stress [217]. As mentioned, the UPR can be an adaptive or maladaptive response depending on stress intensity and duration, that can impact mitochondrial morphology and function [172, 218, 219].
The UPRER effects on mitochondrial morphology go through different stages: early ER stress (30 min) induces mitochondrial fragmentation, MERCS formation and Ca2+ influx into mitochondria; adaptive ER stress (6 h) promotes mitochondrial elongation and network establishment, improving oxidative phosphorylation efficiency [228], known as stress-induced mitochondrial hyperfusion [172] (Fig. 5). Maladaptive ER stress (24 h or more) triggers apoptosis through mitochondrial fragmentation and opening of the mPTP [172, 220, 229]. Inhibition of PERK or p-eIF2α during the adaptive UPRER stage induced the blockage of mitochondrial hyperfusion and fragmentation of the mitochondrial network [172], indicating that communication between the ER and mitochondria is mediated by the PERK-eIF2α axis.
Adaptive UPRER protects the cells against oxidative damage though the activation of PERK, which can boost the production of ATP [218] and activation of the antioxidant response [223]. The modulation of mitochondrial metabolism by PERK results in improved cristae formation, assembly of the ETC and oxidative phosphorylation efficiency [220]. During adaptive UPR, PERK phosphorylates N-acetyl-glucosamine transferase OGT, which can activate TOM70 stimulating the import and assembly of the mitochondrial contact site and cristae-organizing system (MICOS) [221] (Fig. 5). The activation of ATF4 by PERK promotes the expression of SCAF1, a protein that mediates the assembly of the ETC [218, 222] (Fig. 5). It has been reported that cells with a missense mutation in complex I NADH ubiquinone oxidoreductase, were able to recover the assembly of the super complexes by pharmacologically activating PERK [218]. As a counter measure to stress, the adaptive UPRER promotes one-carbon metabolism, in a process mediated by PERK [223]. One-carbon metabolism links the methionine and folate pathways through the interconversion of Serine and Glycine providing one carbon units for biosynthesis and reducing power in the form of NADH and NADPH [230] (Fig. 5).
PERK can promote cell survival by increasing antioxidant capacity through the activation of nuclear factor erythroid 2-related factor 2 (Nrf2) [224] (Fig. 5). PERK phosphorylation of Nrf2, releases it from Keap1 and subsequent translocation to the nucleus, initiating the transcription of numerous antioxidant genes, including thioredoxins, glutathione synthetase, glutathione S-transferase, and ferritin [231]. PERK silencing resulted in disrupted Nrf2 activation, an increase in ROS and an impairment of mitochondrial bioenergetics [232]. Key interactions of PERK that help determine mitochondrial capacity are established with TFEB, ERO1⍺ and the UPRmt [90, 139, 219]. During the adaptive UPRER response, there is an upregulation and nuclear translocation of TFEB [225]. TFEB can activate the ISR via ATF4 and CHOP, promotes the activation of mitophagy machinery and boost mitochondrial biogenesis by the expression of PGC1α, TFAM and NRF1 [219] (Fig. 5). The formation of a PERK-ERO1⍺ complex can restore mitochondrial homeostasis and promote the formation of MERCS by increasing tethering via GRP75 and MFN2 [188, 226] and stimulating Ca2+ transfer to increase mitochondrial capacity [90] (Fig. 5). PERK is essential for ATF5 activation and UPRmt [139], and can reduce mitochondrial protein import by promoting the degradation of mitochondrial translocase TIM17A by phosphorylation of eIF2α [227] (Fig. 5).
MERCS in ageing and disease
The dynamic nature of MERCS in terms of assembly and disassembly are determined by intracellular cues, allowing adaptation to the intracellular environment for both cell survival associated with increased metabolism but also potentially triggering the collapse of mitochondrial membrane potential resulting in apoptosis or senescence. MERCS can regulate Ca2+ homeostasis, redox signalling and lipid transfer, providing signalling hubs that can modulate mitochondrial dynamics, apoptosis, protein homeostasis and inflammation [40]. As a result, disruption of MERCS assembly and disassembly is thought to play a key role in pathophysiological conditions particularly in cancers and age-related diseases. In proliferating cells with high anabolic demand, mitochondrial fission predominates over mitochondrial fusion, MERCS can help determine mitochondrial morphology and allow efficient transfer of Ca2+ and other metabolites to mitochondria during proliferation. The accumulation of cells that have entered cell cycle arrest or senescence in ageing tissues is well documented [233]. MERCS assembly and disassembly provide a regulatory role in determining cell fate. Disrupted Ca2+ flow from the ER to the mitochondria can result in mitochondrial dysfunction with loss of mitochondrial membrane potential and increased mitochondrial ROS generation, resulting in activation of apoptotic pathways or senescence [40]. Senescent cells accumulate during ageing, an increase in the cell capacity to remove senescent cells results in delayed aging and improves both lifespan and health-span [234]. It has been reported that the exposure to pro-senescent stressors or other stimuli can alter the number of MERCS [40, 235]. An aberrant increase in MERCS, during ageing, can result in the accumulation of Ca2+ in the mitochondria, activation of the p53/p21 and p16/Rb pathways, leading to cell cycle arrest and Senescence-Associated Secretory Phenotype (SASP) partially driven by NF-κB [40, 235]. Senescence of endothelial cells is considered to be a risk factor related to the development of cardiovascular disease and can contribute to disrupted vascular tone and angiogenesis [236]. It has been demonstrated in an in vitro model of endothelial cell ageing that increased MERCS formation result in an increase in Ca2+ transfer, altering mitochondrial bioenergetics and cell senescence [237]. Most studies would indicate senescence is associated with increased MERCS formation and elevated mitochondrial Ca2+, however decreased MERCS formation could also be a pro-senescent signal [40]. However, it is clear that not only the abundance of MERCS is important but also the width of the interface between the ER and OMM, where loose junctions (~ 25–40 nm) promote Ca2+ transfer and tight junctions (~ 10 nm) inhibit Ca2+ transfer between the organelles [121].
Changes in MERCS formation is context dependent and distinct between cell types, with a number of pathologies reporting increased MERCS formation and others decreased MERCS formation. In cancerous cells, increased Ca2+ uptake in the mitochondria can promote metabolism and tumorigenesis, however excessive Ca2+ uptake can induce cell death [238]. In neurodegenerative diseases such as Alzheimer disease and Parkinson disease, increased MERCS have been reported [239]. Mitochondrial dysfunction in neurodegenerative diseases, are associated with the loss of neuron structure and function and altered protein composition of MAMs, required for the scaffolding of MERCS and ultimately disrupted mitochondrial turnover [240,241,242].
Skeletal muscle and adaptive UPR signalling
In almost all eukaryotic cells the ER is an essential organelle for protein synthesis and folding, lipid and sterol synthesis, as well as a depot for the storage of Ca2+. The contraction and relaxation of skeletal muscle depends on the on the release and uptake of Ca2+ from the sarcoplasmic reticulum (SR). The SR has been described as a fully differentiated domain of the muscle ER and it is recognised that the ER and SR are a continuous membrane system of different specialised regions [243, 244]. The SR contains a number of recognised ER proteins, although at a relatively lower concentration and it was proposed that during myogenic differentiation there is ER expansion that is engulfed by myofibrils [243, 245].
UPRER activation during myoblast differentiation
UPRER activation is crucial for muscle stem cell homeostasis, myogenic differentiation, exercise adaptation and skeletal muscle regeneration after injury [246]. Myogenesis is a complex and tightly regulated process that involves the selection of multipotent mesodermal cells to produce myoblasts, their exit from the cell cycle and differentiation into myotubes [247]. During muscle differentiation a population of myoblasts, that are differentiation-incompetent or less resistant to stress, will undergo selective apoptosis [248]. This process is thought to be mediated by the UPRER and it is crucial for skeletal muscle development [247]. The UPRER plays an essential role in this process by controlling the induction of caspase-12, promoting a caspase signalling cascade that results in selective apoptosis [249]. Markers of the UPR, such as ATF6, CHOP, and BiP, are upregulated during myogenesis and it has also been demonstrated that pharmacological induction of ER stress increased apoptosis in myoblasts, leading to improved myogenesis [249, 250]. Pharmacological induction of ER stress (using the N-glycosylation inhibitor tunicamycin and the SERCA inhibitor thapsigargin) in myoblasts lead to an increase in cell apoptosis, however the remaining myoblasts differentiated more efficiently into myotubes [250].
Redox and adaptive UPRER in skeletal muscle
Exercise is one of the most effective and beneficial interventions for overall health. Exercise can improve insulin sensitivity, cardiovascular health and help maintain muscle mass and function [251]. Regular exercise has been shown to reduce oxidative stress, inflammation, and reverse mitochondrial and ER dysfunction [252]. The changes in Ca2+ flux during muscle contractions has been associated with the formation of contact sies between the sarcoplasmic reticulum and mitochondria [244]. During contractile activity there is localised endogenous ROS generation that is required for the activation of specific signalling pathways required for the adaptive response to exercise [253, 254]. Temporal endogenous ROS generation is also necessary for the repair and activation of quiescent satellite cells following muscle injury [255]. Fluctuations in Ca2+ homeostasis, together with an altered redox environment are linked to the activation of all 3 branches of the UPRER following exercise with downstream signalling effects on mitochondrial dynamics [113, 256].
Although chronic ER stress can activate cell death pathways, recent research suggests that low levels of ER stress may potentially benefit cells by inducing an adaptive UPR that can reduce the harmful consequences of accumulating misfolded proteins [231]. Physical exercise generates a physiological stress and activation of UPRER pathways, several studies have demonstrated that acute exercise is characterised by an increase in BiP translation and eIF2 phosphorylation [257,258,259]. As a result, regular exercise can inhibit the activation of pro-apoptotic pathways, maintaining or decreasing the levels of BiP, PERK, IRE1a and CHOP including downstream UPRER components such as ATF4 and XBP1 [260, 261]. Mitochondria are also affected by contractile activity in skeletal muscle, it has been demonstrated that exercise plays a key role in mitochondrial adaptation to stress, promoting mitochondrial biogenesis and mitophagy [254]. PGC-1α is activated in skeletal muscle in response to exercise, promoting mitochondrial biogenesis and the adaptative response to exercise [256]. It has also been reported that PGC-1α regulates the expression of ATF5 [262], providing a link between activation of the UPR and mitochondrial biogenesis.
In skeletal muscle there are distinct populations of mitochondria, subsarcolemmal and intermyofibrillar, providing the ATP required for sustaining contractions and membrane potential. Mitochondria are in close contact with the SR and it has been proposed that MERCS are essential for maintaining muscle homeostasis [263]. MERCS impairment in skeletal muscle is associated with ageing and muscle wasting, caused by the downregulation of SR-mitochondria Ca2+ transport proteins IP3R, VDAC, and GRP75 [264]. Disruption of Ca2+ transits between the SR and the mitochondria may contribute to the decline in muscle performance during ageing [264,265,266]. In single adult skeletal muscle fibres, pharmacologically opening of the mPTP resulted in increased mtROS and caspase activation, leading to muscle fibre atrophy [267]. In striated muscle, the partitioning of ER/SR and mitochondria is highly organised and as a result MERCS formation are considered more ordered compared to proliferating cells [264]. Disrupted Ca2+ homeostasis is thought to play a role in the age-related loss of skeletal muscle function and muscular pathologies. Decreased MERCS formation has been reported with age [268] and depletion of MERCS are associated with muscular dystrophy [263]. In pathophysiological conditions, disrupted inter-organelle communication between mitochondria and ER results in altered contact sites, potentially resulting in a resistance to mitochondrial degradation, accumulation of dysfunctional mitochondria, release of proinflammatory mtDNA and an amplification of the pathophysiological response. Energetic stress and subsequent AMPK activation has been demonstrated in cell models to promote autophagy and MERCS formation [269]. From a physiological perspective introducing an exercise protocol that involves extensive cytoskeletal remodelling and energetic stress, that can promote UPR activation and induce mitochondrial remodelling, would ultimately result in an improved bioenergetic profile. This beneficial adaptive response may be facilitated by increased formation of MERCS.
Conclusions
The intricate crosstalk between the ER and mitochondria can be mediated by MERCS, providing an effective conduit for cell signalling and facilitating the exchange of information and metabolites. There are still a large number of outstanding questions in the field in relation to how the activation of the UPR following ER stress mediates the assembly and disassembly of MERCS. Similarly, it is still uncertain how MERCS influence the UPR and how alterations in MERCS may impact the cell's ability to respond to ER stress. It is clear from studies using a variety of tissues that MERCS directly impact and determine mitochondrial function and dynamics. As a result, MERCS are critical regulators of cell fate under conditions of stress, determining whether the cell will undergo an adaptive response, proliferate, initiate apoptosis or undergo cell cycle arrest and senescence. Disruption of MERCS formation could result in ER stress response dysfunction, where there is impaired UPR activation and failure to activate the appropriate arms of the UPR and subsequent downstream signalling effects. Modulation of MERCS formation could potentially be a valuable therapeutic approach in order to exacerbate mitochondrial Ca2+, increased ROS formation to potentially sensitise senescent cells to apoptosis [270].
Availability of data and material
Not applicable.
References
Li X, Yang T, Sun Z (2019) Hormesis in health and chronic diseases. Trend Endocrinol Metabol 30:944–958
Merry TL, Ristow M (2016) Mitohormesis in exercise training. Free Radic Biol Med 98:123–130
Done AJ, Traustadottir T (2016) Nrf2 mediates redox adaptations to exercise. Redox Biol 10:191–199
Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24(10):R453–R462
Rowland AA, Voeltz GK (2012) Endoplasmic reticulum–mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13(10):607–615
Wang M, Kaufman RJ (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529(7586):326–335
Csordás G, Weaver D, Hajnóczky G (2018) Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell Biol 28(7):523–540
Schwarz DS, Blower MD (2016) The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 73(1):79–94
Hetz C, Zhang K, Kaufman RJ (2020) Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21:21–438
Senft D, Ronai ZEA (2015) UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trend Biochem Sci 40:141–148
Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334(6059):1081–1086
Kopp MC et al (2019) UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat Struct Mol Biol 26(11):1053–1062
Bertolotti A et al (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2(6):326–332
Kopp MC et al (2018) In vitro FRET analysis of IRE1 and BiP association and dissociation upon endoplasmic reticulum stress. Elife 7:e30257
Carrara M et al (2015) Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 4:03522
Yoshida H et al (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107(7):881–891
Calfon M et al (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415(6867):92–96
Shen X et al (2001) Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107(7):893–903
Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13:89–102
Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313(5783):104–107
Jiang HY et al (2003) Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol 23(16):5651–5663
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397(6716):271–274
Scheuner D et al (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7(6):1165–1176
Vattem KM, Wek RC (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 101(31):11269–11274
Novoa I et al (2001) Feedback Inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol 153(5):1011–1022
Jousse CL et al (2003) Inhibition of a constitutive translation initiation factor 2α phosphatase, CReP, promotes survival of stressed cells. J Cell Biol 163(4):767–775
Haze K et al (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10(11):3787–3799
Ye J et al (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6(6):1355–1364
Lee K et al (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16(4):452–466
Urano F et al (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287(5453):664–666
Zhu X et al (2014) Ubiquitination of Inositol-requiring enzyme 1 (IRE1) by the E3 ligase CHIP mediates the IRE1/TRAF2/JNK pathway. J Biol Chem 289(44):30567–30577
Shi L et al (2018) Bax inhibitor-1 is required for resisting the early brain injury induced by subarachnoid hemorrhage through regulating IRE1-JNK pathway. Neurol Res 40(3):189–196
Wei MC et al (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292(5517):727–730
Lerner AG et al (2012) IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metabol 16(2):250–264
Oslowski CM et al (2012) Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metabol 16(2):265–273
Marciniak SJ et al (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18(24):3066–3077
Song B et al (2008) Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Investig 118(10):3378–3389
Timmins JM et al (2009) Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Investig 119(10):2925–2941
Li G et al (2009) Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress–induced apoptosis. J Cell Biol 186(6):783–792
Ziegler DV, Martin N, Bernard D (2021) Cellular senescence links mitochondria-ER contacts and aging. Commun Biol 4(1):1323–1323
Vilas-Boas EA et al (2023) Goldilocks calcium concentrations and the regulation of oxidative phosphorylation: too much, too little, or just right. J Biol Chem 299(3):102904
Mori K (2009) Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem 146(6):743–750
Zhou J et al (2006) The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci 103(39):14343–14348
Credle JJ et al (2005) On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci 102(52):18773–18784
Oikawa D et al (2010) Identification of a consensus element recognized and cleaved by IRE1α. Nucleic Acids Res 38(18):6265–6273
Upton J-P et al (2012) IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic caspase-2. Science 338(6108):818–822
Hetz C, Glimcher LH (2009) Fine-tuning of the unfolded protein response: assembling the IRE1α interactome. Mol Cell 35(5):551–561
Nguyên DT et al (2004) Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol Biol Cell 15(9):4248–4260
Hu P et al (2006) Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol 26(8):3071–3084
Yoshida H et al (1998) Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. J Biol Chem 273(50):33741–33749
Shen J et al (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev Cell 3(1):99–111
Bommiasamy H et al (2009) ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci 122(Pt 10):1626–1636
Sriburi R et al (2004) XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol 167(1):35–41
Shaffer AL et al (2004) XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21(1):81–93
Hwang J, Qi L (2018) Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trend Biochem Sci 43:593–605
Travers KJ et al (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101(3):249–258
Malhotra JD, Kaufman RJ (2007) Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 9(12):2277–2293
Mendes CS et al (2009) ER stress protects from retinal degeneration. EMBO J 28(9):1296–1307
Salminen A, Kaarniranta K (2010) ER stress and hormetic regulation of the aging process. Ageing Res Rev 9:211–217
Mattson MP (2008) Hormesis defined. Ageing Res Rev 7(1):1–7
Naidoo N et al (2008) Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J Neurosci 28(26):6539–6548
Sheng Y et al (2021) Distinct temporal actions of different types of unfolded protein responses during aging. J Cell Physiol 236(7):5069–5079
Matai L et al (2019) Dietary restriction improves proteostasis and increases life span through endoplasmic reticulum hormesis. Proc Natl Acad Sci USA 116(35):17383–17392
Kozlowski L et al (2014) The Caenorhabditis elegans HP1 family protein HPL-2 maintains ER homeostasis through the UPR and hormesis. Proc Natl Acad Sci U S A 111(16):5956–5961
Taylor RC, Dillin A (2013) XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153(7):1435–1447
Rzechorzek NM et al (2015) Hypothermic preconditioning of human cortical neurons requires proteostatic priming. EBioMedicine 2(6):528–535
Molenaars M et al (2020) A conserved mito-cytosolic translational balance links two longevity pathways. Cell Metab 31(3):549-563.e7
Statzer C et al (2022) ATF-4 and hydrogen sulfide signalling mediate longevity in response to inhibition of translation or mTORC1. Nat Commun 13(1):967
Steffen KK et al (2008) Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell 133(2):292–302
Han D et al (2009) IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138(3):562–575
Li J, Lee B, Lee AS (2006) Endoplasmic reticulum stress-induced apoptosis. J Biol Chem 281(11):7260–7270
Puthalakath H et al (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129(7):1337–1349
Lam M et al (2018) Confirming a critical role for death receptor 5 and caspase-8 in apoptosis induction by endoplasmic reticulum stress. Cell Death Differ 25(8):1530–1531
Muñoz-Pinedo C, López-Rivas A (2018) A role for caspase-8 and TRAIL-R2/DR5 in ER-stress-induced apoptosis. Cell Death Differ 25(1):226–226
Back SH et al (2009) Translation attenuation through eIF2α phosphorylation prevents oxidative stress and maintains the differentiated state in β cells. Cell Metab 10(1):13–26
Eletto D et al (2014) Redox controls UPR to control redox. J Cell Sci 127:3649–3658
Gogala M et al (2014) Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 506(7486):107–110
Zhang Z et al (2019) Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol 25:101047
Hwang C, Sinskey AJ, Lodish HF (1992) Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257(5076):1496–1502
Cao SS, Kaufman RJ (2014) Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 21(3):396–413
Appenzeller-Herzog C, Ellgaard L (2008) The human PDI family: versatility packed into a single fold. Biochim Biophys Acta 1783(4):535–548
Bulleid NJ, Freedman RB (1988) Defective co-translational formation of disulphide bonds in protein disulphide-isomerase-deficient microsomes. Nature 335(6191):649–651
Tu BP, Weissman JS (2004) Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 164(3):341–346
Kirstein J et al (2015) Proteotoxic stress and ageing triggers the loss of redox homeostasis across cellular compartments. EMBO J 34(18):2334–2349
Bhattarai KR et al (2020) Endoplasmic reticulum (ER) stress response failure in diseases. Trends Cell Biol 30(9):672–675
Eletto D et al (2014) Protein Disulfide isomerase A6 controls the decay of ire1α signaling via disulfide-dependent association. Mol Cell 53(4):562–576
Higa A et al (2014) Endoplasmic reticulum stress-activated transcription factor ATF6α requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol Cell Biol 34(10):1839–1849
Coelho JPL, Feige MJ (2020) In case of stress, hold tight: phosphorylation switches PDI from an oxidoreductase to a holdase, tuning ER proteostasis. EMBO J 39(10):e104880
Kranz P et al (2017) PDI is an essential redox-sensitive activator of PERK during the unfolded protein response (UPR). Cell Death Dis 8(8):e2986
Bassot A et al (2023) The endoplasmic reticulum kinase PERK interacts with the oxidoreductase ERO1 to metabolically adapt mitochondria. Cell Rep 42(1):111899
Hourihan JM et al (2016) Cysteine sulfenylation directs IRE-1 to activate the SKN-1/Nrf2 antioxidant response. Mol Cell 63(4):553–566
Kaufman RJ, Malhotra JD (2014) Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim Biophys Acta 1843(10):2233–2239
Lukyanenko V et al (2001) Potentiation of Ca(2+) release by cADP-ribose in the heart is mediated by enhanced SR Ca(2+) uptake into the sarcoplasmic reticulum. Circ Res 89(7):614–622
Bezprozvanny I, Watras J, Ehrlich BE (1991) Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351(6329):751–754
Young KW et al (2000) Lysophosphatidic acid-induced Ca2+ mobilization requires intracellular sphingosine 1-phosphate production. Potential involvement of endogenous EDG-4 receptors. J Biol Chem 275(49):38532–38539
Cardozo AK et al (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 54(2):452–461
Fu S et al (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473(7348):528–531
Moore CE et al (2011) PERK activation at low glucose concentration is mediated by SERCA pump inhibition and confers preemptive cytoprotection to pancreatic β-cells. Mol Endocrinol 25(2):315–326
Kiviluoto S et al (2013) Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim Biophys Acta 1833(7):1612–1624
Adachi T et al (2004) S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 10(11):1200–1207
Higo T et al (2005) Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 120(1):85–98
Xu L et al (1998) Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279(5348):234–237
Raturi A, Ortiz-Sandoval C, Simmen T (2014) Redox dependence of endoplasmic reticulum (ER) Ca2+ signaling. Histol Histopathol 29(5):543–552
Seimon TA et al (2006) Combinatorial pattern recognition receptor signaling alters the balance of life and death in macrophages. Proc Natl Acad Sci 103(52):19794–19799
Nakagawa T et al (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403(6765):98–103
Wu H et al (2020) Copper sulfate-induced endoplasmic reticulum stress promotes hepatic apoptosis by activating CHOP, JNK and caspase-12 signaling pathways. Ecotoxicol Environ Saf 191:110236
Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148(6):1145–1159
Ngo J et al (2023) Mitochondrial morphology controls fatty acid utilization by changing CPT1 sensitivity to malonyl-CoA. EMBO J 42(11):111901
Campos JC et al (2023) Exercise preserves physical fitness during aging through AMPK and mitochondrial dynamics. Proc Natl Acad Sci U S A 120(2):e2204750120
Chen H, Chan DC (2017) Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab 26(1):39–48
Friedman JR et al (2011) ER tubules mark sites of mitochondrial division. Science 334(6054):358–362
Murley A, Nunnari J (2016) The emerging network of mitochondria-organelle contacts. Mol Cell 61(5):648–653
Bravo R et al (2011) Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 124(Pt 13):2143–2152
Rainbolt TK, Saunders JM, Wiseman RL (2014) Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trend Endocrinol Metabol 25:528–537
Ryan MT, Hoogenraad NJ (2007) Mitochondrial-nuclear communications. Annu Rev Biochem 76:701–722
Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521(7553):525–528
Palikaras K, Lionaki E, Tavernarakis N (2015) Coupling mitogenesis and mitophagy for longevity. Autophagy 11(8):1428–1430
Chen M et al (2016) Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12(4):689–702
Laker RC et al (2017) Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun 8(1):548
Giacomello M, Pellegrini L (2016) The coming of age of the mitochondria–ER contact: a matter of thickness. Cell Death Differ 23(9):1417–1427
Cieri D et al (2018) SPLICS: a split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ 25(6):1131–1145
Wolf C et al (2020) Redox modifications of proteins of the mitochondrial fusion and fission machinery. Cells 9(4):815
Cao W et al (2004) p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol 24(7):3057–3067
Anderson S et al (1981) Sequence and organization of the human mitochondrial genome. Nature 290(5806):457–465
Bykov YS et al (2020) Cytosolic events in the biogenesis of mitochondrial proteins. Trends Biochem Sci 45(8):650–667
Chacinska A et al (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138(4):628–644
Eckl E-M et al (2021) Sensing, signaling and surviving mitochondrial stress. Cell Mol Life Sci 78(16):5925–5951
Anderson NS, Haynes CM (2020) Folding the mitochondrial UPR into the integrated stress response. In Trends in cell biology. Elsevier Ltd. pp 428–439
Costa-Mattioli M, Walter P (2020) The integrated stress response: from mechanism to disease. Science. https://doi.org/10.1126/science.aat5314
Pakos-Zebrucka K et al (2016) The integrated stress response. EMBO Rep 17(10):1374–1395
Münch C (2018) The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. https://doi.org/10.1186/s12915-018-0548-x
Shpilka T, Haynes CM (2018) The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol 19:109–120
Fusakio ME et al (2016) Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol Biol Cell 27(9):1536–1551
Horibe T, Hoogenraad NJ (2007) The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2(9):e835
Aldridge JE, Horibe T, Hoogenraad NJ (2007) Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2(9):e874
Haynes CM et al (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Develop Cell 13(4):467–480
Fiorese CJ et al (2016) The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol 26(15):2037–2043
Wang G et al (2022) Insight into the mitochondrial unfolded protein response and cancer: opportunities and challenges. Cell Biosci 12(1):18
Zhou D et al (2008) Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J Biol Chem 283(11):7064–7073
Wrobel L et al (2015) Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524(7566):485–488
Wang X, Chen XJ (2015) A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 524(7566):481–484
Mouchiroud L et al (2013) The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154(2):430–441
Papa L, Germain D (2014) SirT3 regulates the mitochondrial unfolded protein response. Mol Cell Biol 34(4):699–710
Papa L, Germain D (2011) Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J Cell Sci 124(Pt 9):1396–1402
Katiyar A et al (2020) HSF1 is required for induction of mitochondrial chaperones during the mitochondrial unfolded protein response. FEBS Open Bio 10(6):1135–1148
Tan K et al (2015) Mitochondrial SSBP1 protects cells from proteotoxic stresses by potentiating stress-induced HSF1 transcriptional activity. Nat Commun 6:6580
Scarpulla RC (2006) Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem 97(4):673–683
Münch C, Harper JW (2016) Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 534(7609):710–713
Bernstein SH et al (2012) The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 119(14):3321–3329
Kang BH et al (2009) Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J Clin Investig 119(3):454–464
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417(1):1–13
Hernansanz-Agustin P, Enriquez JA (2021) Generation of reactive oxygen species by mitochondria. Antioxidants (Basel) 10(3):415
Chouchani ET et al (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515(7527):431–435
Robb EL et al (2018) Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem 293(25):9869–9879
Quinlan CL et al (2012) Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem 287(32):27255–27264
Burger N et al (2022) ND3 Cys39 in complex I is exposed during mitochondrial respiration. Cell Chem Biol 29(4):636-64914 e14
Hernansanz-Agustin P et al (2020) Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature 586(7828):287–291
Povea-Cabello S, Brischigliaro M, Fernandez-Vizarra E (2024) Emerging mechanisms in the redox regulation of mitochondrial cytochrome c oxidase assembly and function. Biochem Soc Trans 52:873–885
Kleele T et al (2021) Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593(7859):435–439
Goljanek-Whysall K et al (2020) miR-181a regulates p62/SQSTM1, parkin, and protein DJ-1 promoting mitochondrial dynamics in skeletal muscle aging. Aging Cell 19(4):e13140
Burte F et al (2015) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11(1):11–24
Lévy E et al (2019) causative links between protein aggregation and oxidative stress: a review. Int J Mol Sci 20(16):3896
Runkel ED et al (2013) Surveillance-activated defenses block the ROS–induced mitochondrial unfolded protein response. PLoS Genet 9(3):e1003346
Nargund AM et al (2015) Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell 58(1):123–133
Schulz TJ et al (2007) Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6(4):280–293
Dancy BM, Sedensky MM, Morgan PG (2014) Effects of the mitochondrial respiratory chain on longevity in C. elegans. Exp Gerontol 56:245–255
Onukwufor JO et al (2022) A reversible mitochondrial complex I thiol switch mediates hypoxic avoidance behavior in C. elegans. Nat Commun 13(1):2403
Scialò F et al (2016) Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab 23(4):725–734
Matsumura T et al (2020) N-acetyl-l-tyrosine is an intrinsic triggering factor of mitohormesis in stressed animals. EMBO Rep 21(5):e49211
Bahar E, Kim H, Yoon H (2016) ER stress-mediated signaling: action potential and Ca2+ as Key players. Int J Mol Sci 17(9):1558
Zheng M et al (2012) Sensing endoplasmic reticulum stress by protein kinase RNA-like endoplasmic reticulum kinase promotes adaptive mitochondrial DNA biogenesis and cell survival via heme oxygenase-1/carbon monoxide activity. FASEB J 26(6):2558–2568
Lebeau J et al (2018) The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep 22(11):2827–2836
Sorge S et al (2020) ATF4-Induced Warburg metabolism drives over-proliferation in Drosophila. Cell Rep 31(7):107659–107659
Shpilka T et al (2021) UPRmt scales mitochondrial network expansion with protein synthesis via mitochondrial import in Caenorhabditis elegans. Nat Commun 12(1):479–479
Zhang Q et al (2021) The memory of neuronal mitochondrial stress is inherited transgenerationally via elevated mitochondrial DNA levels. Nat Cell Biol 23(8):870–880
van Vliet AR, Sassano ML, Agostinis P (2018) The unfolded protein response and membrane contact sites: tethering as a matter of life and death? Contact 1:2515256418770512
Wilson EL, Metzakopian E (2021) ER-mitochondria contact sites in neurodegeneration: genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ 28(6):1804–1821
Naon D et al (2016) Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc Natl Acad Sci 113(40):11249–11254
Muñoz JP et al (2013) Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J 32(17):2348–2361
De Vos KJ et al (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet 21(6):1299–1311
Stoica R et al (2016) ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB–PTPIP51 interaction and ER–mitochondria associations. EMBO Rep 17(9):1326–1342
Stoica R et al (2014) ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun 5(1):3996
Iwasawa R et al (2011) Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J 30(3):556–568
Namba T (2019) BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci Adv 5(6):eaaw1386
De Stefani D et al (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476(7360):336–340
Baughman JM et al (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476(7360):341–345
Liu Y et al (2019) DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc Natl Acad Sci 116(50):25322–25328
Szabadkai GR et al (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175(6):901–911
Rizzuto R et al (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280(5370):1763–1766
Rizzuto R et al (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13(9):566–578
Wenzel EM et al (2022) ER as master regulator of membrane trafficking and organelle function. J Cell Biol 221(10):202205135
Chakrabarti R et al (2018) INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol 217(1):251–268
Korobova F, Ramabhadran V, Higgs HN (2013) An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339(6118):464–467
Guo Y et al (2018) visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 175(5):1430-1442.e17
Kornmann B, Osman C, Walter P (2011) The conserved GTPase Gem1 regulates endoplasmic reticulum–mitochondria connections. Proc Natl Acad Sci 108(34):14151–14156
McLelland GL et al (2018) Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife 7:e32866
Carreras-Sureda A et al (2019) Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat Cell Biol 21(6):755–767
Khan AA et al (1996) Lymphocyte apoptosis: mediation by increased type 3 inositol 1,4,5-trisphosphate receptor. Science 273(5274):503–507
Abrisch RG et al (2020) Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J Cell Biol 219(4):e201911122
Koch A et al (2005) A Role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 16(11):5077–5086
Ilamathi HS et al (2023) Contact sites between endoplasmic reticulum sheets and mitochondria regulate mitochondrial DNA replication and segregation. iScience 26(7):107180
Lewis SC, Uchiyama LF, Nunnari J (2016) ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353(6296):5549
Irazoki A et al (2023) Disruption of mitochondrial dynamics triggers muscle inflammation through interorganellar contacts and mitochondrial DNA mislocation. Nat Commun 14(1):108
Victorelli S et al (2023) Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 622(7983):627–636
Hamasaki M et al (2013) Autophagosomes form at ER–mitochondria contact sites. Nature 495(7441):389–393
Gelmetti V et al (2017) PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 13(4):654–669
Cao Y-L et al (2017) MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 542(7641):372–376
Chen H et al (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160(2):189–200
Song Z et al (2009) Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20(15):3525–3532
Wang X et al (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147(4):893–906
Leadsham JE et al (2013) Loss of cytochrome c oxidase promotes RAS-dependent ROS production from the ER resident NADPH oxidase, Yno1p. Yeast. Cell Metabolism 18(2):279–286
Yoboue ED, Sitia R, Simmen T (2018) Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis 9(3):331
Booth DM et al (2016) Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol Cell 63(2):240–248
van Vliet AR, Agostinis P (2016) When under pressure, get closer: PERKing up membrane contact sites during ER stress. Biochem Soc Trans 44(2):499–504
Verfaillie T et al (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19(11):1880–1891
Liu Z-W et al (2013) Protein kinase RNA- like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)- mediated endoplasmic reticulum stress- induced apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol 12(1):158–158
Kang Z et al (2022) UPRmt and coordinated UPRER in type 2 diabetes. Frontiers in Cell Develo Biol 10:974083
Balsa E et al (2019) ER and nutrient stress promote assembly of respiratory chain supercomplexes through the PERK-eIF2α axis. Mol Cell 74(5):877-890.e6
Kim HJ et al (2018) Carbon monoxide-induced TFEB nuclear translocation enhances mitophagy/mitochondrial biogenesis in hepatocytes and ameliorates inflammatory liver injury. Cell Death Dis 9(11):1060
Almeida LM et al (2022) The PERKs of mitochondria protection during stress: insights for PERK modulation in neurodegenerative and metabolic diseases. Biol Rev 97(5):1737–1748
Latorre-Muro P et al (2021) A cold-stress-inducible PERK/OGT axis controls TOM70-assisted mitochondrial protein import and cristae formation. Cell Metab 33(3):598-614.e7
Cogliati S, Enriquez JA, Scorrano L (2016) Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci 41(3):261–273
Rendleman J et al (2018) New insights into the cellular temporal response to proteostatic stress. Elife 7:39054
Cullinan SB et al (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23(20):7198–7209
Martina JA et al (2016) TFEB and TFE3 are novel components of the integrated stress response. EMBO J 35(5):479–495
De Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456(7222):605–610
Rainbolt KT et al (2013) Stress-regulated translational attenuation adapts mitochondrial protein import through tim17A degradation. Cell Metabol 18(6):908–919
Tondera D et al (2009) SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J 28(11):1589–1600
Hom JR et al (2007) Thapsigargin induces biphasic fragmentation of mitochondria through calcium-mediated mitochondrial fission and apoptosis. J Cell Physiol 212(2):498–508
Celardo I et al (2017) dATF4 regulation of mitochondrial folate-mediated one-carbon metabolism is neuroprotective. Cell Death Differ 24(4):638–648
Mollereau B, Manié S, Napoletano F (2014) Getting the better of ER stress. J Cell Commun Signal 8(4):311–321
Mohamed E et al (2020) The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity 52(4):668-682.e7
Kumari R, Jat P (2021) Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol 9:645593
Mchugh D, Gil J (2018) Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol 217(1):65–77
Ziegler DV et al (2021) Calcium channel ITPR2 and mitochondria–ER contacts promote cellular senescence and aging. Nat Commun 12(1):720
Jia G et al (2019) Endothelial cell senescence in aging-related vascular dysfunction. Biochim Biophys Acta Mol Basis Dis 1865(7):1802–1809
Madreiter-Sokolowski CT et al (2019) Enhanced inter-compartmental Ca. Redox Biol 20:458–466
Dejos C, Gkika D, Cantelmo AR (2020) The two-way relationship between calcium and metabolism in cancer. Front Cell Dev Biol 8:573747
Twyning MJ et al (2024) Partial loss of MCU mitigates pathology in vivo across a diverse range of neurodegenerative disease models. Cell Rep 43(2):113681
Paillusson S et al (2016) There’s something wrong with my MAM; the ER–mitochondria axis and neurodegenerative diseases. Trends Neurosci 39(3):146–157
Paillusson S et al (2017) α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol 134(1):129–149
Cherubini M, Lopez-Molina L, Gines S (2020) Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca. Neurobiol Dis 136:104741
Volpe P et al (1992) The endoplasmic reticulum-sarcoplasmic reticulum connection: distribution of endoplasmic reticulum markers in the sarcoplasmic reticulum of skeletal muscle fibers. Proc Natl Acad Sci 89(13):6142–6146
Rossi D et al (2022) The sarcoplasmic reticulum of skeletal muscle cells: a labyrinth of membrane contact sites. Biomolecules 12(4):488
Kaisto T, Metsikko K (2003) Distribution of the endoplasmic reticulum and its relationship with the sarcoplasmic reticulum in skeletal myofibers. Exp Cell Res 289(1):47–57
Bohnert KR, McMillan JD, Kumar A (2018) Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease. J Cell Physiol 233:67–78
Afroze D, Kumar A (2019) ER stress in skeletal muscle remodeling and myopathies. FEBS J 286(2):379–398
Fidziańska A, Goebel HH (1991) Human ontogenesis. Acta Neuropathol 81(5):572–577
Nakanishi K, Sudo T, Morishima N (2005) Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol 169(4):555–560
Nakanishi K, Dohmae N, Morishima N (2007) Endoplasmic reticulum stress increases myofiber formation in vitro. FASEB J 21(11):2994–3003
Kirwan JP, Sacks J, Nieuwoudt S (2017) The essential role of exercise in the management of type 2 diabetes. Clevel Clin J Med 84(7 suppl 1):S15–S21
Estébanez B et al (2018) Endoplasmic reticulum unfolded protein response, aging and exercise: an update. Front Physiol 9:1744
Bouviere J et al (2021) Exercise-stimulated ROS sensitive signaling pathways in skeletal muscle. Antioxidants 10(4):537
Xia Q et al (2023) Peroxiredoxin 2 is required for the redox mediated adaptation to exercise. Redox Biol 60:102631
Le Moal E et al (2017) Redox control of skeletal muscle regeneration. Antioxid Redox Signal 27(5):276–310
Wu J et al (2011) The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell Metab 13(2):160–169
Jamart C et al (2013) Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J Physiol Endocrinol Metab 305(8):E964–E974
West DWD et al (2018) Normal ribosomal biogenesis but shortened protein synthetic response to acute eccentric resistance exercise in old skeletal muscle. Front Physiol 9:1915
Ogborn DI et al (2014) The unfolded protein response is triggered following a single, unaccustomed resistance-exercise bout. Am J Physiol Regul Integr Comp Physiol 307(6):R664–R669
Deldicque L et al (2010) The unfolded protein response is activated in skeletal muscle by high-fat feeding: potential role in the downregulation of protein synthesis. Am J Physiol Endocrinol Metab 299(5):E695-705
Rieusset J et al (2012) Reduction of endoplasmic reticulum stress using chemical chaperones or Grp78 overexpression does not protect muscle cells from palmitate-induced insulin resistance. Biochem Biophys Res Commun 417(1):439–445
Slavin MB, Kumari R, Hood DA (2022) ATF5 is a regulator of exercise-induced mitochondrial quality control in skeletal muscle. Mol Metabol 66:101623–101623
Zhang S-S et al (2021) A review of the role of endo/sarcoplasmic reticulum-mitochondria Ca2+ transport in diseases and skeletal muscle function. Int J Environ Res Public Health 18(8):3874–3874
Morgado-Cáceres P et al (2022) The aging of ER-mitochondria communication: A journey from undifferentiated to aged cells. Front Cell Develop Biol 10:946678
Pietrangelo L et al (2015) Age-dependent uncoupling of mitochondria from Ca2+ release units in skeletal muscle. Oncotarget 6(34):35358–35371
Umanskaya A et al (2014) Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc Natl Acad Sci 111(42):15250–15255
Skinner SK et al (2021) Mitochondrial permeability transition causes mitochondrial reactive oxygen species- and caspase 3-dependent atrophy of single adult mouse skeletal muscle fibers. Cells 10(10):2586
Tubbs E et al (2018) Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 67(4):636–650
Hu Y et al (2021) The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 17(5):1142–1156
Cavinato M et al (2021) Targeting cellular senescence based on interorganelle communication, multilevel proteostasis, and metabolic control. FEBS J 288(12):3834–3854
Bulteau AL et al (2005) Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc Natl Acad Sci U S A 102(17):5987–5991
Murata H et al (2003) Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J Biol Chem 278(50):50226–50233
McStay GP, Clarke SJ, Halestrap AP (2002) Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem J 367(Pt 2):541–548
Nadanaka S et al (2007) Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol Cell Biol 27(3):1027–1043
Giangregorio N, Palmieri F, Indiveri C (2013) Glutathione controls the redox state of the mitochondrial carnitine/acylcarnitine carrier Cys residues by glutathionylation. Biochim Biophys Acta 1830(11):5299–5304
Hurd TR et al (2008) Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J Biol Chem 283(36):24801–24815
Chouchani ET et al (2013) Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med 19(6):753–759
Chen YR et al (2007) Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J Biol Chem 282(45):32640–32654
Wang SB et al (2013) Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc Med 23(1):14–18
Nguyen TT et al (2011) Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem 286(46):40184–40192
Sutandy FXR et al (2023) A cytosolic surveillance mechanism activates the mitochondrial UPR. Nature 618(7966):849–854
Cho DH et al (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324(5923):102–105
Benham AM et al (2013) Ero1-PDI interactions, the response to redox flux and the implications for disulfide bond formation in the mammalian endoplasmic reticulum. Philos Trans R Soc Lond B Biol Sci 368(1617):20110403
Kozlov G et al (2010) A structural overview of the PDI family of proteins. FEBS J 277(19):3924–3936
Jiang KL et al (2023) Discovery of toxoflavin, a potent IRE1alpha inhibitor acting through structure-dependent oxidative inhibition. Acta Pharmacol Sin 44(1):234–243
Liu CY, Xu Z, Kaufman RJ (2003) Structure and intermolecular interactions of the luminal dimerization domain of human IRE1alpha. J Biol Chem 278(20):17680–17687
Wang L et al (2014) Glutathione peroxidase 7 utilizes hydrogen peroxide generated by Ero1α to promote oxidative protein folding. Antioxid Redox Signal 20(4):545–556
Kanemura S et al (2020) Characterization of the endoplasmic reticulum-resident peroxidases GPx7 and GPx8 shows the higher oxidative activity of GPx7 and its linkage to oxidative protein folding. J Biol Chem 295(36):12772–12785
Wei PC et al (2012) Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol Cell 48(5):747–759
Kil IS, Park JW (2005) Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J Biol Chem 280(11):10846–10854
Joseph SK et al (2018) Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J Biol Chem 293(45):17464–17476
Dong Z et al (2017) Mitochondrial Ca. Mol Cell 65(6):1014-1028.e7
Shutt T et al (2012) The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep 13(10):909–915
Zhao J et al (2011) Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J 30(14):2762–2778
McLain AL, Szweda PA, Szweda LI (2011) α-Ketoglutarate dehydrogenase: a mitochondrial redox sensor. Free Radic Res 45(1):29–36
Yan LJ et al (2013) Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic Res 47(2):123–133
Cox AG et al (2009) Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem J 421(1):51–58
Konno T et al (2015) ERO1-independent production of H2O2 within the endoplasmic reticulum fuels Prdx4-mediated oxidative protein folding. J Cell Biol 211(2):253–259
Baković J et al (2019) A key metabolic integrator, coenzyme A, modulates the activity of peroxiredoxin 5 via covalent modification. Mol Cell Biochem 461(1–2):91–102
Lee SR et al (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 277(23):20336–20342
Heckler EJ et al (2008) Generating disulfides with the Quiescin-sulfhydryl oxidases. Biochim Biophys Acta 1783(4):567–577
Aracena-Parks P et al (2006) Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem 281(52):40354–40368
Tong X et al (2008) High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J Mol Cell Cardiol 44(2):361–369
De Pinto V et al (2016) Role of cysteines in mammalian VDAC isoforms’ function. Biochim Biophys Acta 1857(8):1219–1227
Wajih N, Hutson SM, Wallin R (2007) Disulfide-dependent protein folding is linked to operation of the vitamin K cycle in the endoplasmic reticulum. A protein disulfide isomerase-VKORC1 redox enzyme complex appears to be responsible for vitamin K1 2,3-epoxide reduction. J Biol Chem 282(4):2626–2635
Funding
Open Access funding provided by the IReL Consortium. JCMM is a recipient of a PhD Hardiman Scholarship from the University of Galway. AS is funded by Precision Oncology Ireland, which is part-funded by the Science Foundation Ireland (SFI) Strategic Partnership Programme (Grant Number 18/SPP/3522), a Science Foundation Ireland grant co-funded under the European Regional Development Fund (grant number 13/RC/2073_P2), and funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant number H2020-MSCA-COFUND-2019-945425 (‘DevelopMed’).
Author information
Authors and Affiliations
Contributions
JCMM and BMcD, conceptualisation, writing, editing and reviewing. AS writing, editing and reviewing.
Corresponding author
Ethics declarations
Conflicts of interests
Not applicable.
Ethical approval and consent to participate
Not applicable.
Consent for publication
All authors have reviewed final draft and consent to publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Casas-Martinez, J.C., Samali, A. & McDonagh, B. Redox regulation of UPR signalling and mitochondrial ER contact sites. Cell. Mol. Life Sci. 81, 250 (2024). https://doi.org/10.1007/s00018-024-05286-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05286-0