
Abstract
Loss of GLI-Similar 3 (GLIS3) function in mice and humans causes congenital hypothyroidism (CH). In this study, we demonstrate that GLIS3 protein is first detectable at E15.5 of murine thyroid development, a time at which GLIS3 target genes, such as Slc5a5 (Nis), become expressed. This, together with observations showing that ubiquitous Glis3KO mice do not display major changes in prenatal thyroid gland morphology, indicated that CH in Glis3KO mice is due to dyshormonogenesis rather than thyroid dysgenesis. Analysis of GLIS3 in postnatal thyroid suggested a link between GLIS3 protein expression and blood TSH levels. This was supported by data showing that treatment with TSH, cAMP, or adenylyl cyclase activators or expression of constitutively active PKA enhanced GLIS3 protein stability and transcriptional activity, indicating that GLIS3 activity is regulated at least in part by TSH/TSHR-mediated activation of PKA. The TSH-dependent increase in GLIS3 transcriptional activity would be critical for the induction of GLIS3 target gene expression, including several thyroid hormone (TH) biosynthetic genes, in thyroid follicular cells of mice fed a low iodine diet (LID) when blood TSH levels are highly elevated. Like TH biosynthetic genes, the expression of cell cycle genes is suppressed in ubiquitous Glis3KO mice fed a LID; however, in thyroid-specific Glis3 knockout mice, the expression of cell cycle genes was not repressed, in contrast to TH biosynthetic genes. This indicated that the inhibition of cell cycle genes in ubiquitous Glis3KO mice is dependent on changes in gene expression in GLIS3 target tissues other than the thyroid.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
CH is one of the most common neonatal endocrine disorders that has been subclassified into thyroid dysgenesis caused by abnormal thyroid development and dyshormonogenesis caused by defects in thyroid hormone (TH; T3 and T4) biosynthesis [1,2,3,4,5,6]. Abnormal thyroid gland organogenesis accounts for 80–85% of patients with primary CH. During embryonic thyroid development, foregut endoderm cells give rise to thyroid progenitors, which subsequently differentiate along the follicular cell lineage leading to the formation of the TH-producing thyroid follicles [7, 8]. Several transcription factors (TFs) that are expressed early during embryonic thyroid development, including paired box 8 (PAX8), NK2 homeobox 1 (NKX2.1; TTF1), NKX2.5, forkhead box E1 (FOXE1; TTF2), and hematopoietically expressed homeobox (HHEX), play a critical role in the regulation of thyroid gland development [7]. Loss-of-function mutations in these genes cause thyroid dysgenesis [3, 6, 9, 10]. Mutations in genes critical for TH biosynthesis, including sodium iodide symporter (NIS; SLC5A5), pendrin (PDS; SLC26A4), thyroglobulin (TG), thyroperoxidase (TPO), thyroid stimulating hormone receptor (TSHR), and dual oxidase 2 (DUOX2), have been causally linked to thyroid dyshormonogenesis [11,12,13].
The Krüppel-like zinc finger transcription factor, GLI-Similar 3 (GLIS3), plays a critical role in the regulation of many biological processes, including thyroid follicular cell functions, and has been implicated in several pathologies [14, 15]. Loss-of-function mutations in the GLIS3 gene are recessive. Biallelic loss-of-function mutations in human GLIS3 leads to a multi-organ phenotype that includes congenital hypothyroidism [16,17,18,19,20,21,22,23,24], while monoallelic, as well as biallelic, single nucleotide polymorphisms in GLIS3 are associated with an increased risk of thyroid dysfunction and CH [25,26,27,28,29,30,31,32]. Ubiquitous Glis3 knockout (Glis3KO) mice exhibit a very similar phenotype as human patients with GLIS3 deficiency, including the development of neonatal diabetes and CH [33, 34].
The development of CH in Glis3KO mice was at least in part due to transcriptional repression of several TH biosynthesis-related genes suggesting that CH in Glis3KO mice is related to dyshormonogenesis [15, 33]. To follow-up on this study [33], we examined whether GLIS3 also plays a role in the regulation of murine thyroid gland development and whether GLIS3 transcriptional activity is modulated by TSH signaling by analyzing the expression of GLIS3 protein during pre- and postnatal murine thyroid gland. These analyses showed that the development of CH in Glis3KO mice is due to dyshormonogenesis rather than thyroid dysgenesis and further indicated a connection between GLIS3 protein and the TSH–TSHR signaling pathway. It is well established that TSH binding to its receptor activates several kinases that subsequently lead to the induction of gene expression, including genes involved in TH biosynthesis and cell proliferation. We provide evidence showing that TSH and activation of protein kinase A (PKA) enhance the transcriptional activity of GLIS3. We propose that the induction of several TH biosynthesis-related genes by TSH, particularly in mice fed a LID, is in part related to the increase in GLIS3 transcriptional activity [14, 15, 35]. Although, TSH also induces the expression of cell cycle-regulatory genes, our study shows that, unlike TH biosynthesis-related genes, they are not directly regulated by GLIS3 but indirectly through changes in gene expression in other GLIS3 target tissues.
Materials and methods
Mice and diet
Glis3-EGFP mice (C57BL/6-Glis3 < tm3(Glis3-EGFP)Amj >) expressing a GLIS3-EGFP fusion protein, and the ubiquitous Glis3-deficient Glis3KO1 (B6.Glis3 < tm1AmJ >) and Glis3KO2 (C57BL/6-Glis3 < m3(mCherry)Amj >) mice were described previously [33, 36,37,38]. Both mouse strains develop neonatal diabetes and CH. Conditional Glis3 knockout mice, Glis3-Pax8Cre (CKO), in which the expression of Cre causes a cell type-specific deletion of exon 5 encoding zinc finger 3 and 4 of GLIS3, were generated by crossing Glis3fl/fl mice [39] with Pax8Cre mice (B6.129P2(Cg)-Pax8tm1.1(cre)Mbu/J; Jackson Laboratory # 028196). Mice were routinely fed an NIH-31 diet (ND; Harlan, Madison, WI). For diet study, 4-week-old WT and Glis3-Pax8Cre mice were fed ND or a low-iodine diet (LID; TD95125 diet, Harlan) for 6 days.
Histology and immunohistochemistry
For histological analysis, thyroid glands from different stages of mouse embryonic development and from postnatal mice were fixed overnight in 4% paraformaldehyde, washed with PBS, and embedded in paraffin. Sections (5 μm) were then stained with hematoxylin and eosin (H&E). For immunohistochemical analysis, fixed thyroid glands were washed, transferred into 30% sucrose for 2–3 days, and then frozen in OCT compound (Tissue-Tek). Frozen Sects. (10 μm) were obtained using a cryostat (Leica Biosystems, Deer Park, IL) and subsequently immunostained with the antibodies indicated. A list of primary and secondary antibodies is shown in Supplementary Table 1. Cell membranes were stained with Alexa Fluor 647-conjugated wheat germ agglutinin (WGA) and F-actin with Alex Fluor 594-conjugated phalloidin. Nuclei were stained with DAPI Prolong Diamond (ThermoFisher). Proliferation of thyroid follicular cells was analyzed by Click-iT EdU assay kit (ThermoFisher) as described previously [33]. Fluorescence was examined with a Zeiss LSM780 confocal microscope. Metamorph Premier Offline version 7.10.3.279 (Molecular Devices LLC, Sunnyvale, CA) was used to quantify GLIS3 and PAX8 positive cells. A median filter was applied to the red and green channels and Multi Wavelength Cell Scoring was used to count the total number of red cells and double positive cells.
Measurement of serum and tissue TSH and TH levels
Serum T3, T4, and TSH levels were measured by radioimmunoassay as described in detail previously [33, 40].
RNA-Seq analysis
Four-week-old WT (n = 4) and Glis3-Pax8Cre (n = 4) mice were fed an ND or LID for 6 days before thyroid glands were collected and RNA isolated with a RNAqueous-Micro total RNA isolation kit (ThermoFisher). mRNA isolation and library generation were carried out with a TruSeq Stranded mRNA and TruSeq RNA Library preparation kits (Illumina Inc., San Diego, CA), respectively. Sequences were read by paired-end sequencing using a NextSeq500 or NovaSeq 6000 Sequencing System (Illumina). Raw reads pairs were filtered to retain only those with average base quality score > 20 for both read ends. Filtered read pairs were mapped to the mm10 reference assembly via STAR v2.5 [41] with parameters "–outSAMattrIHstart 0 –outFilterType BySJout –alignSJoverhangMin 8 –limitBAMsortRAM 55000000000 –outSAMstrandField intronMotif –outFilterIntronMotifs RemoveNoncanonical". Counts per gene were extracted via featureCounts (Subread v1.5.0-p1) [42] with parameters "-s0 -Sfr -p" for RefSeq gene models as downloaded from the UCSC Table Browser on 11-07-2017. Differential gene expression analysis was performed using DESeq2 v1.14.1 [43]. For the purposes of pathway analysis, differentially expressed genes are defined at FDR threshold 0.01 and fold change > 2 or < -2. The RNA-seq expression heatmap was generated by R package ComplexHeatmap (v2.0.0) using rlog transformed values as reported by DESeq2 (v1.14.1).
qRT-PCR analysis
RNA from thyroid glands was extracted using RNAqueous-Micro total RNA isolation kit (ThermoFisher). RNA (1 μg) was reverse-transcribed using a high-capacity cDNA reverse transcription kit (ThermoFisher) and analyzed by qRT-PCR using the TaqMan or SYBR system. QRT-PCR reactions were carried out in triplicate using a StepOnePlus Real-time PCR system (Applied Biosystem). Primer sequences are listed in Supplementary Table 2.
Plasmids and reagents
The expression plasmid encoding constitutively active PKA (PKA catalytic subunit Calpha) was obtained from Addgene (#15310). p3xGLISBS-Luc and p3XFlag-Glis3 were described previously [33, 44]. Forskolin, FK506, and 8-Bromo-cAMP (8-Br-cAMP) were obtained from Millipore Sigma (Burlington, MA), H89 and Gö6976 from BioVision (Waltham, MA), Trametinib from Cayman Chemical Company (Ann Arbor, MI), and ZSTK474 from Enzo Life Science (Farmingdale, NY).
Cell culture and reporter assay
Rat thyrocyte PCCl3 cells were cultured in F12 medium supplemented with 5% FBS, 1 mIU/ml TSH, 10 µg/ml Insulin, 10 ng/ml somatostatin, 10 ng/ml 1-glycyl-histidyl-lysine, 5 µg/ml apo-transferrin, and 10 nM hydrocortisone. To examine GLIS3 transcription activity, PCCl3 cells were plated in 12-well plates at 150,000 cells/well and cultured for 72 h to reach 80% confluent. Cells were grown for an additional 72 h in the absence of TSH and subsequently co-transfected with pCMV-βGal (control), p3XFlag-Glis3, and the luciferase reporter p3xGLISBS-Luc using Lipofectamine 2000 (ThermoFisher). After 24 h, cells were treated with TSH or with the compound indicated and assayed 24 h later for luciferase and β-galactosidase activity using a Luciferase assay system (Promega) and Luminescent β-gal detection kit (Takara), respectively. Luciferase activity was normalized to β-gal activity. Assays were performed in triplicate.
Western blot analysis
PCCl3-pIND20-Glis3 cells, expressing a doxycycline (Dox)-inducible GLIS3, tagged with Flag and HA at its N- and C-terminus, respectively, were generated by infecting cells with pIND20-Flag-Glis3-HA lentivirus as described [45]. To evaluate the expression of GLIS3, PCCl3-pIND20-Glis3 cells were seeded in six-well-plate (300,000 cells/well) and cultured for 72 h, then treated with 100 ng/ml Dox for 96 h in the absence of TSH. After 24 h treatment with TSH, GLIS3-HA protein was examined by Western blot analysis and confocal microscopy. To examine degradation by proteasomes and GLIS3 protein stability, cells treated with Dox for 96 h in the absence of TSH, were subsequently treated with the proteasome inhibitor MG132 (0.5 μM) for 5 h or with the protein synthesis inhibitor cycloheximide (10 μg/ml) and TSH for the times indicated. Nuclear extracts were prepared for Western blot analysis. Briefly, cells were washed twice with cold PBS, then scraped and collected in 1.5 ml tubes. After centrifugation at 13,000 rpm for 1 min, cells were resuspended in 500 μl lysis buffer (0.5% NP-40, 100 mM Tris pH 8.0) and incubated on ice for 5 min. After a 5 min centrifugation at 13,000 rpm, nuclear pellets were resuspended in 50 μl extraction buffer (20 mM Hepes pH 8.0, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT) containing 1X Halt protease and phosphatase inhibitor cocktail (ThermoFisher), and then incubated on ice for 20 min. After centrifugation for 20 min at 13,000 rpm, supernatant was analyzed for GLIS3-HA protein by Western blot analysis with an HA antibody. Histone H3 (H3) was used as internal control of nuclear protein. Ribosomal protein S6 (RPS6), pRPS6, and NIS expression in whole cell lysates from thyroid glands of mice fed an ND or LID was examined by Western blot analysis. β-actin was used as internal control.
Statistical analysis
P values were calculated by one-way ANOVA.
Results
Expression of GLIS3 during thyroid folliculogenesis
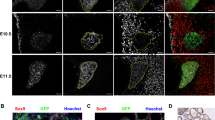
Previously, we reported that GLIS3 protein is expressed in postnatal murine thyroid follicular cells and that it functions as a critical transcriptional regulator of several genes required for thyroid hormone biosynthesis [33]; however, whether GLIS3 plays a role in the regulation of mouse thyroid organogenesis, has not been clearly established. To investigate this further, we monitored the expression of GLIS3 protein in Glis3-EGFP mice at different stages of embryonic development in comparison to PAX8, which is critical for early thyroid organogenesis, and NIS, a marker of differentiated thyroid follicular cells [4, 7, 46]. In contrast to PAX8, which is already expressed at embryonic day 13.5 (E13.5), GLIS3 protein was undetectable in the thyroid gland at E13.5 and E14.5 (Fig. 1A, B, and Supplementary Fig. 1). Weak expression of GLIS3 protein was first observed at E15.5 in the nucleus of PAX8+ cells (Fig. 1A) and the intensity of GLIS3 immunostaining was significantly increased at E16.5. The appearance of GLIS3 protein expression at E15.5–16.5 coincided with that of the follicular cell marker NIS, which is directly regulated by GLIS3 (Fig. 1B).

GLIS3 protein is first detectable during E15.5 of mouse embryonic thyroid gland development. A, B GLIS3 expression was examined in thyroid glands from Glis3-EGFP mice at E15.5, E16.5, E17.5 and PND1 by immunofluorescent staining using antibodies for GFP (GLIS3, green) and PAX8 (red) (A), and for GFP (GLIS3, green) and NIS (red) (B). Nuclei were stained with DAPI. Scale bars: 50 μm
To determine whether loss of GLIS3 function had any major effect on thyroid gland morphology and folliculogenesis, thyroid glands from Glis3KO1, in which the coding sequence of the 5th zinc finger of the DNA-binding domain was deleted [36], and Glis3KO2, in which the mCherry coding sequence with stop codon was inserted into exon 3 causing termination of GLIS3 translation before the DNA binding domain [33, 37], were examined by (immuno)histochemistry. Both GLIS3-deficient mouse strains develop CH. H&E histochemical staining of sections of E17.5 WT and Glis3KO1 thyroid glands revealed no significant differences in thyroid gland morphology (Fig. 2A) and showed a comparable pattern of PAX8 and NKX2.1 immunostaining (Fig. 2B, C). Similar observations were made with thyroid glands from postnatal day 3 (PND3) Glis3KO2 mice (Supplementary Fig. 2A–C).

Loss of GLIS3 function does not cause major changes in mouse thyroid gland development. A H&E-stained sections of thyroid glands from E17.5 WT and ubiquitous Glis3KO1 mouse embryos. B–G Representative images of sections of thyroid glands from E17.5 WT and Glis3KO1 embryos immunostained with antibodies against PAX8 and CDH1 (B), NKX2.1 and CDH1 (C), CDH16 (D), ZO-1 and PECAM1 (E), β-Catenin and F-actin (F), and NIS (G). PECAM1 was used to stain endothelial cells. Nuclei were stained with DAPI. All scale bars are 50 μm, except for 20 μm in E and F. (H) The expression of several polarity and thyroid gland transcription factor genes was examined in thyroid gland of WT and Glis3KO1 from E18.5 embryos by qRT-PCR. WT n = 3, Glis3KO1 n = 4. *p < 0.05, **p < 0.01
Immunostaining further demonstrated no differences in the basolateral localization of the planar cell polarity markers, CDH1 and CDH16, and the localization of ZO-1 to apical tight junctions (Fig. 2B–E, Supplementary Fig. 2B-E) [47,48,49] and no change was observed in the expression of Cdh1 and Cdh16 mRNA (Fig. 2H). In addition, the staining patterns of β-Catenin and F-actin, which are localized to the basolateral and apical membrane, respectively, were also not altered in Glis3KO1 thyroid gland (Fig. 2F). These data indicate that loss of GLIS3 function did not significantly alter thyroid follicular cell polarity. Together, these observations suggest that GLIS3 does not play a major role in mouse embryonic thyroid morphogenesis. This conclusion is consistent with data showing that the expression of transcription factors that are critical for early embryonic thyroid development, were not significantly altered (Foxe1 and Nkx2.1) in thyroid glands from E18.5 Glis3KO1, while Pax8 expression was somewhat increased (25%) (Fig. 2H). In contrast, Slc5a5 expression was decreased by 83% in thyroid follicular cells in E18.5 Glis3KO1 embryos (Fig. 2H). This was accompanied with reduced expression of NIS protein in E18.5 Glis3KO1 embryos (Fig. 2G) as well as in PND3 Glis3KO2 mice (Supplementary Fig. 2F). The observed synchronized induction of GLIS3 and NIS protein expression during embryonic development, the suppression of Slc5a5 expression in GLIS3-deficient thyroids, and the observation that GLIS3 is expressed at E15.5, much later than PAX8 and NKX2.1 [4, 8, 50], are consistent with the hypothesis that in GLIS3-deficient mice the development of congenital hypothyroidism is due to dyshormonogenesis rather than thyroid dysgenesis [33].
Correlation between GLIS3 protein expression and TSH levels
Analysis of GLIS3 protein expression during the first 2 months of postnatal thyroid development showed that GLIS3 is localized to the nucleus of thyroid follicular cells at all ages examined (Figs. 1A and 3A) consistent with previous observations [33]. However, both the intensity of GLIS3 protein staining in follicular cells and the percentage of PAX8+GLIS3+ cells in Glis3-EGFP mice gradually decreased over this period (Fig. 3A, B). This paralleled the steady decline in postnatal blood TSH levels (Fig. 3C). This raised the question whether there is a connection between the regulation of GLIS3 protein levels and TSH signaling. The relationship between GLIS3 protein and TSH levels was strengthened by observations showing that GLIS3 staining and the percentage of PAX8+GLIS3+ cells were greatly increased in Glis3-EGFP mice fed a LID, a condition in which blood TSH level is greatly elevated (Fig. 3D, E). These changes in GLIS3 protein did not show a strong correlation with alterations in Glis3 mRNA expression (Supplementary Fig. 3) suggesting that the higher levels of GLIS3 protein expression under conditions of elevated TSH, might be due to an increase in protein stability or rate of translation rather than increased transcription.

GLIS3 protein expression in thyroid follicular cells correlated with serum TSH levels. A, B GLIS3 protein expression is greatly decreased during early postnatal development. Expression of endogenous GLIS3 and PAX8 protein was examined in thyroid glands from 1-, 2-, 4-, 6-, and 8-week-old Glis3-EGFP mice by immunofluorescence staining with GFP (green) and PAX8 (red) antibodies. Nuclei were stained with DAPI (A). The percentage of PAX8+ cells that were GLIS3+ (from A) was calculated and plotted (B). n ≥ 3 for each group. Scale bars: 50 μm. C Serum TSH levels in WT mice were examined at PND5, 5- and 10-weeks. **p < 0.01. D, E GLIS3 protein expression in thyroid follicular cells is increased in mice fed a LID (high serum TSH). 8-week-old Glis3-EGFP mice were fed an ND and LID for 2 weeks before the expression of GLIS3 and PAX8 was examined in thyroid glands by immunofluorescence staining (D). The percentage of PAX8+ cells (from D) that are GLIS3+ was calculated and plotted (E). n ≥ 3 for each group. Scale bars: 50 μm
Link between PKA and GLIS3 stability and transcriptional activity
To obtain further support for the hypothesis that GLIS3 protein expression is regulated by TSH signaling, we examined the effect of TSH on GLIS3 protein and Glis3 mRNA expression in Dox-treated PCCl3-pIND20-Glis3 cells by immunofluorescence staining, Western blot and qRT-PCR analyses. These analyses showed that addition of TSH significantly increased GLIS3 protein expression in Dox-treated PCCl3-pIND20-Glis3 cells without changing Glis3 mRNA expression (Fig. 4A–C). This raised the possibility that TSH might enhance GLIS3 expression by increasing GLIS3 protein stability. To examine the effect of TSH on GLIS3 protein stability, we analyzed GLIS3 protein levels in Dox-treated PCCl3-pIND20-Glis3 cells that were subsequently treated with cycloheximide in the presence or absence of TSH. Western blot analysis indicated that addition of TSH increased GLIS3 protein stability (Fig. 4D). We further showed that proteolytic degradation of GLIS3 is inhibited by the proteasome inhibitor MG132 (Fig. 4E). These data support the concept that TSH enhance GLIS3 protein stability.

TSH and PKA activation enhances GLIS3 protein stability and transcription activity. A–C TSH enhanced GLIS3-HA protein expression in Dox-treated PCCl3-pIND20-Glis3 cells without increasing Glis3 mRNA expression. PCCl3-pIND20-Glis3 cells were treated with Dox in the absence of TSH, then treated with or without TSH for 24 h as described in Materials and methods. GLIS3-HA protein was examined by confocal microscopy (A, Scale bars: 50 μm) and Western blot analysis (B), and Glis3-HA mRNA by qRT-PCR analysis (C). D PCCl3-pIND20-Glis3 cells were treated with Dox for 96 h in the absence of TSH to induce GLIS3 and subsequently with 10 μg/ml cycloheximide (CHX, to block protein synthesis) in the presence or absence of TSH. Cells were harvested at the times indicated, nuclear extracts prepared, and GLIS3-HA levels examined by Western blot analysis. E GLIS3 degradation by proteasomes is stabilized by MG132. PCCl3-pIND20-Glis3 cells were treated with Dox for 96 h in the absence of TSH and subsequently with or without 0.5 μM of MG132 for 5 h before cells were harvested, and nuclear extracts examined for GLIS3-HA expression by Western blot analysis. H3 was used as an internal nuclear protein marker. F GLISBS-dependent transcriptional activation of the Luc reporter by GLIS3 was examined in PCCl3 cells as described in Materials and Methods. Addition of TSH stimulated GLIS3-mediated transcriptional activation. This increase was inhibited by the PKA inhibitor H89, whereas treatment with the mTOR inhibitor rapamycin (Rapa), the PKC inhibitor Gö6976, or the ERK inhibitor Trametinib (Trmt) had little or slight effect on the TSH-stimulated transcriptional activation by GLIS3. G Addition of 8-Br-cAMP or the adenylyl cyclase activator forskolin enhanced transcriptional activation by GLIS3. The increase by forskolin was inhibited by the PKA inhibitor H89. Expression of constitutively active PKA (CA_PKA) enhanced GLISBS-dependent transcriptional activation of the Luc reporter by GLIS3. Cells were transfected with either 0.1 ( +) or 0.2 μg (+ +) PKA plasmid DNA/well. *p < 0.05, **p < 0.01, ***p < 0.001
Next, we investigated whether TSH also affected GLIS3-mediated transcriptional activity. The results in Fig. 4F show that addition of TSH increased GLIS3-mediated transcriptional activation of a GLISBS-dependent luciferase reporter in rat thyrocyte PCCl3 cells by 60–300% depending on the experiment; however, in the absence of exogenous GLIS3, TSH did not increase GLISBS-dependent activation (Supplementary Fig. 4). It is well established that interaction of TSH with TSHR induces the activation of several protein kinases, including PKA, phospholipase C (PLC), PI3K, mTOR, ERK, and Ca++-mediated signaling [7, 51,52,53,54]. To examine whether any of these downstream kinase pathways are involved in the regulation of GLIS3 activity, we analyzed the effect of several kinase inhibitors on GLIS3-mediated transcriptional activation. Addition of the PKA inhibitor H89 almost totally abolished the TSH-induced increase in GLIS3-mediated transactivation (Fig. 4F). Rapamycin, ZSTK474, and trametinib, which inhibit the mTOR, PI3K, and ERK, respectively, had little effect, while the PKC inhibitor Gö6976 reduced transactivation by about 20% (Fig. 4F and Supplementary Fig. 4). A role for PKA activation in the regulation of GLIS3-mediated transcriptional activation by TSH was supported by data showing that co-expression of a constitutively active form of PKA increased GLIS3 activity by 76% and 280% when, respectively, 0.1 and 0.2 µg PKA plasmid DNA/well was transfected into cells. Treatment with 8-Br-cAMP or the adenylyl cyclase agonist, forskolin, enhanced GLIS3-dependent transcriptional activation by 133% and 144%, respectively; this increase was significantly inhibited by H89 (Fig. 4G). Together, these results are consistent with the concept that activation of PKA is part of the mechanism by which by TSH enhances GLIS3 protein stability and transcriptional activity.
Analysis of thyroid follicular cell proliferation in thyroid-selective Glis3-Pax8Cre mice
It is well established that in addition to increasing the expression of several TH biosynthetic genes, elevated TSH levels in mice fed a LID greatly induce the expression of cell cycle genes and increase thyroid follicular cell proliferation [55, 56]. Although this increase in cell proliferation and cell cycle gene expression is suppressed in ubiquitous Glis3KO mice [33], very few cell cycle genes were found to be directly regulated by GLIS3 [57]. This raised the question whether the repression of cell cycle genes is caused by changes in gene expression in other GLIS3 target tissues. Because insulin-like growth factors (IGFs) and insulin play a critical role in the positive regulation of thyroid follicular cell proliferation [52, 58,59,60,61,62], the development of severe hypoinsulinemia in ubiquitous Glis3KO mice might suppress cell proliferation [15, 38, 39]. To investigate whether hypoinsulinemia contributed to the inhibition of thyroid follicular cell proliferation in ubiquitous Glis3KO thyroid phenotype, we analyzed the thyroid gland phenotype in thyroid-selective Glis3 knockout mice, Glis3-Pax8Cre (referred as conditional knockout or CKO in the Figures), in which Pax8Cre efficiently (> 90%) deleted exon 5 in Glis3 in the thyroid gland, but not in the pancreas (Fig. 5A and Supplementary Fig. 5A). We demonstrated that in contrast to ubiquitous Glis3KO mice, pancreatic insulin expression and non-fasting blood glucose levels were not changed in Glis3-Pax8Cre mice (Supplementary Fig. 5B, C) confirming that these mice did not develop hypoinsulinemia/hyperglycemia [63]. However, serum T4 levels were still significantly decreased (44%) in Glis3-Pax8Cre(ND) mice, while those of TSH and T3 were increased by 300% and 28%, respectively (Fig. 5B). Serum T4 and T3 levels were greatly decreased in both WT(LID) and Glis3-Pax8Cre(LID) mice fed a LID, whereas TSH was greatly increased but to a significantly greater extent in Glis3-Pax8Cre(LID) mice (Fig. 5B). The inconsistent change in T3 levels between Glis3KO(ND) and Glis3-Pax8Cre(ND) mice might be related to altered expression of Dio1, 2 or 3 in ubiquitous Glis3KO mice in tissues other than the thyroid gland. Consistent with this is that little change in serum T3 level was also observed in Slc5a5 knockout mice fed a ND [64].

Loss of GLIS3 function does not suppress thyroid follicular cell proliferation in thyroid-selective Glis3-Pax8Cre(LID) mice. A QRT-PCR analysis of Glis3 mRNA expression confirming the specific deletion of exon 5 (E5) in the thyroid gland of Glis3-Pax8Cre mice (CKO). Exon 9–10 (E9−10), a non-deleted region of Glis3, served as control. n ≥ 4 for each group. B Serum levels of T3, T4, and TSH in 5-week-old WT(ND), Glis3-Pax8Cre(ND) (referred to as CKO(ND)), WT(LID), and Glis3-Pax8Cre(LID) (referred to as CKO(LID)). Each dot represents one mouse. *p < 0.05, **p < 0.01, ***p < 0.001. C Cell proliferation in thyroid glands from WT and Glis3-Pax8Cre mice fed a ND or LID was analyzed by EdU incorporation (green) as described in Material and Methods. Thyroid follicular cells were stained with a PAX8 antibody (red). In contrast to ubiquitous Glis3-knockout mice [33], thyroid follicular cell proliferation was not suppressed in Glis3-Pax8Cre(LID) mice. Scale bars: 50 μm. D The percentages of the number of PAX8+ cells that stained EdU+ were calculated and plotted. n = 3 for WT(ND), CKO(ND), CKO(LID), and n = 2 for WT(LID). E Activation of the mTOR pathway, as indicated by pRPS6 immunofluorescent staining, was not suppressed in thyroids from Glis3-Pax8Cre(LID) mice. pRBS6 (green), WGA (red), DAPI (blue). Scale bars: 50 μm. ** p < 0.01, **p < 0.01, ***p < 0.001. F. The expression of pRBS6, RBS6 was examined in thyroid gland from WT and Glis3-Pax8Cre mice fed a ND or LID by Western blot analysis. β-actin was used as internal control. G The relative weight of thyroid glands (TG) in male and female WT(LID) and Glis3-Pax8Cre(LID) were increased compared to mice fed a ND, while total body weights were not changed. Each dot represents one mouse
However, in contrast to ubiquitous Glis3KO2(LID) mice, the percentage of EdU+PAX8+ cells in Glis3-Pax8Cre(LID) mice was increased compared to Glis3-Pax8Cre(ND) mice and was similar to that in WT(LID) mice (Fig. 5C, D). These observations indicated that thyroid follicular cell proliferation was not repressed in Glis3-Pax8Cre(LID) mice. This was supported by data showing that mTOR activation, a major signaling pathway driving TSH-induced proliferation of thyroid follicular cells, was increased to a similar extent in Glis3-Pax8Cre(LID) and WT(LID) thyroid glands as indicated by pRSP6 immunofluorescence staining and Western blot analysis (Fig. 5E, F). The increase in thyroid follicular cell proliferation correlated with the development of thyroid gland hypertrophy in both male and female Glis3-Pax8Cre(LID) mice (Fig. 5G). In fact, in both male and female mice, thyroid hypertrophy was more pronounced in Glis3-Pax8Cre(LID) than in WT(LID) mice as indicated by the larger increase in thyroid weight (Fig. 5G). No significant difference in total body weights was observed between Glis3-Pax8Cre and WT mice (Fig. 5G). Together, these results suggested that GLIS3 does not play a direct role in the regulation of TSH-stimulated thyroid follicular cell proliferation and that the repression of cell proliferation in ubiquitous Glis3KO(LID) mice appears to be related to changes in other cell types that indirectly affect thyroid follicular cell proliferation.
Gene expression analysis in Glis3-Pax8Cre thyroid gland
To determine to what extent differences in the thyroid phenotype between ubiquitous Glis3 knockout mice and thyroid-selective Glis3-Pax8Cre knockout mice fed a LID correlated to changes in gene expression, we performed RNA-Seq analysis (Supplementary Table 3). The transcriptome and qRT-PCR analyses were carried out with thyroid glands from Glis3-Pax8Cre female mice. Changes in gene expression in thyroids of female mice were like those in male Glis3-Pax8Cre mice and did not reveal gender differences (Supplementary Fig. 6). Analysis of up-regulated genes identified transcriptional-misregulation-in-cancer, calcium and AMPK signaling among the top pathways (Fig. 6A and Supplementary Table 4). KEGG and heatmap analysis of genes down-regulated in Glis3-Pax8Cre(LID) thyroids identified ECM-receptor interaction, focal adhesion, and thyroid hormone synthesis among the top pathways (Fig. 6A, B), but no cell proliferation-regulatory pathways, in contrast to ubiquitous Glis3KO(LID) mice (Fig. 6B) [33]. The latter was supported by qRT-PCR analysis showing that the expression of cell cycle regulatory genes, including Ccnb1, Ccnb2, and Cdca2, was not suppressed in Glis3-Pax8Cre(LID) thyroid glands (Fig. 6C) consistent with our data showing little difference in the percentage of PAX8+EdU+ cells between Glis3-Pax8Cre(LID) and WT(LID) thyroids (Fig. 5D). These data support the concept that the inhibition of thyroid follicular cell proliferation in ubiquitous Glis3KO mice appears to be related to changes in gene expression in other cell types that subsequently affect the proliferation of these cells.

Transcriptome analysis of thyroid glands from female WT(LID) and thyroid-selective Glis3-Pax8Cre(LID) mice. A KEGG pathway analysis of genes up-regulated (red, fold change > 2; FDR < 0.01) and down-regulated (blue) in Glis3-Pax8Cre(LID) thyroid glands compared to those of WT(LID). B The relative expression of genes associated with TH synthesis, TSH induction, thyroid function, thyroid TFs, cell cycle, ECM, inflammation from RNA-Seq was visualized by heatmap. n = 4 for each group. C QRT-PCR analysis of several cell cycle-related genes. The expression of cell cycle-related genes was not repressed in Glis3-Pax8Cre(LID) consistent with data in Fig. 3. n ≥ 4 for each group. *p < 0.05, **p < 0.01, ***p < 0.001. In contrast to ubiquitous Glis3-knockout mice [33], cell proliferation-related genes were not suppressed in Glis3-Pax8Cre(LID) mice
Analysis of the expression of several TH biosynthetic and other TSH-induced genes, including Slc5a5, Slc26a4, Adm2, Sod3, Cdh13, Duox2, and Duoxa2, which are greatly increased in WT(LID) compared to WT(ND), were significantly repressed in thyroids from Glis3-Pax8Cre(LID) mice (Figs. 6B, and 7A), consistent with our previous study of ubiquitous Glis3 knockout mice [33]. NIS protein expression was dramatically increased in WT(LID), but not in Glis3-Pax8Cre(LID) thyroids by immunofluorescence staining and Western blot analysis (Fig. 7B, C). Similarly, the increase in the expression of several ECM-related and inflammatory genes, including Col18a1, Col6a2, Col4a1, Ccl7, Ccl2, and Itga2, observed in WT(LID) thyroids was significantly suppressed in Glis3-Pax8Cre(LID) thyroid glands (Figs. 6B and 7D). Analysis of thyroid TF expression showed that Pax8 expression was increased in both Glis3-Pax8Cre(ND) and Glis3-Pax8Cre(LID) mice compared to that of WT(ND) and WT(LID), whereas Nkx2.1 and Foxe1 RNA expression was not changed (Supplementary Fig. 7). This is consistent with the conclusion that the suppression of gene expression in Glis3-Pax8Cre(LID) thyroid is independent of changes in expression of these thyroid TFs. Together, these observations indicate that in contrast to TH biosynthetic genes, which transcription is directly regulated by GLIS3, GLIS3 does not play a major role in the direct transcriptional regulation of cell proliferation-regulatory genes in thyroid follicular cells in mice fed a LID. These findings are consistent with the concept that TSH regulates proliferation and TH biosynthesis in thyroid follicular cells through activation of different signaling pathways (Fig. 8) [7, 52, 62, 65].

The expression of TH biosynthetic genes, ECM- and inflammation-related genes was suppressed in thyroid glands from thyroid-selective Glis3-Pax8Cre(LID) mice. (A) Comparison of the expression of several TSH-inducible genes in thyroid glands from WT(ND), WT(LID), Glis3-Pax8Cre(ND), and Glis3-Pax8Cre(LID) mice by QRT-PCR analysis. n ≥ 4 for each group. (B) The expression of SLC5A5 (NIS, green) was examined in thyroid glands from WT(ND), WT(LID), Glis3-Pax8Cre(ND), and Glis3-Pax8Cre(LID) mice by immunofluorescence staining. DAPI (blue). Scale bars: 50 μm. (C) The expression of NIS was examined by Western blot analysis. β-actin was used as internal control. (D) QRT-PCR analysis of the expression of several collagen and inflammatory genes in thyroid glands from WT(ND), WT(LID), Glis3-Pax8Cre(ND), and Glis3-Pax8Cre(LID) mice. n ≥ 4 for each group. *p < 0.05, **p < 0.01, ***p < 0.001

Schematic showing a model of the mechanistic connection between TSH signaling, PKA, and regulation of GLIS3 transcriptional activity. Binding of TSH to TSHR leads to activation of several protein kinase pathways. Activation of PKA by TSH, the PKA activator 8-Br-cAMP, the adenylyl cyclase activator forskolin, and expression of constitutively active PKA increases GLIS3 transcriptional activity resulting in the induction of several GLIS3 target genes, including several TH-biosynthetic genes. The PKA antagonist H89 suppressed the TSH-mediated increase, whereas inhibition of PI3K, mTOR, PKC, or ERK pathways by, respectively, ZSTK474, rapamycin, Gö6976, or trametinib, had little effect. The stimulation in GLIS3 transcriptional activity by the TSH-TSHR-PKA pathway appears to be at least in part due to increased GLIS3 protein stability. This mechanism of regulation provides an explanation for the dramatic increase in GLIS3 protein expression and the subsequent induction of GLIS3 target genes, including several thyroid hormone biosynthetic genes, in thyroid follicular cells of mice fed a LID. In contrast to TH-biosynthetic genes, cell cycle genes and cell proliferation are not directly regulated by GLIS3
Discussion
Loss of GLIS3 function in both humans and mice causes CH [15, 16, 18, 33]. However, whether this is related to thyroid dysgenesis or dyshormonogenesis has not been clearly established. To determine whether GLIS3 plays a major role in thyroid morphogenesis, the expression of GLIS3 protein was analyzed at different stages of mouse thyroid development. This analysis demonstrated that GLIS3 protein was first detectable in thyroid follicular cells at E15.5, a time during which thyroid follicles are being formed (Fig. 1 and Supplementary Fig. 1) [4, 8]. We further show that thyroid gland morphology and the formation of thyroid follicles are not significantly altered in E17.5 and neonatal Glis3KO mice (Fig. 2A, Supplementary Fig. 2). These data indicate that GLIS3 is not required for early thyroid development in mice and that the development of CH in Glis3KO mice is due to dyshormonogenesis rather than thyroid dysgenesis. This contrasts the role of glis3 in zebrafish, in which glis3 has been shown to play an important role in thyroid development [66]. Moreover, unlike glis3 knockdown in zebrafish, the expression of NKX2.1 and PAX8 was not impaired in thyroid gland in Glis3KO mice (Fig. 2B, C). Whether the development of CH in human patients with GLIS3 deficiency is related to thyroid dysgenesis or dyshormonogenesis has been inconclusive and shown to vary among patients [16, 18, 20,21,22, 24, 25, 29, 67, 68]. This variability might be attributed to the highly oligogenic nature of CH [27, 29, 32].
Our study further shows that the onset of GLIS3 expression is distinct from that of the thyroid TFs, PAX8, NKX2.1, FOXE1, and HHEX, which are expressed much earlier than GLIS3 during embryonic thyroid development [4, 7, 8, 50, 69,70,71]. These TFs are critical for early thyroid gland development and their loss of function causes thyroid dysgenesis [3, 4, 6, 9, 10, 12, 72]. In addition to their developmental function, several of these TFs also play a role in the regulation of several thyroid follicular cell functions, including TH biosynthesis. This is supported by our recent study showing that GLIS3 regulates the transcription of several TH biosynthesis-related genes in coordination with PAX8 and NKX2.1 [57]. This study also demonstrated that PAX8 and NKX2.1 are bound to regulatory regions of the Glis3 gene, which would be consistent with the concept that these factors have a role in the transcriptional activation of Glis3 during thyroid development and act upstream of GLIS3 [33, 57]. Our analysis of GLIS3 protein during thyroid development, further indicated that the time of GLIS3 expression during E15.5 and 16.5 parallels that of NIS protein (Figs. 1B and 2G). This is in accordance with our data showing that Slc5a5 is a major GLIS3 target gene and that GLIS3 is required for NIS expression [33, 57]. NIS repression in Glis3-deficient thyroids may play a major role in the impairment in iodide transport and the development of CH in Glis3KO mice [33] like CH development in NIS knockout mice [64]. Together, the timing of GLIS3 expression during thyroid development and its role in directly regulating TH biosynthesis are consistent with our conclusion that CH in Glis3KO mice is due to dyshormonogenesis rather than thyroid dysgenesis.
It is well established that TSH not only regulates TH biosynthesis but also thyroid follicular cell proliferation [11, 52, 65]. We previously demonstrated that under LID conditions, when TSH levels are high, not only the activation of thyroid hormone biosynthetic genes was suppressed in ubiquitous GLIS3-deficient mice, but also cell cycle gene expression, thyroid follicular cell proliferation, and the activation of the mTOR pathway [15, 33]. We also reported that, in addition to congenital hypothyroidism, these mice develop neonatal diabetes and hypoinsulinemia [39, 63]. IGF-1 and insulin, together with TSH, have been reported to play a critical role in the regulation of thyroid follicular cell gene expression and proliferation [52, 58,59,60,61, 65, 73] raising the possibility that hypoinsulinemia in ubiquitous Glis3-deficient mice might influence the function and gene expression in thyroid follicular cells. Analysis of thyroid-selective Glis3-Pax8Cre(LID) mice, which do not develop hyperinsulinemia, showed that in contrast to ubiquitous Glis3-KO(LID) mice, thyroid follicular cell proliferation, expression of cell cycle genes, including Ccnb1, Ccnb2, and Cdca2, and activation of the mTOR, a pathway that promotes cell proliferation, were not repressed in Glis3-Pax8Cre(LID) mice but induced to a similar degree as in WT(LID) mice (Figs. 5C– F and 6C). These data indicate that GLIS3 does not play a major role in the regulation of thyroid follicular cell proliferation and suggest that the suppression of thyroid follicular cell proliferation in ubiquitous Glis3-deficient mice does not involve direct transcriptional regulation of cell cycle-related genes by GLIS3, but is related to abnormalities in other tissues, such as hypoinsulinemia that indirectly affect thyroid follicular cell proliferation. This conclusion is consistent with our cistrome analysis showing that GLIS3 binds to very few cell cycle-related genes [57].
In contrast to cell proliferation-regulatory genes, the expression of several TH-inducible genes, including Slc5a5, Slc26a4, Adm2, Sod3, and Cdh13, remain dramatically suppressed in thyroid-selective Glis3-Pax8Cre(LID) thyroid as we observed in ubiquitous GLIS3-deficient mice [33], consistent with the conclusion that in thyroid follicular cells their transcription is directly regulated by GLIS3 [57]. Although, IGF-1/insulin through activation of PI3K has been reported to repress TSH-induced expression of Slc5a5 [74, 75], since GLIS3 is required for its transcription, its repression in Glis3-Pax8Cre(LID) and ubiquitous Glis3-KO(LID) thyroids is independent of IGF1/insulin levels [33]. These findings are consistent with reports showing that the regulation of thyroid follicular cell proliferation and TH biosynthesis by TSH involve different mechanisms [7, 67, 76].
It was interesting to note that during postnatal development, GLIS3 protein expression steadily decreased together with blood TSH levels (Fig. 3A, C). Most strikingly, GLIS3 expression was greatly increased in mice fed a LID (Fig. 3D), a condition when TSH levels are highly elevated and TSH/TSHR-dependent genes are highly induced. This is also the condition when the largest repression of GLIS3 target genes was observed in thyroids from ubiquitous Glis3KO mice as well as thyroid-selective Glis3-Pax8Cre mice (Fig. 6B[33]. Together, these observations suggested a possible link between the TSH/TSHR signaling pathway, the regulation of GLIS3 protein, and the transcriptional activation of GLIS3 target genes. This was supported by data showing that TSH enhanced GLIS3-mediated transcriptional activation and GLIS3 protein expression in PCCl3 cells (Fig. 4A, B, and F). This induction was not due to a change in the level of Glis3 mRNA expression (Fig. 4C) suggesting that it might be due to increased GLIS3 protein stability or rate of translation. We provided evidence indicating that the stimulation of GLIS3 transcriptional activity is at least in part due to increased GLIS3 protein stability (Fig. 4B, D). GLIS3 protein stability and GLIS3-dependent transcriptional activation of target genes might be controlled by posttranslational modification(s) of GLIS3 that are mediated by TSH/TSHR-induced activation of (a) downstream kinase pathway(s) [11, 52, 65, 76]. Study of the effect of several kinase inhibitors on TSH-induced stimulation of GLIS3 transcriptional activity revealed that the PKA antagonist H89 suppressed this increase, but that inhibition of several other kinase pathways (ERK, PKC, AKT, mTOR) had relatively little effect (Fig. 4F and Supplementary Fig. 4). A role for PKA in mediating the effect of TSH was supported by data showing that the PKA agonist, 8-Br-cAMP, and the adenylyl cyclase agonist, forskolin, similarly enhanced GLIS3-mediated transcriptional activation and that this stimulation was inhibited by H89. A role for PKA was further strengthened by observations demonstrating that expression of a constitutively active PKA stimulated GLIS3-mediated transcriptional activation (Fig. 4G). It is interesting to note that previous studies found a relationship also between PKA and its regulation of PAX8 and NKX2.1 transcriptional activity [77, 78]. Mass spectrometric analysis showed that GLIS3 is phosphorylated at multiple sites (unpublished data) and further studies are needed to identify the amino acid(s) within GLIS3 that are phosphorylated by PKA and critical for regulating GLIS3 protein stability and activity. Together, our study reveals a link between TSH signaling and its regulation of GLIS3 protein activity and transcriptional activation GLIS3 target genes, including several TH biosynthesis-related genes. We further provide evidence for a role of the PKA signaling pathway in mediating the transcriptional regulation of several TSH-inducible genes by GLIS3 in thyroid follicular cells (Fig. 8). Greater insights into the upstream pathways that regulate GLIS3 expression and/or its transcription activity might provide new therapeutic approaches in the management of hypo- and hyperthyroidism.
Data availability
RNA-Seq data were deposited in the NCBI Gene Expression Omnibus (GEO) database https://www.ncbi.nlm.nih.gov/geo/ under accession GSE207775.
Abbreviations
- GLIS3:
-
Gli-similar 3
- CH:
-
Congenital hypothyroidism
- TH:
-
Thyroid hormone
- GLISBS:
-
GLIS-binding site
- TF:
-
Transcriptional factor
- TSH:
-
Thyroid stimulating hormone
- TSHR:
-
Thyroid stimulating hormone receptor
- ECM:
-
Extracellular matrix
- NDH:
-
Neonatal diabetes and congenital hypothyroidism
- PND:
-
Postnatal day
- KO:
-
Knockout
- CKO:
-
Conditional knockout
- PKA:
-
Protein kinase A
- ND:
-
Normal diet
- LID:
-
Low iodine diet
- NIS:
-
Sodium-iodide symporter
- DUOX2:
-
Dual oxidase 2
- FOXE1:
-
Forkhead box E1
- HHEX:
-
Hematopoietically expressed homeobox
- NKX2.1:
-
NK2 homeobox 1
- PAX8:
-
Paired box 8
- PDS:
-
Pendrin
- TPO:
-
Thyroid peroxidase
- TG:
-
Thyroglobulin
- cAMP:
-
Cyclic AMP
- ChIP:
-
Chromatin immunoprecipitation
- EGFP:
-
Enhanced green fluorescent protein
References
Targovnik HM, Scheps KG, Rivolta CM (2020) Defects in protein folding in congenital hypothyroidism. Mol Cell Endocrinol 501:110638. https://doi.org/10.1016/j.mce.2019.110638
Wassner AJ (2018) Congenital hypothyroidism. Clin Perinatol 45(1):1–18. https://doi.org/10.1016/j.clp.2017.10.004
Mio C, Grani G, Durante C, Damante G (2020) Molecular defects in thyroid dysgenesis. Clin Genet 97(1):222–231. https://doi.org/10.1111/cge.13627
Fernandez LP, Lopez-Marquez A, Santisteban P (2015) Thyroid transcription factors in development, differentiation and disease. Nat Rev Endocrinol 11(1):29–42. https://doi.org/10.1038/nrendo.2014.186
Nilsson M, Fagman H (2013) Mechanisms of thyroid development and dysgenesis: an analysis based on developmental stages and concurrent embryonic anatomy. Curr Top Dev Biol 106:123–170. https://doi.org/10.1016/B978-0-12-416021-7.00004-3
De Felice M, Di Lauro R (2004) Thyroid development and its disorders: genetics and molecular mechanisms. Endocr Rev 25(5):722–746. https://doi.org/10.1210/er.2003-0028
Lopez-Marquez A, Carrasco-Lopez C, Fernandez-Mendez C, Santisteban P (2021) Unraveling the complex interplay between transcription factors and signaling molecules in thyroid differentiation and function from embryos to adults. Front Endocrinol (Lausanne) 12:654569. https://doi.org/10.3389/fendo.2021.654569
Nilsson M, Fagman H (2017) Development of the thyroid gland. Development 144(12):2123–2140. https://doi.org/10.1242/dev.145615
Mansouri A, Chowdhury K, Gruss P (1998) Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet 19(1):87–90. https://doi.org/10.1038/ng0598-87
Macchia PE, Lapi P, Krude H, Pirro MT, Missero C, Chiovato L et al (1998) PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet 19(1):83–86. https://doi.org/10.1038/ng0598-83
Citterio CE, Targovnik HM, Arvan P (2019) The role of thyroglobulin in thyroid hormonogenesis. Nat Rev Endocrinol 15(6):323–338. https://doi.org/10.1038/s41574-019-0184-8
Kostopoulou E, Miliordos K, Spiliotis B (2021) Genetics of primary congenital hypothyroidism-a review. Hormones (Athens) 20(2):225–236. https://doi.org/10.1007/s42000-020-00267-x
Grasberger H, Refetoff S (2021) Genetic causes of congenital hypothyroidism due to dyshormonogenesis. Curr Opin Pediatr 23(4):421–428. https://doi.org/10.1097/MOP.0b013e32834726a4
Jetten AM (2018) GLIS1–3 transcription factors: critical roles in the regulation of multiple physiological processes and diseases. Cell Mol Life Sci 75(19):3473–3494. https://doi.org/10.1007/s00018-018-2841-9
Scoville DW, Kang HS, Jetten AM (2020) Transcription factor GLIS3: Critical roles in thyroid hormone biosynthesis, hypothyroidism, pancreatic beta cells and diabetes. Pharmacol Ther. https://doi.org/10.1016/j.pharmthera.2020.107632
Dimitri P (2017) The role of GLIS3 in thyroid disease as part of a multisystem disorder. Best Pract Res Clin Endocrinol Metab 31(2):175–182. https://doi.org/10.1016/j.beem.2017.04.007
Dimitri P, Habeb AM, Garbuz F, Millward A, Wallis S, Moussa K et al (2015) Expanding the clinical spectrum associated with GLIS3 mutations. J Clin Endocrinol Metab 100(10):E1362-1369. https://doi.org/10.1210/jc.2015-1827
Senee V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC et al (2006) Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet 38(6):682–687. https://doi.org/10.1038/ng1802
Habeb AM, Al-Magamsi MS, Eid IM, Ali MI, Hattersley AT, Hussain K et al (2012) Incidence, genetics, and clinical phenotype of permanent neonatal diabetes mellitus in northwest Saudi Arabia. Pediatr Diabetes 13(6):499–505. https://doi.org/10.1111/j.1399-5448.2011.00828.x
London S, De Franco E, Elias-Assad G, Barhoum MN, Felszer C, Paniakov M et al (2021) Case report: neonatal diabetes mellitus caused by a novel GLIS3 mutation in twins. Front Endocrinol (Lausanne) 12:673755. https://doi.org/10.3389/fendo.2021.673755
Splittstoesser V, Vollbach H, Plamper M, Garbe W, De Franco E, Houghton JAL et al (2021) Case report: extended clinical spectrum of the neonatal diabetes with congenital hypothyroidism syndrome. Front Endocrinol (Lausanne) 12:665336. https://doi.org/10.3389/fendo.2021.665336
Sarikaya E, Kendirci M, Demir M, Dundar M (2022) Neonatal diabetes, congenital hypothyroidism, and congenital glaucoma coexistence: a case of GLIS3 mutation. J Clin Res Pediatr Endocrinol. https://doi.org/10.4274/jcrpe.galenos.2022.2021-12-19
Boddu PK, Velumula PK, Sharif S, Monika B (2022) A neonate with diabetes mellitus, congenital hypothyroidism, and congenital glaucoma. Cureus 14(9):e29488. https://doi.org/10.7759/cureus.29488
Perdas E, Gadzalska K, Hrytsiuk I, Borowiec M, Fendler W, Mlynarski W (2022) Case report: neonatal diabetes mellitus with congenital hypothyroidism as a result of biallelic heterozygous mutations in GLIS3 gene. Pediatr Diabetes 23(6):668–674. https://doi.org/10.1111/pedi.13341
Fu C, Luo S, Long X, Li Y, She S, Hu X et al (2018) Mutation screening of the GLIS3 gene in a cohort of 592 Chinese patients with congenital hypothyroidism. Clin Chim Acta 476:38–43. https://doi.org/10.1016/j.cca.2017.11.011
Porcu E, Medici M, Pistis G, Volpato CB, Wilson SG, Cappola AR et al (2013) A meta-analysis of thyroid-related traits reveals novel loci and gender-specific differences in the regulation of thyroid function. PLoS Genet 9(2):e1003266. https://doi.org/10.1371/journal.pgen.1003266
de Filippis T, Gelmini G, Paraboschi E, Vigone MC, Di Frenna M, Marelli F et al (2017) A frequent oligogenic involvement in congenital hypothyroidism. Hum Mol Genet 26(13):2507–2514. https://doi.org/10.1093/hmg/ddx145
Teumer A, Chaker L, Groeneweg S, Li Y, Di Munno C, Barbieri C et al (2018) Genome-wide analyses identify a role for SLC17A4 and AADAT in thyroid hormone regulation. Nat Commun 9(1):4455. https://doi.org/10.1038/s41467-018-06356-1
Yamaguchi T, Nakamura A, Nakayama K, Hishimura N, Morikawa S, Ishizu K et al (2020) Targeted next-generation sequencing for congenital hypothyroidism with positive neonatal TSH screening. J Clin Endocrinol Metab. https://doi.org/10.1210/clinem/dgaa308
Zhang RJ, Zhang JX, Du WH, Sun F, Fang Y, Zhang CX et al (2021) Molecular and clinical genetics of the transcription factor GLIS3 in Chinese congenital hypothyroidism. Mol Cell Endocrinol 528:111223. https://doi.org/10.1016/j.mce.2021.111223
Park KS (2021) Analysis of worldwide carrier frequency and predicted genetic prevalence of autosomal recessive congenital hypothyroidism based on a general population database. Genes (Basel) 12(6):863. https://doi.org/10.3390/genes12060863
Li L, Li X, Wang X, Han M, Zhao D, Wang F et al (2023) Mutation screening of eight genes and comparison of the clinical data in a Chinese cohort with congenital hypothyroidism. Endocrine 79(1):125–134. https://doi.org/10.1007/s12020-022-03188-4
Kang HS, Kumar D, Liao G, Lichti-Kaiser K, Gerrish K, Liao XH et al (2017) GLIS3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J Clin Invest 127(12):4326–4337. https://doi.org/10.1172/JCI94417
Watanabe N, Hiramatsu K, Miyamoto R, Yasuda K, Suzuki N, Oshima N et al (2009) A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett 583(12):2108–2113. https://doi.org/10.1016/j.febslet.2009.05.039
Jetten AM, Scoville DW, Kang HS (2022) GLIS1–3: Links to Primary cilium, reprogramming, stem cell renewal, and disease. Cells 11(11):1833. https://doi.org/10.3390/cells11111833
Kang HS, Beak JY, Kim YS, Herbert R, Jetten AM (2009) Glis3 is associated with primary cilia and Wwtr1/TAZ and implicated in polycystic kidney disease. Mol Cell Biol 29(10):2556–2569. https://doi.org/10.1128/MCB.01620-08
Kang HS, Chen LY, Lichti-Kaiser K, Liao G, Gerrish K, Bortner CD et al (2016) Transcription factor GLIS3: a new and critical regulator of postnatal stages of mouse spermatogenesis. Stem Cells 34(11):2772–2783. https://doi.org/10.1002/stem.2449
Kang HS, Takeda Y, Jeon K, Jetten AM (2016) The spatiotemporal pattern of glis3 expression indicates a regulatory function in bipotent and endocrine progenitors during early pancreatic development and in beta PP and Ductal Cells. PLoS ONE 11(6):e0157138. https://doi.org/10.1371/journal.pone.0157138
Scoville D, Lichti-Kaiser K, Grimm S, Jetten A (2019) GLIS3 binds pancreatic beta cell regulatory regions alongside other islet transcription factors. J Endocrinol. https://doi.org/10.1530/JOE-19-0182
Di Cosmo C, Liao XH, Dumitrescu AM, Philp NJ, Weiss RE, Refetoff S (2010) Mice deficient in MCT8 reveal a mechanism regulating thyroid hormone secretion. J Clin Invest 120(9):3377–3388. https://doi.org/10.1172/JCI42113
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21. https://doi.org/10.1093/bioinformatics/bts635
Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7):923–930. https://doi.org/10.1093/bioinformatics/btt656
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550. https://doi.org/10.1186/s13059-014-0550-8
ZeRuth GT, Williams JG, Cole YC, Jetten AM (2015) HECT E3 ubiquitin ligase itch functions as a novel negative regulator of gli-similar 3 (Glis3) transcriptional activity. PLoS ONE 10(7):e0131303. https://doi.org/10.1371/journal.pone.0131303
Jeon K, Kumar D, Conway AE, Park K, Jothi R, Jetten AM (2019) GLIS3 transcriptionally activates WNT genes to promote differentiation of human embryonic stem cells into posterior neural progenitors. Stem Cells 37(2):202–215. https://doi.org/10.1002/stem.2941
Postiglione MP, Parlato R, Rodriguez-Mallon A, Rosica A, Mithbaokar P, Maresca M et al (2002) Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc Natl Acad Sci U S A 99(24):15462–15467. https://doi.org/10.1073/pnas.242328999
Cali G, Gentile F, Mogavero S, Pallante P, Nitsch R, Ciancia G et al (2012) CDH16/Ksp-cadherin is expressed in the developing thyroid gland and is strongly down-regulated in thyroid carcinomas. Endocrinology 153(1):522–534. https://doi.org/10.1210/en.2011-1572
Koumarianou P, Gomez-Lopez G, Santisteban P (2017) Pax8 controls thyroid follicular polarity through cadherin-16. J Cell Sci 130(1):219–231. https://doi.org/10.1242/jcs.184291
Stevenson BR, Anderson JM, Bullivant S (1988) The epithelial tight junction: structure, function and preliminary biochemical characterization. Mol Cell Biochem 83(2):129–145. https://doi.org/10.1007/BF00226141
Plachov D, Chowdhury K, Walther C, Simon D, Guenet JL, Gruss P (1990) Pax8, a murine paired box gene expressed in the developing excretory system and thyroid gland. Development 110(2):643–651. https://doi.org/10.1242/dev.110.2.643
Andrade BM, Araujo RL, Perry RL, Souza EC, Cazarin JM, Carvalho DP et al (2011) A novel role for AMP-kinase in the regulation of the Na+/I–symporter and iodide uptake in the rat thyroid gland. Am J Physiol Cell Physiol 300(6):C1291-1297. https://doi.org/10.1152/ajpcell.00136.2010
Morgan SJ, Neumann S, Marcus-Samuels B, Gershengorn MC (2016) Thyrotropin and insulin-like growth factor 1 receptor crosstalk upregulates sodium-iodide symporter expression in primary cultures of human thyrocytes. Thyroid 26(12):1794–1803. https://doi.org/10.1089/thy.2016.0323
Chu YD, Yeh CT (2020) The molecular function and clinical role of thyroid stimulating hormone receptor in cancer cells. Cells 9(7):1730. https://doi.org/10.3390/cells9071730
Ohno M, Zannini M, Levy O, Carrasco N, di Lauro R (1999) The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol 19(3):2051–2060. https://doi.org/10.1128/MCB.19.3.2051
Nagasaki S, Fukui M, Asano S, Ono K, Miki Y, Araki S et al (2014) Induction of adrenomedullin 2/intermedin expression by thyroid stimulating hormone in thyroid. Mol Cell Endocrinol 395(1–2):32–40. https://doi.org/10.1016/j.mce.2014.07.008
Kolaja KL, Petrick JS, Klaassen CD (2000) Inhibition of gap-junctional-intercellular communication in thyroid-follicular cells by propylthiouracil and low iodine diet. Toxicology 143(2):195–202. https://doi.org/10.1016/s0300-483x(99)00172-9
Kang HS, Grimm SA, Jothi R, Santisteban P, Jetten AM (2023) GLIS3 regulates transcription of thyroid hormone biosynthetic genes in coordination with other thyroid transcription factors. Cell Biosci 13(1):32. https://doi.org/10.1186/s13578-023-00979-8
Ock S, Ahn J, Lee SH, Kang H, Offermanns S, Ahn HY et al (2013) IGF-1 receptor deficiency in thyrocytes impairs thyroid hormone secretion and completely inhibits TSH-stimulated goiter. FASEB J 27(12):4899–4908. https://doi.org/10.1096/fj.13-231381
Zhang X, Sheng X, Miao T, Yao K, Yao D (2019) Effect of insulin on thyroid cell proliferation, tumor cell migration, and potentially related mechanisms. Endocr Res 44(1–2):55–70. https://doi.org/10.1080/07435800.2018.1522641
Kushchayeva Y, Kushchayev S, Jensen K, Brown RJ (2022) Impaired glucose metabolism, anti-diabetes medications, and risk of thyroid cancer. Cancers (Basel) 14(3):555. https://doi.org/10.3390/cancers14030555
Smith TJ (2021) Insulin-like growth factor pathway and the thyroid. Front Endocrinol (Lausanne) 12:653627. https://doi.org/10.3389/fendo.2021.653627
Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP (2001) Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocr Rev 22(5):631–656. https://doi.org/10.1210/edrv.22.5.0444
Kang HS, Kim YS, ZeRuth G, Beak JY, Gerrish K, Kilic G et al (2009) Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol Cell Biol 29(24):6366–6379. https://doi.org/10.1128/MCB.01259-09
Ferrandino G, Kaspari RR, Reyna-Neyra A, Boutagy NE, Sinusas AJ, Carrasco N (2017) An extremely high dietary iodide supply forestalls severe hypothyroidism in Na(+)/I(−) symporter (NIS) knockout mice. Sci Rep 7(1):5329. https://doi.org/10.1038/s41598-017-04326-z
Brewer C, Yeager N, Di Cristofano A (2007) Thyroid-stimulating hormone initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer Res 67(17):8002–8006. https://doi.org/10.1158/0008-5472.CAN-07-2471
Rurale G, Marelli F, Duminuco P, Persani L (2020) Glis3 as a critical regulator of thyroid primordium specification. Thyroid 30(2):277–289. https://doi.org/10.1089/thy.2019.0196
Alghamdi KA, Alsaedi AB, Aljasser A, Altawil A, Kamal NM (2017) Extended clinical features associated with novel Glis3 mutation: a case report. BMC Endocr Disord 17(1):14. https://doi.org/10.1186/s12902-017-0160-z
Taha D, Barbar M, Kanaan H, Williamson Balfe J (2003) Neonatal diabetes mellitus, congenital hypothyroidism, hepatic fibrosis, polycystic kidneys, and congenital glaucoma: a new autosomal recessive syndrome? Am J Med Genet A 122A(3):269–273. https://doi.org/10.1002/ajmg.a.20267
Lazzaro D, Price M, de Felice M, Di Lauro R (1991) The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development 113(4):1093–1104. https://doi.org/10.1242/dev.113.4.1093
Zannini M, Avantaggiato V, Biffali E, Arnone MI, Sato K, Pischetola M et al (1997) TTF-2, a new forkhead protein, shows a temporal expression in the developing thyroid which is consistent with a role in controlling the onset of differentiation. EMBO J 16(11):3185–3197. https://doi.org/10.1093/emboj/16.11.3185
Thomas PQ, Brown A, Beddington RS (1998) Hex: a homeobox gene revealing peri-implantation asymmetry in the mouse embryo and an early transient marker of endothelial cell precursors. Development 125(1):85–94. https://doi.org/10.1242/dev.125.1.85
Martinez Barbera JP, Clements M, Thomas P, Rodriguez T, Meloy D, Kioussis D et al (2000) The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development 127(11):2433–2445. https://doi.org/10.1242/dev.127.11.2433
Oglio R, Salvarredi L, Rossich L, Copelli S, Pisarev M, Juvenal G et al (2019) Participation of NADPH 4 oxidase in thyroid regulation. Mol Cell Endocrinol 480:65–73. https://doi.org/10.1016/j.mce.2018.10.012
Garcia B, Santisteban P (2002) PI3K is involved in the IGF-I inhibition of TSH-induced sodium/iodide symporter gene expression. Mol Endocrinol 16(2):342–352. https://doi.org/10.1210/mend.16.2.0774
Saji M, Kohn LD (1991) Insulin and insulin-like growth factor-I inhibit thyrotropin-increased iodide transport in serum-depleted FRTL-5 rat thyroid cells: modulation of adenosine 3’,5’-monophosphate signal action. Endocrinology 128(2):1136–1143. https://doi.org/10.1210/endo-128-2-1136
Kero J, Ahmed K, Wettschureck N, Tunaru S, Wintermantel T, Greiner E et al (2007) Thyrocyte-specific Gq/G11 deficiency impairs thyroid function and prevents goiter development. J Clin Invest 117(9):2399–2407. https://doi.org/10.1172/JCI30380
Poleev A, Okladnova O, Musti AM, Schneider S, Royer-Pokora B, Plachov D (1997) Determination of functional domains of the human transcription factor PAX8 responsible for its nuclear localization and transactivating potential. Eur J Biochem 247(3):860–869. https://doi.org/10.1111/j.1432-1033.1997.00860.x
Velasco JA, Acebron A, Zannini M, Martin-Perez J, Di Lauro R, Santisteban P (1998) Ha-ras interference with thyroid cell differentiation is associated with a down-regulation of thyroid transcription factor-1 phosphorylation. Endocrinology 139(6):2796–2802. https://doi.org/10.1210/endo.139.6.6076
Acknowledgements
We would like to acknowledge Laura Miller Degraff and Fredrick Chang at NIEHS for, respectively, managing the mouse colonies and RNA-Seq analyses, Dr. Min Shi at NIEHS for the advice with the statistical analyses.
Funding
This research was supported by the Intramural Research Program of the NIEHS, NIH Z01-ES-101585 (AMJ) and RO1 DK15070 from NIH (XHL).
Author information
Authors and Affiliations
Contributions
HSK designed and performed experiments, data analysis, and wrote the manuscript. SAG performed data analysis and edited the manuscript. XHL provided TH analysis data. AMJ, designed experiments, analyzed data, and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Ethical approval
All animal studies followed guidelines outlined by the NIH Guide for the Care and Use of Laboratory Animals and protocols were approved by the Institutional Animal Care and Use Committee at the NIEHS.
Consent to publish
All authors have approved the content of this manuscript and provided consent for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, H.S., Grimm, S.A., Liao, XH. et al. GLIS3 expression in the thyroid gland in relation to TSH signaling and regulation of gene expression. Cell. Mol. Life Sci. 81, 65 (2024). https://doi.org/10.1007/s00018-024-05113-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05113-6