
Abstract
DExD/H-box helicases play critical roles in multiple cellular processes, including transcription, cellular RNA metabolism, translation, and infections. Several seminal studies over the past decades have delineated the distinct functions of DExD/H-box helicases in regulating antiviral innate immune signaling pathways, including Toll-like receptors, retinoic acid-inducible gene I-like receptors, cyclic GMP-AMP synthase-the stimulator of interferon gene, and NOD-like receptors signaling pathways. Besides the prominent regulatory roles, there is increasing attention on their functions as nucleic acid sensors involved in antiviral innate immunity. Here we summarize the complex regulatory roles of DExD/H-box helicases in antiviral innate immunity. A better understanding of the underlying molecular mechanisms of DExD/H-box helicases’ regulatory roles is vital for developing new therapeutics targeting DExD/H-box helicases and their mediated signaling transduction in viral infectious diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most putative RNA helicases fall into two families, DExD-box helicases (DDX) and DExH-box helicases (DHX) [1]. Both families bear the Asp-Glu-X-Asp/His signature. A highly conserved helicase core domain consists of two linked RecA-like domains, harboring at least twelve conserved motifs involved in Adenosine triphosphate (ATP) binding/hydrolysis and Ribonucleic acid (RNA) binding [2,3,4]. RNAs interact with both RecA-like domains along the sugar-phosphate backbone, explaining why DExD/H-box helicases’ binding to RNA is generally sequence-independent but structure-dependent. Typically, the helicase core exists in an ‘open conformation’ in the absence of RNA and ATP, where the two Rec-A-like domains can rotate flexibly around the linker. However, both ATP and RNA’s binding causes the enzyme to transition to a ‘close conformation’ and facilitate efficient ATP binding and hydrolysis [5,6,7]. Apart from the conserved functional core, most DEAD/H-box helicases contain highly variable N- and C-terminal regions responsible for the functional specificity to individual helicases through interactions with specific RNA substrates or a variety of proteins [8]. Therefore, DExD/H-box helicases can participate in multiple biological processes, and some of these do not rely on their helicase activity.
There is growing appreciation for the roles of DExD/H-box helicases in antiviral innate immune responses, either by acting as sensors for viral nucleic acids or by regulating signal transduction downstream of various pattern recognition receptors (PRRs) that recognize highly conserved structures present in invading pathogens known as pathogen-associated molecular patterns (PAMPs) [9]. There are several distinct PRRs, including Toll-like receptors (TLRs), retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), cyclic GMP-AMP synthase (cGAS), and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), that activate sophisticated signal transduction cascades following PAMP recognition and result in the production of type I interferon (IFN-I) and inflammatory cytokines [10, 11]. The induction of the IFN-I ultimately triggers the expression of hundreds of interferon-stimulated genes (ISGs) and a tailored adaptive immune response that eventually controls or eradicates the infection [12]. Intriguingly, several DExD/H-box helicases can be exploited by viruses lacking RNA helicase to facilitate their replication cycles. In this review, we outline the recent advances in the roles of DExD/H-box helicases in regulating antiviral innate signaling pathways.
DExD/H-box helicases regulate the TLR signaling pathway
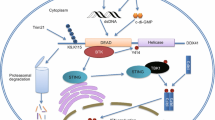
The first identified and best characterized PRRs, TLRs sense a wide range of PAMPs derived from viruses, bacteria, fungi, and parasites, initiating the host’s innate immunity [13, 14]. To date, 10 TLRs have been identified in humans. TLR1, TLR2, TLR4, TLR5, and TLR6 are localized on the cell surface and primarily recognize microbial membrane components, including bacterial lipids and proteins [13, 14]. TLR3, TLR7, TLR8, and TLR9 are mainly expressed within the endosomes and recognize nucleic acids [15]. Upon PAMP recognition, TLRs recruit two main adaptors, myeloid differentiation factor 88 (MyD88) (except TLR3) and TIR domain-containing adaptor inducing interferon-β (TRIF) (only TLR3 and TLR4). These adaptors initiate a series of downstream signaling events that eventually eliminate the infected pathogens [14]. Several DExD/H-box helicases have been implicated in regulating TLR-mediated innate immune responses (Fig. 1).

DExD/H-box helicases regulate the TLR signaling pathway. TLRs located at the plasma membrane and endosomes recognize nucleic acids like dsRNA, ssRNA, and DNA to transduce signals through MyD88 and TRIF and then drive the activation of NF-κΒ to induce IFN-I production. DExD/H-box helicases play regulatory roles in the TLR-mediated signaling pathways. The green/T-shaped arrow indicates the interaction between DExD/H-box helicases and adaptors and promotes/inhibits signaling. MyD88, Myeloid differentiation primary response protein 88; TRIF, TIR-domain-containing adaptor protein inducing IFN-β; TRAF, TNF-receptor-associated factor; IKK, IκB kinase; NEMO, IKKγ, an IKK regulatory subunit; TRAM, TRIF-related adaptor molecule; IκBα, nuclear factor-kappa B inhibitor alpha; P, phosphate; ISRE, IFN-stimulated response element
Recently, Dempsey et al. found that DHX9 is involved in TLR2- and TLR8-dependent cytokine induction [16]. TLR2 forms a heterodimer with TLR1 or TLR6 on the cell surface and recognizes the viral components such as lipoprotein-lipid polypeptide and polysaccharide, whereas TLR8 senses single-stranded RNA (ssRNA) [13, 17]. Knockdown of DHX9 expression results in reduced production of TLR2- and TLR8-stimulated IL-6 and other TLR8-stimulated cytokines, including TNF-ɑ, IP10, and RANTES. It has been subsequently shown that DHX9 enhances NF-κB-dependent promoter transactivation, which probably contributes to increased IL-6 induction. However, the mechanism whereby DHX9 impacts TLR8-dependent cytokine induction remains unclear. A study by Kim et al. showed that DHX9 could function as a cytosolic nucleic acid sensor, recognized CpG-B, and utilized MyD88 as a downstream adaptor [18]. Therefore, a plausible explanation could be that the interaction between DDX and MyD88 only facilitates intracellular TLR signaling.
TRIF
It has been reported that complete TRIF deficiency confers a predisposition to childhood herpes simplex encephalitis, and TRIF−/− mice are highly susceptible to SARS-CoV infection with the aberrant cellular responses that eventually lead to a lethal phenotype [19, 20]. These findings reflect the crucial role of TRIF in host antiviral defense. Not surprisingly, several DExD/H-box helicases function as TRIF’s enhancers to initiate downstream antiviral signaling. In myeloid dendritic cells, pulldown with biotinylated poly(I:C) followed by LC–MS analysis identified three potential RNA sensors, DDX1 as well as DDX21 and DHX36. It has been subsequently shown that these three helicases form a complex where DDX1 directly binds poly(I:C) while both DDX21 and DHX36 serve as bridges to TRIF. Knockdown of each helicase or TRIF results in impaired ability of mDCs to produce IFN-I and cytokines in response to poly(I:C), influenza A virus, and Reovirus [21].
Despite the PAMPs mentioned above, several cellular-derived molecular patterns such as endogenous damage-associated molecular patterns (DAMPs) could trigger TLRs activation. During the influenza A virus infection, the DDX21-TRIF pathway is required for the expression/secretion of S100A9, a DAMP molecule also known as Calgranulin B or MRP-14, which subsequently activates the TLR4/MyD88 pathway for induction of innate antiviral responses against influenza A virus [22]. Given that DDX21 interacts with both DDX1 and DHX36 in mDCs, it would be interesting to see whether these two DDXs are also involved in this process.
Another study by Ruan et al. has identified DDX23 as a nuclear double-stranded RNA (dsRNA) sensor in amphioxus. Further investigation showed that human DDX23 also selectively bound poly(I:C), which subsequently led to its translocation to the cytoplasm, where DDX23 formed a complex with TRIF or MAVS to initiate innate antiviral responses [23].
MyD88
Despite its critical role for all TLRs except for TLR3, MyD88 also serves as a key adaptor molecule in pDCs for DHX36 and DHX9, which have been identified as viral DNA sensors that bind CpG-A and CpG-B, respectively [18]. Both DHX36 and DHX9 directly interact with MyD88 in the cytosol through the helicase-associated 2 (HA2) domain and the unknown function (DUF) domain of DHX, and the Toll-IL receptor domain of MyD88. Knockdown of DHX36 or DHX9 in pDCs leads to significantly reduced IFN-I or TNF-α responses when challenged with DNA virus (HSV) but not with RNA virus (influenza A virus) [18]. Intriguingly, the same group later demonstrated that DHX9 acts as a vital dsRNA sensor in mDCs, and DHX9 knockdown results in decreased IFN-α/β production in response to both poly (I:C) and RNA viruses (reovirus and influenza A virus) [24]. In addition, DHX36 could also recognize viral RNAs [25]. The activities of DHX36 and DHX9 are quite distinct in different cell types (Table 1). It could be that certain innate immunity pathways are triggered in a cell-type-dependent manner. However, further investigations are required to elucidate the function of DExD/H-box helicases in different anatomical regions.
DExD/H-box helicases modulate the RLR signaling pathway
There are three members in the RLR family: retinoic acid-inducible gene I (RIG-I, also known as DDX58), melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2), all of which harbor a central DExD/H-box helicase domain and a C-terminal regulatory domain (CTD), whereas RIG-I and MDA5 contain two additional N-terminal tandem caspase recruitment domains (CARDs) [10, 11]. The helicase domain and CTD work together to recognize cognate dsRNAs, while CARDs act as a signaling module and mediate the interaction with downstream adaptor mitochondrial antiviral signaling (MAVS) for signaling activation [26]. In contrast, LGP2 lacks the N-terminal CARDs but has a stronger RNA binding affinity; thus, functioning as a major RIG-I and MDA5 signaling regulator. Increasing evidence indicates that LGP2 negatively regulates RIG-I’s signaling activity while synergizing with MDA5 to promote antiviral signaling [26,27,28,29,30] (Fig. 2).

DExD/H-box helicases regulate the RLR signaling pathway. MDA5 and RIG-I recognize distinct RNA structures and signal through the adaptor protein MAVS to trigger IRF3 and NF-κΒ activation. The green/T-shaped arrow indicates the interaction between DExD/H-box helicases and adaptors and promotes/inhibits signaling. RIG-I, retinoic acid-inducible gene I; MDA-5, melanoma differentiation-associated protein 5; LGP2, laboratory of genetics and physiology 2; MAVS, mitochondrial antiviral signaling; TBK1, TANK-binding kinase 1; P, phosphate; Ub, ubiquitin; ISRE, IFN-stimulated response element
RIG-I/MDA5
RIG-I and MDA5 are critical cytoplasmic sensors that recognize RNA derived from RNA and DNA viruses, leading to the induction of IFN-I and inflammatory cytokines. DDX3, DDX60, and DDX60L have been identified as ligand-specific sentinels for RLR activation [31] (Fig. 2). Oshiumi et al. demonstrated an interaction between DDX3 with MAVS as well as RIG-I and MDA5, and knockdown of DDX3 led to reduced poly (I:C)-mediated IFN-promoter activation; thus, proposing a model in which DDX3 acts as an enhancer for initial RNA sensing by RLR at early stages of infection when cytosolic RIG-I levels are still low [32]. It has been confirmed that DDX60 interacts with RIG-I and MDA5 and is required for RIG-I- or MDA5-dependent IFN-I and ISGs expression in response to viral infection [33]. Production of IFN-β and ISGs is impaired in cells after DDX60 knockdown in response to Sendai virus, vesicular stomatitis virus, poliovirus, or HSV-1 infection. Another study by the same group found that DDX60 functions upstream of RIG-I as a ligand-specific sentinel that promotes RIG-I signaling in a cell-type-specific manner [31]. DDX60L, a homolog of DDX60, shows similarity to DDX60, also contributes to RIG-I-dependent activation of innate immunity [34].
DDX6, DHX15, and DHX29 act as cofactors and are required for the full-blown activation of RLR-mediated antiviral responses against RNA viruses (Fig. 2). DDX6 interacts with both RIG-I and influenza viral RNA in infected cells and potentiates RIG-I-dependent production of IFN-I [35]. DHX15 is required for virus-induced RLR signaling via interacting with RIG-I CARDs and facilitating signaling to MAVS [36]. DHX29 preferentially enhances dsRNA-MDA5 binding affinity in the encephalomyocarditis virus-infected cells and promotes MDA5-, but not RIG-I-, dependent antiviral signaling [37]. In the human airway epithelial cells and fibroblasts, DHX29 is identified as a co-sensor and implicates in RIG-I-MAVS-associated innate antiviral signaling [38]. On the contrary, DDX24 acts as a negative regulator and enhances viral replication by competing with RIG-I to bind viral RNA in VSV-infected cells [39]. Collectively, these studies have recapitulated that the DExD/H-box helicases govern the RIG-I/MDA5-mediated antiviral innate signaling pathway [31].
LGP2
LGP2 plays a critical role in regulating MDA5- and RIG-I-mediated antiviral responses [40, 41]. DDX39A was identified as a binding partner of LGP2 by a yeast two-hybrid system screen and further confirmed by co-immunoprecipitation assay. DDX39A and LGP2 synergistically inhibit Sendai virus-induced IFN-β promoter activity, indicating that DDX39A interacts with LGP2 to downregulate RLR-mediated immune responses [42]. Interestingly, LGP2 is also required for DDX39B mediated inhibition of NF-κB activity in response to dsRNA [43]. DDX39 directly interacts with LGP2 and exerts this inhibitory effect in a MAVS/TRIF-dependent manner. It is important to investigate how LGP2 coordinates the positive and negative modulation of RLR-dependent responses and whether other DExD/H-box helicases involve LGP2-mediated signaling pathways.
MAVS
The mitochondrial outer membrane-located MAVS contains the N-terminal CARD domain, which is crucial for RIG-I/MDA5 to transduce signaling, the middle proline-rich region (PRR) responsible for the interaction with TRAFs to activate NF-κB or TANK-binding kinase 1 (TBK1)/IRF3 signaling, and the C-terminal transmembrane domain [44, 45]. Recent studies have shown that DExD/H-box helicases, such as DDX3, DHX9, DHX15, DDX23, DHX33, and DDX46, regulate antiviral innate immunity by interacting with MAVS (Fig. 2).
In addition to its role as an enhancing factor for initial RNA-sensing by the RIG-I-MAVS complex, DDX3 also contributes to forming the MAVS-TRAF3 complex [46]. DHX9 is a vital sensor in mDCs, and it has been found to pair with MAVS to trigger the early IFN production independent of RIG-I [24]. MAVS forms functional prion-like aggregates to activate antiviral innate immune responses. However, MAVS aggregation is barely detected in DHX15 knockdown cells after poly(I:C) stimulation, indicating a role of DHX15 for MAVS aggregation [47]. Additionally, DHX15 is required for poly(I:C)-induced and MAVS-mediated apoptosis [48]. In mDCs, DHX33 interacts with and signals through MAVS independent of RIG-I/MDA5, implying a novel way of sensing RNA viruses to initiate antiviral innate immune responses [49].
In contrast to promoting antiviral immunity, as mentioned above, Cao et al. have identified DDX46 as a negative regulator of IFN-I production in response to viral infection. DDX46 binds transcripts that encode molecules involved in antiviral responses, such as MAVS, TRAF3, and TRAF6, via their conserved CCGGUU element and recruits ALKBH5, an ‘eraser’ of the RNA modification N6-methyladenosine (m6A), to erase the m6A modification of these antiviral transcripts and thus to induce their retention in the nucleus [50]. DDX39A also preferentially binds to transcripts of MAVS, TRAF3, and TRAF6 and inhibits their nuclear export. Upon viral infection, this binding capacity is enhanced and leads to impaired antiviral signaling [51].
TRAF3
TRAF3 is another central molecule that could activate NF-κB and TBK1/IRF3 to induce IFN-I production [52]. It has been well established that the K63-linked ubiquitination of TRAF3 is required for the induction of IFN-I. GST-pull down assay has confirmed that DDX3 directly binds N-terminal RING-finger and zinc-finger domains of TRAF3. DDX3 knockdown strongly reduces TRAF3 ubiquitination. While reconstitution of knockdown cells with wild-type DDX3 restores TRAF3 ubiquitination, suggesting that DDX3 is required for TRAF3 ubiquitination [46] (Fig. 2). However, little is known about DExD/H-box helicases’ regulatory roles in the post-modification of diverse molecules involved in RLRs, TLRs, and cGAS-STING pathways, which are worth investigating.
DExD/H-box helicases involve in the DNA-sensing signaling pathway
DNA sensors play a pivotal role in the innate immune responses against DNA viruses, and several DNA sensors have been well characterized in mammal cells, such as cGAS, IFI16, AIM2, and DDX41. These cytosolic DNA sensors depend on an ER-resident adaptor molecule called a stimulator of interferon genes (STING) to activate two canonical pathways, NF-κB and TBK1-IRF3 pathways [53,54,55]. Liu et al. have revealed that DDX41 is an early sensor mediating the activation of STING-associated downstream signaling in DCs, whereas IFI16 is upregulated at the late phase of viral infection in a DDX41-dependent manner forming a DDX41-STING-IFI16 positive feedback regulation [54]. Phosphorylation of DDX41 by Bruton’s Tyrosine Kinase (BTK) increases its binding affinity to foreign dsDNA and strengthens its interaction with STING [56]. The trafficking of proteins is critical for innate immunity. For example, upon DNA viruses activating cGAS, STING traffics from ER to Golgi, inducing innate immunity [57]. Interestingly, DDX41 can interact with Sorting Nexin-2, which is involved in protein trafficking in the trans-Golgi network, endosome, and/or lysosome compartments [58]. Therefore, it is worth investigating whether DDX41 also participates in modulating STING trafficking.
DExD/H-box helicases regulate the TBK1-IRF axis
TBK1 sits at the crossroads of multiple IFN-inducing pathways; therefore, it is vital for host defense against viral infection. IRF3 phosphorylated by activated TBK1 results in homodimerization and translocation into the nucleus, driving IFN-I production [59] (Figs. 1, 2, 3).

DExD/H-box helicases regulate the DNA and the JAK-STAT signaling pathway. cGAS, DDX41, and IFIT16 are cytosolic DNA sensors that activate the ER-resident adaptor STING, which subsequently translocates to Golgi, resulting in TBK1/IRF3 and NF-κΒ signaling and the production of IFN-I and inflammatory cytokines. IFN-I binds to their cognate receptor IFNAR1 and IFNAR2 and triggers the JAK-STAT signaling pathway. The green/T-shaped arrow indicates the interaction between DExD/H-box helicases and adaptors and promotes/inhibits signaling. IFI16, Interferon Gamma Inducible Protein 16; cGAS, Cyclic GMP–AMP synthase; STING, Stimulator of interferon genes; IFNAR, interferon-α/β receptor; STAT, signal transducer and activator of transcription; P, phosphate; ISRE, IFN-stimulated response element; ISGs, interferon-stimulated genes; RNA polymerase II (RNAP II)
DDX3 interacts with TBK1 and acts as a positive regulator of TBK1-mediated IRF activation [60, 61]. Besides, DDX3 augments IFN-I production by directly interacting with IκB kinase ε (IKKε) and IKKα to promote the phosphorylation of IRF3 [62, 63]. SNRNP200 helicase, another member of DExD/H-box helicases, also promotes antiviral signaling. SNRNP200 binds viral RNA upon infection and interacts with TBK1 to promote IRF3 activation and IFN-I production [64]. On the contrary, several DExD/H-box helicases have been reported to regulate TBK1-IRF3 signaling negatively. DDX19 disrupts the interaction between TBK1 and IRF3 to inhibit TBK1-mediated phosphorylation of IRF3 and suppress SeV and poly(I:C)-induced IFN-I production. DDX19 also recruits endosomal/lysosomal adaptor and MAPK and mTOR activator 2 to the TBK1- IKKε -IRF3 complex and facilitates TBK1 and IKKε degradation [65]. DDX25 promotes RNA virus infection and inhibits IFN antiviral responses by interfering with the nuclear translocation of IRF3 and NF-κB [66]. DDX56 disrupts the interaction between IRF3 and importin-β3 to inhibit the nuclear translocation of IRF3 [67].
As the closest family member to IRF3, IRF7 also functions as a principal mediator of IFN induction downstream of TLRs as well as cytosolic RNA and DNA sensors [68]. Evidence has demonstrated that DDX proteins play prominent roles in regulating IRF7-mediated IFN-I production (Fig. 1). Phosphorylation of IRF7 is significantly reduced after the shRNA knockdown of DDX3, confirming that DDX3 is a critical upstream regulator of IRF7 activation [63]. Conversely, DDX24 negatively affects IRF7 function as overexpression of DDX24 suppresses the ability of IRF7 to activate the IFNα4 promoter [39]. The underlying mechanism could be that DDX24 competes with receptor-interacting protein 1 to bind IRF7, thereby disturbing the induction of IFN-I [39].
DExD/H-box helicases regulate NF-κB
The NF-κB complex contains RelA (p65), c-Rel, NF-κB1 (p50 and its precursor p105), NF-κB2 (p52 and its precursor p100), and RelB, and is expressed in virtually all cell types [69]. DExD/H-box helicases have been shown to regulate NF-κB tightly (Figs. 1, 2, 3). DDX1 is associated with p65 in the nucleus and acts as a coactivator to enhance NF-κB-mediated transcription activation [70]. In contrast, DDX3 interacts with p65 and suppresses p65/p50-mediated transcriptional activity [71]. DHX9 interacts with p65 in the nucleus but not the cytosol. DHX9 is recruited to the p65-dependent promoters of antiviral genes during DNA virus infection and is required to recruit RNA polymerase II [72, 73]. Interestingly, in all three cases, the ATP-dependent RNA helicase domain of DExD/H-box helicases is involved in the interaction with p65 and required for mediating transcriptional activation/inhibition. In addition, DDX5 promotes phosphorylation of the p65 subunit via interacting with diacylglycerol kinase ζ, a known NF-κB suppressor [74, 75]. Nevertheless, a detailed mechanism requires further investigation.
DExD/H-box helicases regulate the NLR signaling pathway
NLRs are primarily distributed in the cytoplasm of mucosal epithelial cells, macrophages, DCs, and neutrophils, triggering the inflammasome’s assemble in response to PAMPs. Inflammasomes are multimeric protein complexes containing sensors (NLRs, AIM2, IFI16, etc.), an apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and zymogen procaspase-1 [76]. Viral infection activates caspase-1 through inflammasomes. The bioactive caspase-1 cleaves pro-IL-1β and pro-IL-18 to induce maturation of IL-1β and IL-18 and eventually leads to a form of cell death called ‘pyroptosis’, which helps to remove damaged host cells [76, 77]. However, inflammasome activation requires tight regulation as aberrant inflammasome activation is linked to many diseases. Here, we summarize current findings on the involvement of DExD/H-box helicases in inflammasome regulation.
The role of NLRP3 inflammasome is well established in controlling viral infection [78]. DDX3X physically interacts with NLRP3 and is required to assemble and activate the NLRP3 inflammasome [79, 80]. DHX33 and DDX19A have been identified as components of the NLRP3 inflammasome and act as upstream viral dsRNA sensors. Both proteins interact with NLRP3 following RNA stimulation and mediate NLRP3 inflammasome formation and activation [81,82,83]. Nlrp6, primarily expressed in mouse primary intestinal epithelial cells, regulates antiviral immune responses [84]. In mouse primary intestinal epithelial cells, Nlrp6 specifically interacts with DHX15 and forms a viral RNA-sensing complex, which uses MAVS as a downstream adaptor to induce IFN production, thus, restricting EMCV infection intestine [84]. A recent study by Xing et al. revealed that DHX15 could interact with downstream NLRP6 to recruit MAVS for inducing the production of type I and type III IFNs in intestinal epithelial cells. In addition, DHX15 could also use NLRP6 to interact with downstream ASC/procaspase-1 to activate the NLRP6 inflammasome, which led to the secretion of mature IL-18 [85]. Nlrp9b is a novel NLR, uniquely expressed in different sections of the small and large intestines. During rotavirus infection, Nlrp9b recognizes viral dsRNA through Dhx9, then binds with Asc and caspase-1 to assemble NLRP9b inflammasome in intestinal epithelial cells [86]. This newly discovered inflammasome requires further characterization to elucidate its contribution to innate antiviral immunity fully. These studies have revealed the diverse roles of DExD/H-box helicases in regulating the NLR signaling pathway.
DExD/H-box helicases modulate the activation and function of ISGs
IFN-I induces a wide range of antiviral effectors through the Janus kinase (JAK)/signal transducer and activator of the transcription (STAT) pathway. The activation of the JAK-STAT signaling pathway begins with IFN-I binding to IFNAR, then activated JAKs phosphorylate STATs, leading to the formation of STAT1/STAT2 heterodimers or STAT2 homodimers [87, 88]. These dimers bind to IRF9 in the cytoplasm and form a complex known as interferon stimulating factor 3, which subsequently translocates to the nucleus where it binds interferon-stimulated response element (ISRE) to trigger the production of various ISGs, such as Viperin, zinc-finger antiviral protein (ZAP), cholesterol 25-hydroxylase, ISG15, and the double-stranded RNA-activated protein kinase R (PKR). These ISGs act as actual effectors and mount an antiviral state within a host cell [87]. It is of note that some DExD/H-box helicases are themselves ISGs, such as DDX60/DDX60L, some act as suppressors to block ISGs aberrant activation, such as DDX6 [89], while others are indispensable for the optimal function of certain ISGs like ZAP and PKR (Fig. 3).
DDX60/DDX60L
DDX60 and DDX60L share approximately 70% amino acid identity, and both have been identified as ISGs [33, 90]. However, they fulfill their antiviral functions through distinct mechanisms. DDX60 expression is increased in response to IFN-β or viral infection and functions as a ligand-specific sentinel to promote RLR-mediated antiviral signaling [33]. A study comparing genes differentially induced by IFN- γ in hepatocyte cell lines identifies DDX60L as an effector gene of the IFN-γ response mediating restriction against HCV replication [34]. Intriguingly, DDX60L knockdown facilitates HCV replication in the absence of IFN-γ, while ectopic expression of DDX60L does not alter the expression of IFN-γ induced ISGs, indicating that DDX60L functions as a direct antiviral effector against HCV infection.
In contrast, no such effect is observed in the HAV infection model [34]. Moreover, DDX60L strongly impairs lentiviral vectors’ production, indicating its potential role as a restriction factor in retroviral replication [34]. Whether DDX60L could restrict other retrovirus remains elusive, further investigation in this area would help understand the role of DDX60L in innate immunity against viral infection (Fig. 4).

DExD/H-box helicases regulate the NLR signaling pathway. Activated inflammasomes could cleave Pro IL-1β and Pro IL-18 into mature IL-1β and IL-18. DExD/H-box helicases could also regulate NLRP3, NLRP9b, and NLRP6 inflammasomes and the NLRP6/MAVS signaling. The green arrow indicates the interaction between DExD/H-box helicases and adaptors and promotes signaling. P, phosphate; ISRE, IFN-stimulated response element; ASC, the apoptosis-associated speck-like protein containing CARD
ZAP
ZAP restricts viral replication by directly binding viral mRNA and recruiting the RNA degradation machinery to eliminate target viral mRNAs in the cytoplasm. It has been shown that ZAP’s N-terminus interacts with the N- and C-terminal domains of DDX17, and this interaction is thought to promote ZAP-mediated RNA degradation [91]. Similarly, DHX30 interacts with ZAP directly via their N-terminal domains. Knockdown of DHX30 reduces ZAP’s antiviral activity, implicating that DHX30 is indispensable for the optimal function of ZAP [92]. However, it is unclear how DExD/H-box helicases and ZAP interaction contribute to its antiviral activity, awaiting further investigation.
PKR
PKR contains a kinase domain and two tandem repeats of dsRNA-binding domains (dsRBDs), which interact with dsRNA in a sequence-independent manner [93]. After binding of viral RNA, PKR undergoes autophosphorylation for its activation. Then activated PKR forms a dimer and phosphorylates the eukaryotic translation initiation factor 2 alpha subunit to shut down global protein synthesis and inhibit viral replication [94]. DHX36 interacts with PKR to form a complex in the presence of dsRNA. Although knockdown DHX36 does not affect the total level of PKR, it significantly diminishes virus-triggered PKR phosphorylation and IFN-I production, which indicates that DHX36 relies on its RNA helicases to regulate PKR and thereby to inhibit host cells’ antiviral innate immune responses [25]. As described earlier, DHX9 plays a crucial role in host antiviral responses through multiple mechanisms. A study by Williams et al. identified DHX9 as a substrate for PKR, where PKR directly associates with and phosphorylates DHX9 to perturb the association of helicase with dsRNA [95].
Concluding remarks
This review offers a comprehensive map of DExD/H-box helicases as critical regulators of host antiviral immunity. Interactome studies have proved the importance of some DExD/H-box helicases emerging as nucleic acid sensors [18, 21, 96]. Due to intensive research over the past decades, we now have a good understanding of how DExD/H-box helicases work in detecting external nucleic acids and triggering signaling pathways against viruses. Meanwhile, Some DExD/H-box helicases have been implicated in antiviral immunity through modulating the TLR-/RLR-/NLR-mediated signaling pathways [22, 33]. Although DExD/H-box helicases are important regulators in the RNA-sensing pathway, little research focuses on DExD/H-box helicases’ regulatory roles in the “relatively novel” DNA-sensing signaling pathway. Therefore, more detailed studies are required to address fundamental questions: (a) Whether DExD/H-box helicases specifically or selectively recognize DNA viruses and retroviruses. (b) Do DExD/H-box helicases associate with cytosolic cGAS to influence the cGAMP formation and cGAS-STING pathway? (c) Whether DExD/H-box helicases act as nuclear sensors, and if so, how do DExD/H-box helicases distinguish self and nonself DNA? (d) Whether DExD/H-box helicases function as DNA sensors detecting abnormal DNA in the cytoplasm to induce autoimmune diseases. (e) Whether posttranscriptional modifications regulate DExD/H-box helicases in the early stages of innate immunity.
Other DExD/H-box helicases exert antiviral effects by promoting cellular processes, such as stress granules, P-bodies, autophagy, ER stress, and DNA damage responses (DDR); however, they are usually targeted by many viruses for persistent infection. In addition to a role in regulating innate antiviral signaling, DExD/H-box helicases also interact with viral genomes and proteins and participate in viral genome transcription, nuclear output, and assembly of virions to aid or restrict viral replication [97,98,99]. DDX5 binds to Rev, and this interaction facilitates HIV replication [98]. DDX3X binds to and unwinds 5’ TR of Japanese encephalitis and Zika viruses, which provides a new therapeutic strategy to inhibit Flavivirus [100]. Cellular DHX30 RNA helicase inhibits influenza A virus replication by binding the viral NS1 protein [99]. Furthermore, the N-terminal of DHX30 containing a dsRNA-binding domain possesses potent antiviral activity, suggesting that DHX30 may function as a new RNA sensor [99]. Thus, exploring the interplay between DExD/H-box helicases and viruses would be exciting, which may help provide new strategies against viral infection.
Data availability
Not applicable.
References
Abdelhaleem M et al (2003) The human DDX and DHX gene families of putative RNA helicases. Genomics 81(6):618–622
De Bortoli F et al (2021) DEAH-Box RNA helicases in Pre-mRNA splicing. Trends Biochem Sci 46(3):225–238
Linder P, Jankowsky E (2011) From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 12(8):505–516
Shen L, Pelletier J (2020) General and target-specific DExD/H RNA helicases in eukaryotic translation initiation. Int J Mol Sci 21(12):4402
Caruthers JM et al (2000) Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc Natl Acad Sci USA 97(24):13080–13085
Hauk G, Bowman GD (2011) Structural insights into regulation and action of SWI2/SNF2 ATPases. Curr Opin Struct Biol 21(6):719–727
Caruthers JM, McKay DB (2002) Helicase structure and mechanism. Curr Opin Struct Biol 12(1):123–133
Fuller-Pace FV (2006) DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res 34(15):4206–4215
Talwar T et al (2017) The DEAD-box protein DDX43 (HAGE) is a dual RNA-DNA helicase and has a K-homology domain required for full nucleic acid unwinding activity. J Biol Chem 292(25):10429–10443
Liu J et al (2016) Post-translational modification control of innate immunity. Immunity 45(1):15–30
Liu G, Gack MU (2020) Distinct and orchestrated functions of RNA sensors in innate immunity. Immunity 53(1):26–42
Levy DE et al (1988) Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev 2(4):383–393
Fitzgerald KA, Kagan JC (2020) Toll-like receptors and the control of immunity. Cell 180(6):1044–1066
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34(5):637–650
Ishii KJ et al (2006) A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol 7(1):40–48
Dempsey A et al (2018) Poxviral protein E3-altered cytokine production reveals that DExD/H-box helicase 9 controls Toll-like receptor-stimulated immune responses. J Biol Chem 293(39):14989–15001
Huang Z et al (2015) Aberrant expression and dysfunction of TLR2 and its soluble form in chronic HBV infection and its regulation by antiviral therapy. Antiviral Res 118:10–19
Kim T et al (2010) Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc Natl Acad Sci USA 107(34):15181–15186
Sancho-Shimizu V et al (2011) Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest 121(12):4889–4902
Totura AL et al (2015) Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. MBio 6(3):e00638-e1615
Zhang Z et al (2011) DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34(6):866–878
Tsai SY et al (2014) DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog 10(1):e1003848
Ruan J et al (2019) DDX23, an evolutionary conserved dsRNA sensor, participates in innate antiviral responses by pairing with TRIF or MAVS. Front Immunol 10:2202
Zhang Z et al (2011) DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J Immunol 187(9):4501–4508
Yoo JS et al (2014) DHX36 enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog 10(3):e1004012
Ablasser A, Hur S (2020) Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat Immunol 21(1):17–29
Rothenfusser S et al (2005) The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol 175(8):5260–5268
Saito T et al (2007) Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci USA 104(2):582–587
Venkataraman T et al (2007) Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol 178(10):6444–6455
Satoh T et al (2010) LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A 107(4):1512–1517
Oshiumi H et al (2015) DDX60 is involved in RIG-I-dependent and independent antiviral responses, and its function is attenuated by virus-induced EGFR activation. Cell Rep 11(8):1193–1207
Oshiumi H et al (2010) DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur J Immunol 40(4):940–948
Miyashita M et al (2011) DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol Cell Biol 31(18):3802–3819
Grünvogel O et al (2015) DDX60L is an interferon-stimulated gene product restricting hepatitis C virus replication in cell culture. J Virol 89(20):10548–10568
Núñez RD, et al. (2018) The RNA Helicase DDX6 Associates with RIG-I to Augment Induction of Antiviral Signaling. Int J Mol Sci 19(7):1877
Pattabhi S et al (2019) DHX15 is a coreceptor for RLR signaling that promotes antiviral defense against RNA virus infection. J Interferon Cytokine Res 39(6):331–346
Zhu Q et al (2018) DHX29 functions as an RNA co-sensor for MDA5-mediated EMCV-specific antiviral immunity. PLoS Pathog 14(2):e1006886
Sugimoto N et al (2014) Helicase proteins DHX29 and RIG-I cosense cytosolic nucleic acids in the human airway system. Proc Natl Acad Sci USA 111(21):7747–7752
Ma Z et al (2013) DDX24 negatively regulates cytosolic RNA-mediated innate immune signaling. PLoS Pathog 9(10):e1003721
Bruns AM, Horvath CM (2015) LGP2 synergy with MDA5 in RLR-mediated RNA recognition and antiviral signaling. Cytokine 74(2):198–206
Murali A et al (2008) Structure and function of LGP2, a DEX(D/H) helicase that regulates the innate immunity response. J Biol Chem 283(23):15825–15833
Stone AEL, et al. (2020) DDX39A interacts with LGP2 to inhibit RLR responses. 204 (1): 683–683
Szymura SJ et al (2020) DDX39B interacts with the pattern recognition receptor pathway to inhibit NF-kappaB and sensitize to alkylating chemotherapy. BMC Biol 18(1):32
Brubaker SW et al (2014) A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell 156(4):800–811
Shi Y et al (2015) An autoinhibitory mechanism modulates MAVS activity in antiviral innate immune response. Nat Commun 6:7811
Gu L et al (2017) DDX3 directly regulates TRAF3 ubiquitination and acts as a scaffold to co-ordinate assembly of signalling complexes downstream from MAVS. Biochem J 474(4):571–587
Lu H et al (2014) DHX15 senses double-stranded RNA in myeloid dendritic cells. J Immunol 193(3):1364–1372
Mosallanejad K et al (2014) The DEAH-box RNA helicase DHX15 activates NF-κB and MAPK signaling downstream of MAVS during antiviral responses. Sci Signal 7(323):ra40
Liu Y et al (2014) The interaction between the helicase DHX33 and IPS-1 as a novel pathway to sense double-stranded RNA and RNA viruses in myeloid dendritic cells. Cell Mol Immunol 11(1):49–57
Zheng Q et al (2017) The RNA helicase DDX46 inhibits innate immunity by entrapping m(6)A-demethylated antiviral transcripts in the nucleus. Nat Immunol 18(10):1094–1103
Shi P et al (2020) SUMOylation of DDX39A alters binding and export of antiviral transcripts to control innate immunity. J Immunol 205(1):168–180
Saha SK, Cheng G (2006) TRAF3: a new regulator of type I interferons. Cell Cycle 5(8):804–807
Parvatiyar K et al (2012) The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat Immunol 13(12):1155–1161
Zhang Z et al (2011) The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12(10):959–965
Briard B et al (2020) DNA Sensing in the innate immune response. Physiology (Bethesda) 35(2):112–124
Lee KG et al (2015) Bruton’s tyrosine kinase phosphorylates DDX41 and activates its binding of dsDNA and STING to initiate type 1 interferon response. Cell Rep 10(7):1055–1065
Saitoh T et al (2009) Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 106(49):20842–20846
Abdul-Ghani M et al (2005) Abstrakt interacts with and regulates the expression of sorting nexin-2. J Cell Physiol 204(1):210–218
Zhao C, Zhao W (2019) TANK-binding kinase 1 as a novel therapeutic target for viral diseases. Expert Opin Ther Targets 23(5):437–446
Soulat D et al (2008) The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J 27(15):2135–2146
Schröder M et al (2008) Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. Embo j 27(15):2147–2157
Gu L et al (2013) Human DEAD box helicase 3 couples IκB kinase ε to interferon regulatory factor 3 activation. Mol Cell Biol 33(10):2004–2015
Fullam A et al (2018) DDX3 directly facilitates IKKα activation and regulates downstream signalling pathways. Biochem J 475(22):3595–3607
Tremblay N et al (2016) Spliceosome SNRNP200 promotes viral RNA sensing and IRF3 activation of antiviral response. PLoS Pathog 12(7):e1005772
Zhang K et al (2019) DDX19 inhibits type I interferon production by disrupting TBK1-IKKε-IRF3 interactions and promoting TBK1 and IKKε degradation. Cell Rep 26(5):1258-1272.e4
Feng T et al (2017) DEAD-box helicase DDX25 is a negative regulator of type I interferon pathway and facilitates RNA virus infection. Front Cell Infect Microbiol 7:356
Li D et al (2019) DDX56 inhibits type I interferon by disrupting assembly of IRF3-IPO5 to inhibit IRF3 nucleus import. J Cell Sci 133(5):jcs230409
Jefferies CA (2019) Regulating IRFs in IFN driven disease. Front Immunol 10:325
Harhaj EW, Dixit VM (2012) Regulation of NF-κB by deubiquitinases. Immunol Rev 246(1):107–124
Ishaq M et al (2009) The DEAD-box RNA helicase DDX1 interacts with RelA and enhances nuclear factor kappaB-mediated transcription. J Cell Biochem 106(2):296–305
Xiang N et al (2016) The DEAD-Box RNA Helicase DDX3 interacts with NF-κB Subunit p65 and suppresses p65-mediated transcription. PLoS ONE 11(10):e0164471
Ng YC et al (2018) A DNA-sensing-independent role of a nuclear RNA helicase, DHX9, in stimulation of NF-κB-mediated innate immunity against DNA virus infection. Nucleic Acids Res 46(17):9011–9026
Tetsuka T et al (2004) RNA helicase A interacts with nuclear factor kappaB p65 and functions as a transcriptional coactivator. Eur J Biochem 271(18):3741–3751
Tanaka K et al (2020) Knockdown of DEAD-box RNA helicase DDX5 selectively attenuates serine 311 phosphorylation of NF-kappaB p65 subunit and expression level of anti-apoptotic factor Bcl-2. Cell Signal 65:109428
Tsuchiya R et al (2015) Downregulation of diacylglycerol kinase zeta enhances activation of cytokine-induced NF-kappaB signaling pathway. Biochim Biophys Acta 2:361–369
Malik A, Kanneganti TD (2017) Inflammasome activation and assembly at a glance. J Cell Sci 130(23):3955–3963
Hornung V (2014) SnapShot: nucleic acid immune sensors, part 2. Immunity 41(6):1066-1066.e1
Liu D et al (2020) Advances in the molecular mechanisms of NLRP3 inflammasome activators and inactivators. Biochem Pharmacol 175:113863
Samir P et al (2019) DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573(7775):590–594
Kesavardhana S et al (2021) DDX3X coordinates host defense against influenza virus by activating the NLRP3 inflammasome and type I interferon response. J Biol Chem 1:100579
Banerjee S (2016) RNase L and the NLRP3-inflammasome: an old merchant in a new trade. Cytokine Growth Factor Rev 29:63–70
Mitoma H et al (2013) The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity 39(1):123–135
Li J et al (2015) DDX19A senses Viral RNA and Mediates NLRP3-dependent inflammasome activation. J Immunol 195(12):5732–5749
Wang P et al (2015) Nlrp6 regulates intestinal antiviral innate immunity. Science 350(6262):826–830
Xing J et al (2021) DHX15 is required to control RNA virus-induced intestinal inflammation. Cell Rep 35(12):109205
Zhu S et al (2017) Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546(7660):667–670
Raftery N, Stevenson NJ (2017) Advances in anti-viral immune defence: revealing the importance of the IFN JAK/STAT pathway. Cell Mol Life Sci 74(14):2525–2535
Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14(1):36–49
Lumb JH et al (2017) DDX6 represses aberrant activation of interferon-stimulated genes. Cell Rep 20(4):819–831
Khsheibun R et al (2014) Gene expression profiling of the response to interferon beta in Epstein-Barr-transformed and primary B cells of patients with multiple sclerosis. PLoS ONE 9(7):e102331
Chen G et al (2008) p72 DEAD box RNA helicase is required for optimal function of the zinc-finger antiviral protein. Proc Natl Acad Sci USA 105(11):4352–4357
Ye P et al (2010) DEXH-Box protein DHX30 is required for optimal function of the zinc-finger antiviral protein. Protein Cell 1(10):956–964
Hur S (2019) Double-stranded RNA sensors and modulators in innate immunity. Annu Rev Immunol 37:349–375
García MA et al (2006) Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70(4):1032–1060
Sadler AJ et al (2009) An antiviral response directed by PKR phosphorylation of the RNA helicase A. PLoS Pathog 5(2):e1000311
Stavrou S et al (2018) DDX41 recognizes RNA/DNA retroviral reverse transcripts and is critical for in vivo control of murine leukemia virus infection. MBio 9(3):e00923-e1918
Boeras I et al (2016) DHX9/RHA binding to the PBS-segment of the genomic RNA during HIV-1 assembly bolsters virion infectivity. J Mol Biol 428(11):2418–2429
Zhou X et al (2013) DDX5 facilitates HIV-1 replication as a cellular co-factor of Rev. PLoS ONE 8(5):e65040
Chen G et al (2020) A double-stranded RNA platform is required for the interaction between a host restriction factor and the NS1 protein of influenza A virus. Nucleic Acids Res 48(1):304–315
Nelson C et al (2021) Human Ddx3x unwinds Japanese encephalitis and Zika Viral 5’ terminal regions. Int J Mol Sci 22(1):413
Acknowledgements
We apologize to all colleagues whose contributions were not discussed and cited owing to space constraints.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
CS and TY wrote the manuscript. CZ reviewed and modified the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
No conflicting interest.
Ethics approval
Not applicable.
Consent for publication
We all agree to publish this review.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Su, C., Tang, Yd. & Zheng, C. DExD/H-box helicases: multifunctional regulators in antiviral innate immunity. Cell. Mol. Life Sci. 79, 2 (2022). https://doi.org/10.1007/s00018-021-04072-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-021-04072-6