
Abstract
Diabetic hyperglycemia induces dysfunctions of arterial smooth muscle, leading to diabetic vascular complications. The CaV1.2 calcium channel is one primary pathway for Ca2+ influx, which initiates vasoconstriction. However, the long-term regulation mechanism(s) for vascular CaV1.2 functions under hyperglycemic condition remains unknown. Here, Sprague–Dawley rats fed with high-fat diet in combination with low dose streptozotocin and Goto-Kakizaki (GK) rats were used as diabetic models. Isolated mesenteric arteries (MAs) and vascular smooth muscle cells (VSMCs) from rat models were used to assess K+-induced arterial constriction and CaV1.2 channel functions using vascular myograph and whole-cell patch clamp, respectively. K+-induced vasoconstriction is persistently enhanced in the MAs from diabetic rats, and CaV1.2 alternative spliced exon 9* is increased, while exon 33 is decreased in rat diabetic arteries. Furthermore, CaV1.2 channels exhibit hyperpolarized current–voltage and activation curve in VSMCs from diabetic rats, which facilitates the channel function. Unexpectedly, the application of glycated serum (GS), mimicking advanced glycation end-products (AGEs), but not glucose, downregulates the expression of the splicing factor Rbfox1 in VSMCs. Moreover, GS application or Rbfox1 knockdown dynamically regulates alternative exons 9* and 33, leading to facilitated functions of CaV1.2 channels in VSMCs and MAs. Notably, GS increases K+-induced intracellular calcium concentration of VSMCs and the vasoconstriction of MAs. These results reveal that AGEs, not glucose, long-termly regulates CaV1.2 alternative splicing events by decreasing Rbfox1 expression, thereby enhancing channel functions and increasing vasoconstriction under diabetic hyperglycemia. This study identifies the specific molecular mechanism for enhanced vasoconstriction under hyperglycemia, providing a potential target for managing diabetic vascular complications.
Similar content being viewed by others

Introduction
As a common metabolic disorder, diabetes mellitus affects 0.5 billion population, representing an increasing challenge to the health care around the world [1]. The most typical metabolic abnormality of diabetes is elevated blood glucose (hyperglycemia). A persistent hyperglycemic condition leads to the production of chemical moieties known as advanced glycation end-products (AGEs), which play a central role in the pathophysiology of diabetic cardiovascular complications [2], including hypertension, coronary heart disease and myocardial infarction [3, 4]. Hyperglycemia induces abnormal vasoconstriction and vasodilation [5], especially in small arteries, leading to the dysregulated blood pressure. Besides endothelial cells [6, 7], dysregulated functions of vascular smooth muscle cells (VSMCs) are known to play an important role in diabetic arteries in human and animals [8,9,10]. However, the long-term regulation mechanisms of VSMCs remain unclear.
The L-type Ca2+ channel CaV1.2 is essential for arterial constriction, which responds to membrane potential and provides sustained influx of Ca2+ ions to activate vascular smooth muscle contraction [11]. Upon acute increases in extracellular glucose and in high fat diet (HFD)-induced diabetic hyperglycemia, PKA-mediated CaV1.2 α1C Ser1928 phosphorylation is correlated with enhanced Ca2+ influx in VSMCs and the reduction in arterial diameter and blood flow in vivo, which has been confirmed as one of key molecular signaling events underlying potentiation of CaV1.2 channel activity and vasoconstriction [12,13,14]. However, compared with control rats, diabetic rats exhibited enhanced myogenic tone when exposed to a normal range of glucose concentration. Persistent hyperglycemic conditions induce aberrant genes transcription, contributing to the metabolic memory in cardiovascular dysfunction [15, 16]. Notably, optimal glycemia control cannot completely prevent the vascular complications in diabetic patients [17], and glycemic levels do not fully explain the association between long-term vascular complications and mortality in type 1 diabetes [18]. Therefore, besides the short-term effect induced by hyperglycemia, we speculated that there is an alternative long-term mechanism mediating the abnormal vascular CaV1.2 channel function in diabetic arteries, leaving a knowledge gap.
Alternative splicing (AS), as one of post-transcriptional modulation mechanisms, is key to eukaryotic gene expression and cellular functions [19, 20]. Over 95% of human multi-exon genes are regulated by AS [21, 22]. The human CaV1.2 pore-forming α1C subunit-encoding gene CACNA1C has at least 20 alternatively spliced exons [23]. Among them, we have previously demonstrated the unique roles for 3 common spliced exons in regulating the electrophysiological and pharmacological functions of the cardiovascular CaV1.2 channel, including the mutually exclusive exon 8/8a, cassette exon 9* and exon 33 [24,25,26,27]. Under pathological conditions, the splicing patterns of CaV1.2 α1C subunit can be altered. For example, in rat [26] and human [27] hypertrophic hearts, the expressions of CaV1.2 alternative exon 9* and exon 33 were upregulated, respectively. Furthermore, in hypertensive arteries, CaV1.2 alternative exon 8a and exon 9* increased, while alternative exon 33 decreased [24, 28]. However, it remains unknown whether the splicing pattern of CaV1.2 channel is altered in diabetic arteries.
As an RNA-binding protein, the splicing factor regulates different splicing events by binding to the motifs of pre-mRNAs. CACNA1C has several binding sequences for different splicing factors, such as PTBP1 and Rbfox1/2 proteins [29]. Rbfox proteins are known to bind to the element of UGCAUG in pre-mRNA to regulate the splicing events [30, 31]. Through bioinformatic analysis, it has been discovered that CACNA1C gene contains multiple UGCAUG elements, which can be bound by Rbfox1/2 to regulate its AS events during neuronal development [32]. Previously, we found that Rbfox1/2 dynamically regulates CaV1.2 exons 9* and 33 in VSMCs, playing important roles in vasoconstriction of hypertensive arteries [28, 33]. However, the roles of Rbfox proteins in modulating the key function of vascular CaV1.2 channels and arterial constriction during diabetic hyperglycemia remain unidentified.
In this work, we hypothesize that the downregulation of Rbfox1 in diabetic arteries leads to aberrant splicing of vascular CaV1.2 channel. This, in turn, results in persistently enhanced vasoconstriction during diabetic hyperglycemia. Thus, we have uncovered the long-term mechanism that mediates the function of vascular CaV1.2 channels and the persistently enhanced vasoconstriction of small resistance arteries during hyperglycemia.
Materials and methods
Animal model and human samples
All animal experiments were approved by the Committee on Animal Care of Nanjing Medical University and were conducted according to the NIH Guidelines for the Care and Use of Laboratory Animals. All studies involved with animals were reported in accordance with the ARRIVE guidelines. Male Sprague–Dawley (SD) rats (≈180 g) and Wistar rats used in this study were purchased from Jiangsu Laboratory Animal Center (Nanjing). Goto-Kakizaki (GK) rats (16-weeks-old) were purchased from Gene and Peace Biotechnology Ltd (Yangzhou, China). The diabetic rat models were established with a HFD in combination with a low dose of streptozotocin (STZ). Briefly, the 10-weeks-old rats were fed with HFD (45%) for 4 weeks before administration of STZ (intraperitoneal injection, 10 mg/kg/day, in 0.1 mol/L citric acid-sodium citrate buffer solution, pH 4.5) for 5 successive days (Fig. 1A), This was followed by feeding with HFD for another 4 weeks. Control rats were fed a standard diet and injected with an equal volume of citric acid-sodium citrate buffer solution. HFD/STZ-treated rats and GK rats with a random blood glucose level of ≥16.7 mmol/L were considered diabetic [34] and included for further analysis. Human cerebral arteries were collected from patients undergoing surgeries at the Affiliated Brain Hospital of Nanjing Medical University. The study was approved by the Institutional Review Board of Nanjing Medical University (Reference No. 2022/550), and each patient signed a written informed consent document. The clinical characteristics of the patients were listed in Table S1 in the Supplementary Information.

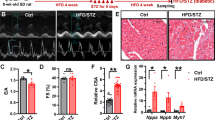
K+‐induced vasoconstriction is increased in mesenteric arteries of diabetic rats. A Diabetic rat models were established using a combination of HFD and STZ. Briefly, 10-week-old rats were fed a HFD for 4 weeks before a low dose of STZ was administered for 5 consecutive days, followed by another 4 weeks of HFD. Control rats were fed a standard diet and administrated with a vehicle. B Random and fasting blood glucose levels were monitored in the rats. ** P < 0.01, 1-way ANOVA followed by a Tukey’s post hoc test. n represents the number of rats. C Traces of mesenteric artery (MA) tension in response to increased KCl extracellular concentration (from 0 to 80 mmol/L KCl) in nondiabetic and diabetic MAs are shown. D Plots of concentration-tension relationship were represented and fitted with Boltzmann equation for the arteries from control (n = 16 arteries from 6 rats) or HFD/STZ-treated rats (n = 18 arteries from 6 rats). * P < 0.05 vs. control rats, 2-way ANOVA followed by Sidak’s multiple comparisons
Preparation of glycated serum
Glycated serum (GS) was used to mimic the AGEs [35, 36]. Briefly, GS was prepared by incubating fetal bovine serum (FBS) (HyClone) and d-glucose (90 g/L) at 37 °C for 3 weeks after filtering and sterilizing. Non-glycated serum (NG) was prepared using the same protocol without adding d-glucose. The concentrations of AGEs in GS were determined by an ELISA assay (Jiancheng Bioengineering Institute, Nanjing, China), which was 84.82 ng/mL in GS and 26.02 ng/mL in NG, respectively. Additionally, the concentration of AGEs was 47.53 ng/mL in the serum from HFD/STZ-treated rats.
Measurement of vascular constriction
The Myograph system (620M, DMT, Denmark) was used to evaluate vasoconstriction by measuring the isometric tension [33]. Briefly, second-order mesenteric arteries (MAs), were isolated in K+-free Hanks’ solution (containing in mmol/L: 1.26 CaCl2, 0.41 MgSO4-7H2O, 137.93 NaCl, 0.34 Na2HPO4, 0.49 MgCl2-6H2O, 4.17 NaHCO3, 5.56 d-Glucose). After removing connective tissues, the arteries were cut into 1-to-1.2-mm segments, then mounted with 20-μm wires and set at a resting tension of 0.1 g. All segments were equilibrated for 45 min with intermittent washes every 15 min using normal Hanks’ solution. We used 60 mmol/L KCl (dissolved in K+-free Hanks’ solution) to perfuse the mounted arteries and check their vascular contractility. After that, the chamber was perfused with normal saline solution. Vasoconstriction was then measured by stepwise increasing extracellular KCl concentration from 20 to 80 mmol/L. The change in vascular constriction (folds) was calculated as the vascular tension under K+-containing bath solution divided by the resting tension. The dosage-effect curves were fitted with the Boltzmann equation. The Ca2+ ionophore ionomycin was used at the end of experiment to induce the maximal contractility and for quality control. Arteries that did not achieve a 10-folds tension change were excluded for further analysis. To check the vasoconstriction of MAs from control or HFD/STZ-treated rats, we only used 2–3 arteries from each rat to measure their vascular contractility.
Isolation of VSMCs
Enzymatic isolation of VSMCs was carried out as previously described [28, 33]. Briefly, rats’ aorta or MAs were isolated rapidly and placed in 4 °C in Hanks’ solution. After cutting into segments, the arteries were digested in 2 steps. First, the tissues were incubated in a solution contained 1 mg/mL albumin, 1 mg/mL papain, and 2 mg/mL dithioerythritol, followed by digestion in a solution contained 1 mg/mL albumin, 5 mg/mL collagenase II, and 1 mg/mL hyaluronidase for 30 min each step. Isolated cells were cultured in DMEM (Gibco) medium and replaced with fresh medium the next day. We only used primary isolated VSMCs for the experiments.
Cell culture and treatments
Isolated VSMCs were cultured in DMEM (Gibco) medium with 10% FBS under a 5% CO2 incubator at 37℃. GS (20%), methylglyoxal (MGO) (500 μmol/L), or mannitol (25 mmol/L) was used to treat VSMCs for 48–72 h. For cell transfection experiments, VSMCs were seeded in 6-well cell culture plates at a density of 60–70%. Then, 50 nmol/L pooled siRNAs (RiboBio, Guangzhou, China) or 2 μg expression plasmids were transfected into cells using the Lipofectamine 3000 kit (Invitrogen) and cultured for another 48 h. The sequence information of siRNAs targeting rat Rbfox1 mRNAs is listed in Table S2.
Reverse-transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from arteries or VSMCs using Trizol reagent (Invitrogen). 2 μg of total RNA was reverse transcribed into cDNA using SuperScript III reverse transcriptase (Invitrogen). The primer sequences used in the present study are listed in Table S3. The gel images were captured using a Gel Imaging System (Tanon, Shanghai).
Western blotting
Total protein was extracted from VSMCs or arterial tissues in RIPA lysis buffer with 1% phenylmethylsulfonyl fluoride. The protein concentration was determined by the Bradford assay. Protein samples were boiled in 5× SDS-sample buffer at 100℃ for 5 min. Equal amounts of the protein samples were separated by 10% SDS-PAGE and then transferred to a PVDF membrane (Millipore, 0.2 μm) under a constant voltage. A solution of 5% milk powder dissolved in Tris-buffered saline with Tween-20 (TBST) was used to block the membrane. The membranes were incubated with primary antibodies overnight at 4℃and then incubated with a horseradish peroxidase-linked secondary antibody (1:10,000) for 1 h at room temperature. Primary antibodies were not reused, and dilutions of the secondary antibody were maintained at 4 °C and reused up to three times. The information of the antibodies was listed in Table S4. The protein bands were visualized using the Immobilon Western Chemiluminescent HRP Substrate Kit (Millipore). All uncropped images of Western blotting were presented in Supplementary Information.
Immunofluorescent staining
The localization of Rbfox1 in cells was determined through immunofluorescence staining. Briefly, freshly isolated MAs were fixed in 4% paraformaldehyde for 24 h at room temperature and then immersed in 15% sucrose and 30% sucrose for 10 min each step. Using a cryostation, the tissues were cut into 20-μm thickness. After permeabilizing with 0.2% Triton X-100 in PBS for 20 min, cells or tissues were blocked with 2% BSA in PBS for 40 min at room temperature. They were then incubated with rabbit anti-Rbfox1 (1 μg/mL, sc-135476, Santa Cruz Biotechnology) and mouse anti-α-smooth muscle actin (SMA) (0.4 μg/mL, NBP2-33006, Novus Biologicals) antibodies dissolved in the blocking solution at room temperature for 12 h. The information of antibodies was listed in Table S4. After washing with PBS, cells or tissues were incubated for 1 h with secondary antibodies labeled with Alexa Fluor 488 or 594 (R37114 or R37119, Molecular Probes) at room temperature. A confocal laser scanning microscope (LSM710, Carl Zeiss, Germany) was used to capture the fluorescence signal.
Introduction of Rbfox1 expression plasmid or siRNAs into rat MAs
Rbfox1 plasmid or siRNAs were introduced into the MAs by a reversible permeabilization (RP) procedure as previously described [25]. First, MAs were incubated for 20 min at 4 °C in a solution contained (in mmol/L) 120 KCl, 2 MgCl2, 10 EGTA, 5 ATP, and 20 TES (pH 6.8). After that, the arteries were incubated in a solution containing 4 μg/mL Rbfox1 plasmid or 40 nmol/L siRNAs, 120 KCl, 2 MgCl2, 5 ATP, and 20 TES (pH 6.8) for 3 h at 4 °C and then in the same solution but with an elevated 10 MgCl2 for 30 min. Permeabilization was reversed by incubating the arteries for 30 min at room temperature in a physiological solution containing (in mmol/L) 140 NaCl, 5 KCl, 10 MgCl2, 5 glucose, and 2 MOPS (pH 7.1). The calcium concentration was gradually increased from 0 to 0.01, 0.1, and 1.8 mmol/L over a period of 45 min. The arteries were cultured in serum-free DMEM-F12 medium (Invitrogen) under 5% CO2 at 37 °C, and their endogenous expression of Rbfox1 was checked by Western blotting after 72 h of culture.
Whole-cell recording of CaV1.2 calcium channel currents
Whole-cell currents were recorded using an Axon 200B amplifier (Molecular Device, Sunnyvale, CA) and the pCLAMP software (version 10.7, Axon Instruments). The pipette solution contained (in mmol/L): 130 CsCl, 5 EGTA, 1 MgCl2, 10 HEPES, 2 Mg-ATP, 0.5 GTP, and 10 d-glucose. The pH was adjusted to 7.2, and the osmolarity was maintained at 290 to 300 mOsm. The bath solution contained (in mmol/L): 122 or 132 TEA-Cl, 10 HEPES, 1 MgCl2, 20 or 10 BaCl2, and 10 d-glucose. The pH was adjusted to 7.2, and the osmolarity was maintained at 300 to 310 mOsm.
The cells were held at −70 mV and then stepped from −50 to +50 mV. The currents were filtered at 1 to 5 kHz and sampled at 5 to 50 kHz, and the series resistance was <5 MΩ after >70% compensation. The P/4 protocol was used to subtract online the leak and capacitive transients. Current size was normalized to cell capacitance (Cm), which was presented as current density. The data were not included if the leaky currents were >100 pA in VSMCs. GS or MGO were applied to the VSMCs for 24–48 h before the recording, and H-89 was added to VSMCs for 4 h prior to the experiments. The current–voltage (I–V) relationship curve was fitted with the equation: I = Gmax(V − Erev)/(1 + exp[V – V0.5]/k), where Gmax represents the maximum conductance, V is testing potential, Erev is the reversal potential, V0.5 is the half-activation potential, and k is the slope rate. The steady-state activation (SSA) curve was derived from an I–V protocol and calculated using the equation G = I/(V – Erev), where G represents the conductance. The curve was fitted with Boltzmann equation. The steady-state inactivation (SSI) curves were obtained from each test pulse, which were normalized to the maximal current amplitude of the normalizing pulse. The SSI curve were fitted with a single Boltzmann equation: Irelative = Imin + (Imax − Imin)/(1 + exp(V0.5,inact − V)/k), where Irelative represents the normalized current; V0.5,inact is the potential for half-inactivation, and k is the slope value.
Measurement of cytosolic Ca2+ concentration
Cytosolic calcium concentration was measured using 4 μmol/L Fluo-4 AM (Molecular Probes). VSMCs should be bathed in Tyrode solution and then transferred to laminin-coated culture dishes prior to imaging on the stage of a confocal microscope (LSM710, Carl Zeiss, German). The images were acquired in time series scanning mode and sampled at 1 fps, and the fluorescence intensity was analyzed offline. To calibrate the fluorescence signal of the intracellular Ca2+ indicator, we used the equation: Δ[Ca2+]i = ΔF/F0 = (F − F0)/F0, where F is the fluorescence signal at any given time and F0 is the mean resting fluorescence, to calculate changes in intracellular calcium concentration.
Statistical analysis
Data are expressed as means ± SEM. The “n” number represents the number of animals in Western blotting, RT-PCR experiments, and arteries during vascular function measurement. It also represents the number of cells in patch clamp study and fluorescence detection from at least 3 independent experiments. Statistical significance was analyzed using Student’s unpaired t test, 1-way ANOVA followed by a Tukey’s post hoc test, or 2-way ANOVA followed by Sidak’s multiple comparisons. P < 0.05 was considered a significant difference.
Results
Hyperglycemia leads to persistently enhanced K+-induced vasoconstriction in diabetic arteries
For the HFD/STZ-induced diabetic model, we used a combination treatment of HFD and STZ to generate the diabetic rat model for ≈9 weeks (Fig. 1A). The peripheral blood was collected to measure the blood glucose levels in HFD/STZ and GK diabetic rats (type 2 diabetic model). Compared to control rats, diabetic rats exhibited significant elevated random and fasting blood glucose levels (Fig. 1B; Fig. S1A), indicating that hyperglycemia was well-established in the diabetic rat models. To determine the vascular tension of the arteries, MAs were isolated from the rats and perfused with K+-free Hanks’ solution. They were then exposed to increasing KCl concentrations to induce vasoconstriction ex vivo (Fig. 1C; Fig. S1C). In comparison to control rats, K+-induced vasoconstriction was significantly increased in the arteries from HFD/STZ-treated rats (Fig. 1D) and GK rats (Fig. S1D), indicating that isolated diabetic arteries have persistently enhanced vasoconstriction. Of significance, the calcium channel blocker (CCB) nifedipine-induced vasodilation was greater in the MAs from diabetic rats compared to those from control rats, indicating that diabetic arteries are more sensitive to CCB (Fig. S2).
Facilitated activation of CaV1.2 channel in VSMCs from diabetic arteries
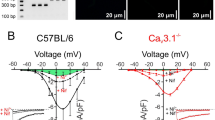
Given that Ca2+ influx from CaV1.2 channels in VSMCs is key for vasoconstriction [11], we next investigated the electrophysiological properties of the CaV1.2 channel in VSMCs during diabetic hyperglycemia. Previously, we performed the experiments using 2 concentrations of nifedipine to assess the levels of expression of CaV1.2 and CaV1.3 currents based on their different sensitivities to dihydropyridine. The results showed that nifedipine inhibits calcium currents similarly at concentrations of 1 μmol/L or 10 μmol/L [25], indicating that the major Ca2+ currents in VSMCs are mediated by CaV1.2, not the CaV1.3 channels. Here, whole-cell patch clamp was applied to record CaV1.2 currents in isolated VSMCs from rats MAs (Fig. 2A). As shown in Fig. 2B, the normalized I–V curve of diabetic VSMCs shifted towards the left (hyperpolarized) by approximately 7.4-mV in V0.5 (half-activation potential) compared to nondiabetic cells. However, the cell capacitance of VSMCs was similar between control and HFD/STZ-treated rats (Fig. S3A). The current densities of CaV1.2 channels in the VSMCs from diabetic rats were lower than those from control rats (Fig. 2C), which may be attributed to diabetic dyslipidemia [37].

CaV1.2 channel activation curve is left-ward shifted (hyperpolarized) in isolated VSMCs from diabetic rats. A VSMCs were freshly isolated from mesenteric arteries from either control or HFD/STZ-treated rats. CaV1.2 channel currents were recorded under various testing potentials, ranging from − 50 to 50 mV over 1500 ms. Subsequently, the currents were recorded by a 200 ms test pulse at 10 mV in a 20 mmol/L Ba2+ bath solution, while holding the cells at − 70 mV. B Plots of CaV1.2 I–V curve of VSMCs were analyzed and fitted with the equation. C Current densities of CaV1.2 channel were calculated by dividing the currents by the cell capacitance (Cm) of the VSMCs from either control or HFD/STZ-treated rats. * P < 0.05, ** P < 0.01, 2-way ANOVA followed by Sidak’s multiple comparisons. D Plots of SSA curves of CaV1.2 channel in VSMCs from either control or HFD/STZ-treated rats were derived from recordings by I–V protocol. E Plots of SSI curves of CaV1.2 channel in VSMCs were recorded by SSI protocol and fitted with the Boltzmann equation. n represents the number of cells from at least 3 rats for each group
To further confirm the detailed electrophysiological properties of CaV1.2 in diabetic rats, we checked the SSA and SSI curves, respectively. As shown in Fig. 2D, the activation curve and V0.5,act of CaV1.2 channel in the VSMCs from diabetic models were shifted towards the left (Table S5), but the inactivation curve (Fig. 2E) and V0.5,inact (half-inactivation potential) remained similar (Table S5). These data suggested that diabetic hyperglycemia facilitates the activation of vascular CaV1.2 channels upon depolarization from the resting membrane potential.
Aberrant splicing of CaV1.2 channel are presented in the arteries of diabetic rats and patients
The splicing pattern of the vascular CaV1.2 channel can be altered under pathological conditions such as atherosclerosis [38] and hypertension [24, 28, 33]. However, it is unknown whether it changes under diabetic hyperglycemia. Hence, we collected the MAs and thoracic arteries (TAs) from nondiabetic and diabetic rats. Using the RT-PCR approach, we found that the proportion of CaV1.2 channels with exon 8a (CaV1.2E8a) did not change in the arteries (Fig. 3A, B). However, the proportions of CaV1.2 channels with exon 9* (CaV1.2E9*) in the arteries from diabetic rats were significantly increased by 18.3% in MAs and 7.9% in TAs compared to those in nondiabetic arteries, respectively (Fig. 3C, D). Particularly, CaV1.2 channels with exon 33 (CaV1.2E33) decreased by 24.2% in MAs and 12.5% in TAs compared to those from nondiabetic rats (Fig. 3E, F). Exceptionally, we observed a more robust change of AS events in MAs than in TAs from rats, indicating that AS of CaV1.2 in the diabetic artery mainly occurs in the small arteries, which are resistance vessels for arterial blood pressure regulation.

CaV1.2 channel is aberrantly spliced in the arteries from diabetic rats. A Specific primers were used to amplify and detect rat Cacna1c mRNA with alternative exon 8/8a, with the upper bands showing the inclusion of exon 8a, and the low bands indicating exon 8. Mesenteric arteries (MAs) and thoracic arteries (TAs) from control or HFD/STZ-treated rats were collected to perform RT-PCR. Gapdh mRNA was detected as loading control. B Combined data showed the proportions of CaV1.2 with alternative exon 8a in MAs or TAs, respectively. C The primers were used to detect rat Cacna1c mRNA with alternative exon 9*, with the upper and low bands showing the inclusion or exclusion of exon 9*, respectively. D The data are presented as bar chart to show the proportions of CaV1.2 with alternative exon 9* in MAs or TAs, respectively. E The specific primers were also used to detect rat Cacna1c mRNA with alternative exon 33. F The data are shown as bar charts to show the proportions of CaV1.2 with alternative exon 33 in mesenteric arteries or thoracic arteries, respectively. * P < 0.05, ** P < 0.01 vs control, unpaired t test. ns indicates no significant differences. n represents the number of rats. G The specific primers were used to detect Cacna1c mRNA with alternative exons. The data are presented as bar charts to show the proportions of CaV1.2 with alternative exon 8a (H), exon 9* (I) or exon 33 (J) in cerebral arteries from the patients with or without diabetes. The ratios between the upper band and the sum of the upper and lower bands present the proportions of CaV1.2 channel with exon 8a, exon 9* or exon 33. * P < 0.05 vs non-diabetic patients, unpaired t test. ns indicates no significant differences. n represents the number of patients
Based on our study using the GK rat as a type 2 diabetic model, we observed a significant increase in the proportion of CaV1.2E9* channel and a decrease in CaV1.2E33 channel (Fig. S4A–D). Importantly, we collected the arteries from the non-hypertensive HFD/STZ rats and examined the expressions of CaV1.2 alternative exons 9* and 33. The results confirmed that the expression of CaV1.2E9* channel was increased, while CaV1.2E33 was decreased (Fig. S4F–I). These findings suggest that hyperglycemia, independent of hypertension, can directly induce aberrant splicing of the CaV1.2 channel in the arteries.
Furthermore, we collected arterial tissues from both diabetic and non-diabetic patients, and found that the proportion of CaV1.2E9* channel was significantly increased in the arteries of diabetic patients. However, the proportion of CaV1.2E8a and CaV1.2E33 remained unchanged (Fig. 3G–I). These results suggest that splice-variants of CaV1.2 channels are aberrantly expressed in diabetic arteries, potentially leading to irregular functions of the CaV1.2 channel.
Expression of Rbfox1 is decreased in diabetic arteries
Rbfox1 is known as a splicing factor that regulates the CaV1.2 alternative exons 9* and 33 during neural development [32], and has also been found to regulate CaV1.2 AS events in hypertensive arteries [33]. To assess the role of Rbfox1 in diabetic arteries, we used Western blotting to detect the expressing levels of Rbfox1 in different rat tissues. We found that Rbfox1 is highly expressed in arterial tissues, including MAs and TAs, compared to brain and cardiac tissues (Fig. 4A, B). Immunofluorescence staining confirmed that Rbfox1 is mainly located in the nucleus of isolated VSMCs and arterial smooth muscle, as stained with α-SMA (Fig. 4C), suggesting that Rbfox1 is an RNA-binding protein. Next, the MAs and TAs of nondiabetic or diabetic rat models were isolated, and Western blotting showed that the expressions of Rbfox1 are remarkably decreased in the arteries from diabetic rat models compared to nondiabetic model (Fig. 4D–F; Fig. S1B). Fluorescence intensity of Rbfox1 in rat VSMCs or tissues also exhibited a downregulation in HFD/STZ rats in comparison to control rats (Fig. S5). Unexpectedly, the arteries from the patients with diabetes showed increased Rbfox1 expression in comparison to those from nondiabetic patients (Fig. S6B, C). We consider this might be due to compensatory effects, though the underlying mechanisms are unknown. Nevertheless, our data indicate that decrease in Rbfox1 might play a crucial role in the regulation of Cacna1c AS events in the diabetic arteries.

Rbfox1 is abundantly expressed in the arterial tissues, but it is downregulated in the arteries from diabetic rats. A Rbfox1 protein was detected via Western blotting in different tissues from rats, with the GAPDH protein serving as an internal control. B Expression levels of Rbfox1 protein were analyzed and presented as a bar chart, ** P < 0.01, 1-way ANOVA followed by a Tukey’s post hoc test. C Immunofluorescence staining was used to determine the localization of Rbfox1 in smooth muscles using anti-Rbfox1 and anti-α-smooth muscle actin in isolated VSMCs or mesenteric arteries (MAs) from rats; the right panel shows the merged images. D MAs and thoracic arteries (TAs) from control or HFD/STZ-treated rats were isolated to determine the expression level of Rbfox1 protein by Western blotting. β-actin was detected as a loading control. The relative Rbfox1 expression levels in the MAs (E) and TAs (F) were analyzed and presented as bar charts comparing HFD/STZ-treated rats to control rats. ** P < 0.01, unpaired t test. n represents the number of rats
GS induces decreased Rbfox1 expression and aberrant CaV1.2 AS in VSMCs and MAs
Since Rbfox1 expression was also reduced in the hypertensive arteries [33], we questioned whether the decreased Rbfox1 in diabetic arteries was due to diabetic hyperglycemia or diabetes-associated hypertension. To evaluate this, we used a cell-based model, treating isolated VSMCs from rat MAs with HG (15 or 25 mmol/L) (Fig. 5A). Unexpectedly, the application of HG did not alter the expression level of Rbfox1 in isolated VSMCs (Fig. 5B), nor the expression of CaV1.2 alternative exon 9* (Fig. 5C) and exon 33 (Fig. 5D). This suggest that HG may not directly affect CaV1.2 AS events of VSMCs in diabetic arteries.

GS, not glucose, decreases Rbfox1 expression in VSMCs and MAs. A Isolated VSMCs were treated with 5.5, 15 or 25 mmol/L d-glucose for 48 h and Western blotting were used to detect the expression of Rbfox1. β-actin was detected as an internal control. B The expression level of Rbfox1 was checked in VSMCs after the application of d-glucose. The proportion of CaV1.2 with alternative exon 9* (C) and exon 33 (D) were detected by RT-PCR. ns indicates no significant differences. 1-way ANOVA followed by a Tukey’s post hoc test. E VSMCs were treated with 20% NG or GS for 48 h and Western blotting was performed to detect the expression of Rbfox1. The proportion of CaV1.2 with alternative exon 9* or exon 33 were detected by RT-PCR. F The relative Rbfox1 expression was normalized in VSMCs after treatments. The values for percent exon 9* (G) or exon 33 inclusion (H) of CaV1.2 channels are presented as bar charts. The ratios between the upper band and the sum of the upper and lower bands present the proportions of CaV1.2 channel with exon 9* or exon 33. I MAs were treated with 20% NG or GS for 48 h and Western blotting was performed to detect the expression of Rbfox1. The proportion of CaV1.2 with exon 9* or exon 33 were detected by RT-PCR. J The relative Rbfox1 expression was normalized to β-actin after treatments. The values for percent exon 9* (K) or exon 33 inclusion (L) of CaV1.2 channels were presented as bar charts. * P < 0.05, ** P < 0.01 vs NG-treated VSMCs and MAs, unpaired t test with or without Welch’s correction
Abnormal vascular functions are known to be driven by AGEs under diabetic hyperglycemic conditions [5]. To explore the role of AGEs in Rbfox1 expression and CaV1.2 AS events, we used GS to mimic AGEs-related modification in diabetes. Isolated VSMCs from rat MAs were treated with 20% GS or NG for 48 h. The expression of Rbfox1 was found to be notably decreased after GS application when compared with NG application (Fig. 5E, F). Significantly, GS treatment increased CaV1.2E9* channels (Fig. 5G) but decreased CaV1.2E33 channels in VSMCs (Fig. 5H). However, mannitol didn’t affect the expression levels of Rbfox1 in VSMCs, eliminating the possible interference of osmotic pressure (Fig. S7A, B). GS decreased Rbfox1 expression (Fig. S7C, D) and regulated CaV1.2 alternative exon 9* in a concentration-dependent manner, but it did not regulate CaV1.2 alternative exon 33 (Fig. S7E–G). CaV1.2 exons 9* and 33 AS events did not change in VSMCs when cultured for 24 or 48 h compared to freshly isolated cells (0 h) (Fig. S8). This indicates the cell culture didn’t alter CaV1.2 AS events. Furthermore, the application of GS in the MAs also induced a reduction in Rbfox1, and increased CaV1.2 exon 9*, but decreased exon 33 (Fig. 5I–L). These results were consistent with the findings that diabetic arteries exhibit decreased Rbfox1 expression and aberrant splicing of the CaV1.2 channel, and imply that GS, not glucose, induces the aberrant splicing of the CaV1.2 channel, which may be related to enhanced channel functions.
In order to eliminate the interference of CaV1.2 phosphorylation induced by GS, we examined the phosphorylation level of CaV1.2 at S1928 in VSMCs treated with GS. Here, we found GS does not increase the expression level of CaV1.2 S1928. However, co-treatment with PKA inhibitor H-89 significantly decreased its expression level (Fig. S9), indicating that Rbfox1-mediated exon splicing is a long-acting mechanism. As the chronic effects of diabetes in enhanced arterial contraction via CaV1.2 had been demonstrated to require phosphorylation at S1928 in a model of STZ-treated mice [14], our observation might be an alternative, compatible mechanism involved in the increased arterial contractility.
Additionally, MGO, a highly reactive dicarbonyl compound that forms AGEs [39], was used to treat VSMCs for 48 h. After that, the expression of Rbfox1 also showed a significant decrease (Fig. S10A, B), while both CaV1.2E9* and CaV1.2E33 channels exhibited an increase when compared with vehicle-treated cells (Fig. S10C, D). These observations further confirmed that the aberrant expressions of Rbfox1 and CaV1.2 AS are indeed due to AGEs.
Overexpression of Rbfox1 reverses aberrant splicing of CaV1.2 channels in GS-treated VSMCs
We then examined the direct effects of Rbfox1 on CaV1.2 AS events. An siRNA approach was used to specifically knockdown the endogenous expression of Rbfox1 in VSMCs (Fig. 6A, B). RT-PCR results showed that the proportion of CaV1.2E9* channel increased (Fig. 6C), but CaV1.2E33 channel decreased (Fig. 6D). Moreover, Rbfox1 siRNA RP in MAs significantly increased CaV1.2E9* channels, but decreased CaV1.2E33 channels (Fig. S11). These results indicated that Rbfox1 could directly regulate AS events of the CaV1.2 channel in VSMCs and in arteries. Furthermore, transfection with Rbfox1 expression plasmids staved off GS-induced Rbfox1 downregulation (Fig. 6E, F), and abolished GS-induced CaV1.2E9* increase (Fig. 6G) and CaV1.2E33 decrease (Fig. 6H), partly reversing the aberrant expressions of exons 9* and 33 induced by GS application in VSMCs. Altogether, these results revealed that the aberrant splicing of CaV1.2 in diabetic arteries is indeed attributed to the decreased Rbfox1.

Rbfox1 dynamically regulates alternative exons 9* and 33 of CaV1.2 channel in VSMCs. A NT or Rbfox1 siRNAs were transfected into isolated VSMCs. After 48 h of culture, the Rbfox1 protein and alternative exon 9* or exon 33 of Cav1.2 were detected by Western blotting and RT-PCR, respectively. B The relative Rbfox1 expression is presented in VSMCs after different treatments. The values for percent exon 9* (C) or exon 33 inclusion (D) are presented as bar charts. * P < 0.05, ** P < 0.01 vs NT siRNA-treated VSMCs, 1-way ANOVA followed by a Tukey’s post hoc test. E VSMCs treated with GS were transfected with Rbfox1 expression plasmids for 48 h, then the Rbfox1 protein and alternative exon 9* or exon 33 of CaV1.2 were detected by Western blotting and RT-PCR, respectively. F Rbfox1 expression level was normalized to β-actin expression in VSMCs after GS treatment. The percentages of exon 9* (G) or exon 33 inclusion (H) are shown as bar charts. The ratios between the upper band and the sum of the upper and lower bands present the proportions of CaV1.2 channel with exon 9* or exon 33. * P < 0.05, ** P < 0.01 vs control, ## P < 0.01 vs GS-treated VSMCs, 1-way ANOVA followed by a Tukey’s post hoc test. n represents the number of cells
GS hyperpolarizes CaV1.2 I–V curve in VSMCs
To determinate whether AGEs affect the functions of vascular CaV1.2 channels, isolated VSMCs from rat aortas were treated with 20% GS or NG for 24–48 h. Then, CaV1.2 channel currents were recorded upon increasing step voltages using a whole-cell patch clamp. As indicated in Fig. 7A and B, GS-treated VSMCs showed an obviously left-shifted I–V relationship curve compared to NG-treated cells (Table S6). However, their current densities of CaV1.2 channels showed no significant differences (Fig. 7C). As the phosphorylation of CaV1.2 α1C at S1928 caused by PKA could increase CaV1.2 currents of VSMCs in diabetic arteries [12], we used PKA specific inhibitor H-89 to block the PKA-mediated phosphorylation of CaV1.2 channels. The data showed that H-89 application didn’t affect Rbfox1 expression and CaV1.2 alternative exon 9* and exon 33 (Fig. S12), and couldn’t restore the left-shifted CaV1.2 I–V relationship curve in GS-treated VSMCs (Fig. 7A, B), indicating PKA signaling doesn’t mediate the CaV1.2 channel kinetics changes in diabetic arteries. GS application increased the cell capacitance of VSMCs (Fig. S3B). Similar results were observed in the MGO-treated VSMCs as shown in Fig. S10E–G and Table S7, but the cell capacitance remained unchanged (Fig. S3C). These observations indicated that AGEs could hyperpolarize CaV1.2 I–V curve in VSMCs, which is not associated with PKA-mediated protein phosphorylation.

Treatment with GS and knockdown of Rbfox1 lead to hyperpolarization of CaV1.2 I–V curve in VSMCs. A CaV1.2 channel currents were recorded under the testing potentials, increasing from − 50 to 50 mV (10-mV increase each step, I–V protocol) in aortic VSMCs when using 10 mmol/L Ba2+ as charger carrier. Representative traces of CaV1.2 currents were presented after NG or GS treatment for 48 h. Additionally, 50 μmol/L PKA inhibitor H-89 was administrated 4 h before the recording in GS-treated VSMCs. B Plots of CaV1.2 I–V curve of VSMCs were analyzed after treating with NG, GS or GS plus H-89. C Current densities of CaV1.2 channel were calculated by dividing currents by cell capacitance (Cm) of the VSMCs after applying with NG, GS or GS plus H-89. D Representative traces of CaV1.2 currents under I–V protocol were shown in NT or Rbfox1 siRNA-transfected VSMCs. E Plots of CaV1.2 I–V curve of VSMCs were analyzed and fitted with the equation. F Current densities of CaV1.2 channel were calculated by dividing by cell capacitance after applying with NT or Rbfox1 siRNAs in VSMCs. ns indicates no significant differences. n represents the number of cells
To explore the electrophysiological relevance of Rbfox1 in CaV1.2 channel, we used siRNAs to knockdown Rbfox1 in isolated VSMCs. Rbfox1 knockdown obviously left-shifted the I–V curve in comparison to NT siRNA-treated VSMCs (Fig. 7D, E; Table S8). However, the cell capacitance (Fig. S3D) and current densities (Fig. 7F) of CaV1.2 channels after Rbfox1 knockdown remained unchanged, indicating that decreased Rbfox1 could directly hyperpolarize CaV1.2 channel I–V kinetics without affecting its current size in VSMCs. Thus, it is reasonable to conclude that the GS-induced hyperpolarized CaV1.2 I–V curve is due to decreased Rbfox1 in VSMCs, and the hyperpolarized I–V and activation curve of CaV1.2 channels in VSMCs from rat diabetic arteries is attributed to decreased Rbfox1 expression.
GS increases K+-induced [Ca2+]i and vascular constriction
Ca2+ influx via the CaV1.2 calcium channel triggers the vasoconstriction and maintain the myogenic tone. Hyperglycemia or GS treatment can induce the hyperpolarization of vascular CaV1.2 I–V kinetics, making the channels open more easily from resting membrane potential, which might increase the [Ca2+]i in VSMCs. To test this possibility, we incubated VSMCs with Fluo-4 AM, and applied 60 mmol/L KCl to trigger the elevation of [Ca2+]i (Fig. 8A). Co-incubation with 20% GS could increase [Ca2+]i in VSMCs. Though the current size of CaV1.2 channel was decreased in VSMCs from HFD/STZ-treated rats (Fig. 2C), the present data supported the result that GS could hyperpolarize CaV1.2 I–V curve, which could also increase the [Ca2+]i in VSMCs (Fig. 8B). To confirm that the [Ca2+]i increase in GS-treated vessels indeed represents a larger Ca2+ influx and is not due to differences in F0, we pretreated the isolated VSMCs with nifedipine, the results showed that application with 10−6 mol/L nifedipine could significantly block the GS-induced increase in [Ca2+]i, indicating the larger [Ca2+]i increase in GS-treated VSMCs is indeed trigged by CaV1.2-mediated Ca2+ influx (Fig. S13).

Application with GS elevates K+-induced [Ca2+]i of VSMCs and enhances vasoconstriction. A VSMCs were treated with 20% NG or GS for 48 h, and [Ca2+]i triggered by 60 mmol/L KCl was measured using Ca2+ fluorescence indicator Fluo-4 AM. The fluorescent intensity was monitored using a time series scanning mode under a confocal microscope. B Δ[Ca2+]i was presented as ΔF/F0 and shown as a line diagram, * P < 0.05, ** P < 0.01 vs NG-treated VSMCs, 2-way ANOVA followed by Sidak’s multiple comparisons. n is the number of cells from at least 3 rats. C Isolated mesenteric arteries were cultured with 20% NG or GS for 48 then the force tension induced by increasing extracellular K+ concentration was recorded using vascular myograph. D Plots of concentration-tension relationship were represented and fitted with the Boltzmann equation for NG or GS-treated arteries. ** P < 0.01 vs NG-treated arteries, 2-way ANOVA followed by Sidak’s multiple comparisons. n represents the number of rats
We next examined the effects of GS application on the vasoconstriction of small arteries. MAs were isolated from the rats and cultured with 20% GS or NG for 48 h. Subsequently, increasing concentrations of KCl were applied to induce vasoconstriction (Fig. 8C). In comparison to NG-treated arteries, GS application significantly increased K+-induced vasoconstriction in MAs (Fig. 8D). However, applying the PKA inhibitor H-89 didn’t change the vascular tension of GS-treated MAs (Fig. S14), indicating that PKA signaling doesn’t mediate GS-induced vasoconstriction. Combined with the finding that GS did not increase the level of CaV1.2S1928 phosphorylation, we confirmed that Rbfox1-mediated CaV1.2 AS is involved in the long-acting mechanisms regulating vasoconstriction. Moreover, we introduced Rbfox1 expression plasmids into isolated MAs from GK rats, and found that the enhanced vasoconstriction in diabetic rats was restored (Fig. S1C, D). Our previous finding showed that the knockdown of Rbfox1 increased K+-induced vasoconstriction [33]. Together, these results indicated that GS application can directly enhance the vascular constriction, which is thought to be mediated by aberrant splicing of CaV1.2 channels induced by decreased Rbfox1.
Discussion
The CaV1.2 channel is indispensable for the excitation–contraction coupling of vascular smooth muscle [40], and its abnormal functions are tightly associated with increased vasotone [41, 42]. The CaV1.2 channel is regulated by many post-transcriptional modulations, including AS [43]. We previously discovered aberrant AS events of the CaV1.2 channel under cardiovascular pathological conditions, such as cardiac hypertrophy [26] and hypertension [24, 28, 33]. In this study, we present, for the first time to our knowledge, that AS of vascular CaV1.2 channel is dysregulated under hyperglycemic condition. This dysregulation facilitates channel function by hyperpolarizing the CaV1.2 channel activation kinetics, leading to persistently enhanced vasoconstriction of diabetic arteries (Fig. 9). Since most of our cell-based experiments were performed in the cultured cells, and AS of vascular CaV1.2 channel may play an important role in the proliferation of smooth muscle cells triggered by calcium influx. Therefore, this AS modulation in CaV1.2 channels might also present in proliferation, not only in contractility of vascular smooth muscle, which needs further investigations. Nevertheless, our findings provide a new perspective on the precise regulatory mechanism of vascular CaV1.2 channel activity in response to diabetic hyperglycemia.

Illustration of main findings in the present study. Schematic diagram illustrates the acute and long-term mechanisms that regulate vascular CaV1.2 functions and vasoconstriction under diabetic hyperglycemia. For the long-lasting mechanism, AGEs significantly decrease Rbfox1 expression, and enhance the function of vascular CaV1.2 channel by dynamically regulating alternative exons 9* and 33, thereby inducing persistent vasoconstriction during diabetic hyperglycemia. NUC, nucleotide; RAGE, receptor for advanced glycation end-product; VSMC, vascular smooth muscle cell
Dysfunctions of smooth muscle are recognized to play significant roles in diabetic vascular complication [44]. In vivo measurements in diabetic animal models have shown increased vasotone [13]. Here, we observed that isolated arteries from HFD/STZ-treated diabetic rats and type 2 diabetic GK rats exhibited persistent constriction in response to K+, reinforcing the correlation between enhanced vasoconstriction and vascular smooth muscle in diabetes. Moreover, GS treatment appeared to induce elevated K+-triggered vasoconstriction, which is believed to be mediated by the long-term regulatory mechanism in the arteries.
The abnormal expression and functioning of the CaV1.2 channel in arteries is associated with enhanced vasotone, which is a crucial factor in the development of hypertension [28, 33]. Under the conditions of diabetic hyperglycemia, the CaV1.2 channel currents are known to increase in response to elevated extracellular glucose. This is dependent on the phosphorylation of CaV1.2S1928, which is mediated by AKAP5/P2Y11/AC5/PKA signaling, thus leading to an increase in vasotone [12, 13, 45, 46]. Considering that phosphorylation process of threonine and serine amino acids required up to dozens of minutes [47], increased CaV1.2 channel function responded to hyperglycemia might be an acute effect. In this work, we found that long-term incubation with GS did not increase the level of CaV1.2S1928 phosphorylation, indicating that facilitated function of vascular CaV1.2 channel is mainly because of Rbfox1-mediated alternative splicing, not PKA-mediated S1928 phosphorylation. Also, we found that the I–V and steady-state activation curve of CaV1.2 channels shifted towards the left (hyperpolarized) in HFD/STZ-treated rat VSMCs. Moreover, GS treatment directly hyperpolarized the CaV1.2 I–V curve in isolated VSMCs, which was independent of PKA signaling, indicating a long-term modulation mechanism in regulating the function of CaV1.2 channel under diabetic hyperglycemia. Therefore, we proposed that the dual mechanisms (acute and long-term) for the regulation of vascular CaV1.2 functions might finely adjust the acute and persistent vasoconstriction of small arteries in response to hyperglycemia, respectively.
Under diabetic hyperglycemia, the vascular CaV1.2 channel shows enhanced function by hyperpolarizing the channel activation kinetics. The CaV1.2 alternative exon 9* increases aberrantly, while exon 33 decreases, generating the upregulation of a specific CaV1.2 isoform, the CaV1.2E9*,ΔE33 channel. We previously found that the electrophysiological properties of the CaV1.2E9*,ΔE33 isoform, when heterologously expressed in human embryonic kidney-293 cells, show hyperpolarized I–V and steady-state activation curves compared to the CaV1.2ΔE9*,E33 isoform [28]. This hyperpolarized CaV1.2 I–V and steady-state activation leads to the channel opening much more easily from the resting membrane potential. Therefore, we conclude that the facilitated function of the vascular CaV1.2 channel is truly attributed to the upregulation of CaV1.2E9*,ΔE33 isoform under diabetic hyperglycemia. As HG could depolarize the membrane potential of VSMCs [14], this may further augment the functions of vascular CaV1.2 channels due to the upregulated CaV1.2E9*,ΔE33 isoforms in diabetes. The electrophysiological properties of CaV1.2 channel differentiate between diabetic rats and VSMCs in vitro. This discrepancies in CaV1.2 channel activity could be associated with the specific animal model and concurrent metabolic abnormalities, such as hypercholesterolemia and hyperinsulinemia, which could affect channel functions [48]. GS and Rbfox1 knockdown did not induce any change in current density, thus we speculated that reduced current densities in smooth muscle cells from HFD/STZ-induced diabetic rats may be attributed to other metabolic abnormalities, such as hypercholesterolemia or hyperinsulinemia. On the other hand, the 48-h serum culture may also have resulted in a phenotypic switch towards a non-contractile/synthetic phenotype. This shift could potentially account for the observed lack of changes in CaV1.2 channel current.
Splicing factors produce different actions by binding to different positions on pre-mRNA (position-dependent) [49]. Several UGCAUG elements surround exon 9* and exon 33 of Cacna1c gene. Rbfox proteins prefer binding upstream of exon 9* to repress its expression, but downstream of exon 33 to enhance its expression [32]. Here, we found that alternative exon 9* is increased, but exon 33 is decreased. This is accompanied by reduced Rbfox1 expression in the arteries of HFD/STZ-induced and GK rats. Moreover, Rbfox1 knockdown in isolated VSMCs directly induces the upregulation of exon 9* and downregulation of exon 33. Although we cannot exclude the possibility that Rbfox1 mediates the AS events of other target genes [50,51,52] in diabetic arteries, our observations are sufficient to conclude that decreased Rbfox1 in VSMCs indeed induces aberrant expression of the vascular CaV1.2E9*,ΔE33 channel under diabetic hyperglycemia, which in turn enhances vasoconstriction. Notably, several variants of RBFOX1 gene are linked to lower blood pressure in humans [53, 54]. Whether these variants influence Rbfox1-mediated CaV1.2 AS in diabetic arteries needs further studies.
The different patterns of increased CaV1.2 exon 33 induced by MGO and decreased exon 33 induced by diabetes and GS suggest that potential factors may modulate the expression of CaV1.2 exon 33 compare with exon 9*. Although we confirmed the important role of Rbfox1 in regulating CaV1.2 functions and vasoconstriction under diabetic hyperglycemia in the present study, we also found another splicing factor, Rbfox2, dynamically regulating CaV1.2 exons 9* and 33 in VSMCs [28]. Besides Rbfox proteins, other splicing factors such as RNA-binding protein motif 20 (RBM20) has also been reported to regulate CaV1.2 alternative exon 8, exon 9*, exon 22 and exon 33, but the magnitude of this regulation is weak [55]. It is not clear whether other splicing factors specifically control the expression of CaV1.2 exon 33, but this requires further clarification.
We initially hypothesized that HG in diabetic condition may mediate the decreased expression of Rbfox1 in the arteries from diabetic rats. Contrary to our expectations, HG didn’t directly affect Rbfox1 expression and the CaV1.2 splicing pattern in VSMCs. AGEs are produced during chronic hyperglycemic condition, causing long-lasting damage to vascular complications through metabolic memory [15]. Treatment with GS, an AGEs mimic, significantly decreased the expression of Rbfox1 and increased the presence of the CaV1.2E9*,ΔE33 isoform in isolated VSMCs. Functionally, the application of GS shifted the CaV1.2 I–V curve towards hyperpolarization in isolated VSMCs, enhancing channel function and increasing K+-induced vasoconstriction. These observations confirm that AGEs can induce a reduction in Rbfox1 protein through metabolic memory, resulting in aberrant splicing and function of the vascular CaV1.2 channel under diabetic hyperglycemia.
Conclusions
In summary, our research shows that AGEs can significantly reduce Rbfox1 expression and enhance the function of vascular CaV1.2 channel by dynamically regulating alternative exons 9* and 33, thereby increasing vasoconstriction in the arteries of diabetic rat. Therefore, targeting Rbfox1 to correct the aberrant splicing of the CaV1.2 channel in vascular smooth muscle may provide a promising approach for the management of diabetic vascular complications.
Data availability
The data sets generated and/or analyzed during this study are available from the corresponding author upon reasonable request.
References
International Diabetes Federation (2021) IDF diabetes atlas 2021. https://diabetesatlas.org/atlas/tenth-edition/. Updated 05 April 2021. Accessed 05 Nov 2021
Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A (2014) Role of advanced glycation end products in cellular signaling. Redox Biol 2:411–429
Raghavan S, Vassy JL, Ho YL, Song RJ, Gagnon DR, Cho K, Wilson PWF, Phillips LS (2019) Diabetes mellitus-related all-cause and cardiovascular mortality in a national cohort of adults. J Am Heart Assoc 8:e011295
Newman JD, Schwartzbard AZ, Weintraub HS, Goldberg IJ, Berger JS (2017) Primary prevention of cardiovascular disease in diabetes mellitus. J Am Coll Cardiol 70:883–893
Matsumoto T, Taguchi K, Kobayashi T (2021) Relationships between advanced glycation end products (AGEs), vasoactive substances, and vascular function. J Smooth Muscle Res 57:94–107
Tousoulis D, Papageorgiou N, Androulakis E, Siasos G, Latsios G, Tentolouris K, Stefanadis C (2013) Diabetes mellitus-associated vascular impairment: novel circulating biomarkers and therapeutic approaches. J Am Coll Cardiol 62:667–676
Meza CA, La Favor JD, Kim DH, Hickner RC (2019) Endothelial dysfunction: is there a hyperglycemia-induced imbalance of NOX and NOS? Int J Mol Sci 20(15):3775
Montero D, Walther G, Perez-Martin A, Vicente-Salar N, Roche E, Vinet A (2013) Vascular smooth muscle function in type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetologia 56:2122–2133
Nieves-Cintron M, Syed AU, Buonarati OR, Rigor RR, Nystoriak MA, Ghosh D, Sasse KC, Ward SM, Santana LF, Hell JW et al (2017) Impaired BKCa channel function in native vascular smooth muscle from humans with type 2 diabetes. Sci Rep 7:14058
Nieves-Cintron M, Syed AU, Nystoriak MA, Navedo MF (2018) Regulation of voltage-gated potassium channels in vascular smooth muscle during hypertension and metabolic disorders. Microcirculation 25:10
Fleischmann BK, Murray RK, Kotlikoff MI (1994) Voltage window for sustained elevation of cytosolic calcium in smooth muscle cells. Proc Natl Acad Sci USA 91:11914–11918
Nystoriak MA, Nieves-Cintron M, Patriarchi T, Buonarati OR, Prada MP, Morotti S, Grandi E, Fernandes JD, Forbush K, Hofmann F et al (2017) Ser 1928 phosphorylation by PKA stimulates the L-type Ca2+ channel CaV1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci Signal 10:eaaf9647
Syed AU, Reddy GR, Ghosh D, Prada MP, Nystoriak MA, Morotti S, Grandi E, Sirish P, Chiamvimonvat N, Hell JW et al (2019) Adenylyl cyclase 5-generated cAMP controls cerebral vascular reactivity during diabetic hyperglycemia. J Clin Invest 129:3140–3152
Martin-Aragon Baudel M, Flores-Tamez VA, Hong J, Reddy GR, Maillard P, Burns AE, Man KNM, Sasse KC, Ward SM, Catterall WA et al (2022) Spatiotemporal control of vascular CaV1.2 by alpha1C S1928 phosphorylation. Circ Res 131:1018–1033
Aschner PJ, Ruiz AJ (2012) Metabolic memory for vascular disease in diabetes. Diabetes Technol Ther 14(Suppl 1):S68–S74
Yao Y, Song Q, Hu C, Da X, Yu Y, He Z, Xu C, Chen Q, Wang QK (2022) Endothelial cell metabolic memory causes cardiovascular dysfunction in diabetes. Cardiovasc Res 118:196–211
Maranta F, Cianfanelli L, Cianflone D (2021) Glycaemic control and vascular complications in diabetes mellitus type 2. Adv Exp Med Biol 1307:129–152
Bebu I, Braffett BH, de Boer IH, Aiello LP, Bantle JP, Lorenzi GM, Herman WH, Gubitosi-Klug RA, Perkins BA, Lachin JM et al (2023) Relationships between the cumulative incidences of long-term complications in type 1 diabetes: the DCCT/EDIC study. Diabetes Care 46:361–368
Marasco LE, Kornblihtt AR (2023) The physiology of alternative splicing. Nat Rev Mol Cell Biol 24:242–254
Rogalska ME, Vivori C, Valcarcel J (2023) Regulation of pre-mRNA splicing: roles in physiology and disease, and therapeutic prospects. Nat Rev Genet 24:251–269
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40:1413–1415
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456:470–476
Tang ZZ, Liang MC, Lu S, Yu D, Yu CY, Yue DT, Soong TW (2004) Transcript scanning reveals novel and extensive splice variations in human L-type voltage-gated calcium channel, Cav1.2 alpha1 subunit. J Biol Chem 279:44335–44343
Lei J, Liu X, Song M, Zhou Y, Fan J, Shen X, Xu X, Kapoor I, Zhu G, Wang J (2020) Aberrant exon 8/8a splicing by downregulated PTBP (polypyrimidine tract-binding protein) 1 increases CaV1.2 dihydropyridine resistance to attenuate vasodilation. Arterioscler Thromb Vasc Biol 40:2440–2453
Wang J, Thio SS, Yang SS, Yu D, Yu CY, Wong YP, Liao P, Li S, Soong TW (2011) Splice variant specific modulation of CaV1.2 calcium channel by galectin-1 regulates arterial constriction. Circ Res 109:1250–1258
Fan J, Fan W, Lei J, Zhou Y, Xu H, Kapoor I, Zhu G, Wang J (2019) Galectin-1 attenuates cardiomyocyte hypertrophy through splice-variant specific modulation of CaV1.2 calcium channel. Biochim Biophys Acta Mol Basis Dis 1865:218–229
Li G, Wang J, Liao P, Bartels P, Zhang H, Yu D, Liang MC, Poh KK, Yu CY, Jiang F et al (2017) Exclusion of alternative exon 33 of CaV1.2 calcium channels in heart is proarrhythmogenic. Proc Natl Acad Sci USA 114:E4288–E4295
Zhou Y, Fan J, Zhu H, Ji L, Fan W, Kapoor I, Wang Y, Wang Y, Zhu G, Wang J (2017) Aberrant splicing induced by dysregulated Rbfox2 produces enhanced function of CaV1.2 calcium channel and vascular myogenic tone in hypertension. Hypertension 70:1183–1192
Hu Z, Liang MC, Soong TW (2017) Alternative splicing of L-type CaV12 calcium channels: implications in cardiovascular diseases. Genes 8(12):344
Zhang C, Zhang Z, Castle J, Sun S, Johnson J, Krainer AR, Zhang MQ (2008) Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev 22:2550–2563
Kim KK, Adelstein RS, Kawamoto S (2009) Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J Biol Chem 284:31052–31061
Tang ZZ, Zheng S, Nikolic J, Black DL (2009) Developmental control of CaV1.2 L-type calcium channel splicing by Fox proteins. Mol Cell Biol 29:4757–4765
Song M, Hou W, Mustafa AU, Li P, Lei J, Zhou Y, Ji L, Sun Y, Zhou H, Xu Y, Wang J (2022) Diminished Rbfox1 increases vascular constriction by dynamically regulating alternative splicing of CaV1.2 calcium channel in hypertension. Clin Sci 136:803–817
Srinivasan K, Viswanad B, Asrat L, Kaul CL, Ramarao P (2005) Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res 52:313–320
Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T, Jerums G, Cooper ME (2001) Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J Clin Invest 108:1853–1863
Li Y, Zhang T, Huang Q, Sun Y, Chang X, Zhang H, Zhu Y, Han X (2016) Inhibition of tumor suppressor p53 preserves glycation-serum induced pancreatic beta-cell demise. Endocrine 54:383–395
Witczak CA, Wamhoff BR, Sturek M (2006) Exercise training prevents Ca2+ dysregulation in coronary smooth muscle from diabetic dyslipidemic yucatan swine. J Appl Physiol 101:752–762
Tiwari S, Zhang Y, Heller J, Abernethy DR, Soldatov NM (2006) Atherosclerosis-related molecular alteration of the human CaV1.2 calcium channel alpha1C subunit. Proc Natl Acad Sci USA 103:17024–17029
Schalkwijk CG, Stehouwer CDA (2020) Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age-related diseases. Physiol Rev 100:407–461
Hofmann F, Flockerzi V, Kahl S, Wegener JW (2014) L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev 94:303–326
Ohya Y, Abe I, Fujii K, Takata Y, Fujishima M (1993) Voltage-dependent Ca2+ channels in resistance arteries from spontaneously hypertensive rats. Circ Res 73:1090–1099
Ohya Y, Tsuchihashi T, Kagiyama S, Abe I, Fujishima M (1998) Single L-type calcium channels in smooth muscle cells from resistance arteries of spontaneously hypertensive rats. Hypertension 31:1125–1129
Liao P, Yong TF, Liang MC, Yue DT, Soong TW (2005) Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovasc Res 68:197–203
Stern C, Schreier B, Nolze A, Rabe S, Mildenberger S, Gekle M (2020) Knockout of vascular smooth muscle EGF receptor in a mouse model prevents obesity-induced vascular dysfunction and renal damage in vivo. Diabetologia 63:2218–2234
Prada MP, Syed AU, Buonarati OR, Reddy GR, Nystoriak MA, Ghosh D, Simo S, Sato D, Sasse KC, Ward SM et al (2019) A G(s)-coupled purinergic receptor boosts Ca2+ influx and vascular contractility during diabetic hyperglycemia. Elife 8:e42214
Prada MP, Syed AU, Reddy GR, Martin-Aragon Baudel M, Flores-Tamez VA, Sasse KC, Ward SM, Sirish P, Chiamvimonvat N, Bartels P et al (2020) AKAP5 complex facilitates purinergic modulation of vascular L-type Ca2+ channel CaV1.2. Nat Commun 11(1):5303
Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127:635–648
Nieves-Cintron M, Flores-Tamez VA, Le T, Baudel MMA, Navedo MF (2021) Cellular and molecular effects of hyperglycemia on ion channels in vascular smooth muscle. Cell Mol Life Sci 78:31–61
Ule J, Blencowe BJ (2019) Alternative splicing regulatory networks: functions, mechanisms, and evolution. Mol Cell 76:329–345
Williams AL, Walton CB, Pinell B, Khadka VS, Dunn B, Lee K, Anagaran MCT, Avelar A, Shohet RV (2021) Ischemic heart injury leads to HIF1-dependent differential splicing of CaMK2gamma. Sci Rep 11:13116
Shen F, Xu X, Yu Z, Li H, Shen H, Li X, Shen M, Chen G (2021) Rbfox-1 contributes to CaMKIIalpha expression and intracerebral hemorrhage-induced secondary brain injury via blocking micro-RNA-124. J Cereb Blood Flow Metab 41:530–545
Wamsley B, Jaglin XH, Favuzzi E, Quattrocolo G, Nigro MJ, Yusuf N, Khodadadi-Jamayran A, Rudy B, Fishell G (2018) Rbfox1 mediates cell-type-specific splicing in cortical interneurons. Neuron 100:846–859
He KY, Li X, Kelly TN, Liang J, Cade BE, Assimes TL, Becker LC, Beitelshees AL, Bress AP, Chang YC et al (2019) Leveraging linkage evidence to identify low-frequency and rare variants on 16p13 associated with blood pressure using TOPMed whole genome sequencing data. Hum Genet 138:199–210
He KY, Wang H, Cade BE, Nandakumar P, Giri A, Ware EB, Haessler J, Liang J, Smith JA, Franceschini N et al (2017) Rare variants in fox-1 homolog A (RBFOX1) are associated with lower blood pressure. PLoS Genet 13:e1006678
Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li SJ, Parrish A et al (2012) RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 18:766–773
Acknowledgements
We would like to thank Professor Douglas L. Black from Department of Microbiology, Immunology, and Molecular Genetics, UCLA for the gift of Rbfox1 expression plasmids.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82270416, 82270483), Scientific Research and Practice Innovation Program of Jiangsu Province (KYCX21_1589), Qing Lan Project of Jiangsu Province (to J.W.) and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Contributions
JW contributed to the study conceptualization, methodology, review and editing of the manuscript, visualization, supervision, and funding acquisition. HW, SY, PL, TC, DQ, LZ, AM, CL, and MS contributed to the study methodology and investigation. HW contributed to the writing the original draft. LZ contributed to the revision of the article. CQ contributed to the resources. XX contributed to the study conceptualization, methodology, reviewing and editing the manuscript, and funding acquisition. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declared no conflict of interest in the present work.
Consent to participate
Human samples were collected from patients undergoing surgeries at the Affiliated Brain Hospital of Nanjing Medical University, which was approved by the Institutional Review Board of Nanjing Medical University (Reference No. 2022/550), and each patient signed a written informed consent document.
Consent to publish
Not applicable.
Ethics approval
All animal works were performed in line with the principles of the Declaration of Helsinki and have been approved by and performed in accordance with the Institutional Animal Care and Use Committee of Nanjing Medical University and conformed to the Guide for the Care and Use of Laboratory Animals (NIH publication, 8th edition, 2011).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hou, W., Yin, S., Li, P. et al. Aberrant splicing of CaV1.2 calcium channel induced by decreased Rbfox1 enhances arterial constriction during diabetic hyperglycemia. Cell. Mol. Life Sci. 81, 164 (2024). https://doi.org/10.1007/s00018-024-05198-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05198-z