
Abstract
Background
SARS-Cov-2 is a single-stranded RNA virus, a Betacoronavirus, composed of 16 non-structural proteins, with specific roles in replication of coronaviruses. The pathogenesis of COVID-19 is not yet fully understood. The virus and host factors interplay among distinct outcomes of infected patients.
Methods
Using MeSH (Medical Subject Headings) in PubMed, authors searched for articles cotaining information on COVID-19 and the skin.
Results
The pathophysiology of the disease is multifactorial: association with innate immune response, hypercoagulability state, lung tissue damage, neurological and/or gastrointestinal tract involvement, monocytic/macrophage activation syndrome, culminating in exaggerated cytokine secretion, called “cytokine storm”, which leads to worsening and death. These systemic conditions may be associated with cutaneous lesions, that have polymorphic aspects, where at histopathological level show involvement in different skin changes. These lesions may be associated with multisystemic manifestations that could occur due to angiotensin-converting enzyme 2 receptor and transmembrane serine protease action, allowing the pulmonary infection and possibly skin manifestation. Several reports in literature show cutaneous lesions similar to chilblain, urticarial eruptions, diffuse or disseminated erythema, livedo racemosa, blue toe syndrome, retiform purpura, vesicle trunk, purpuric exanthema or exanthema with clinical aspects of symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) and others.
Conclusions
This review describes the complexity of Covid-19, pathophysiological and clinical aspects, dermatological finding and other dermatological conditions associated with SARS-CoV-2 infection or COVID-19.
Similar content being viewed by others


Introduction
The 2019 novel beta-coronavirus (2019-nCoV) or the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a new worldwide public health crisis has rapidly spread from its origin in Wuhan City of Hubei Province of China, in December 2019 [1]. So far, May 12-2020 a data chart of Coronavirus Resource Center of the John Hopkins University (USA) at 12:34:40 PM has totalized 4,210,079 COVID-19 cases around the world, with 287,156 deaths, and 1,470,598 recovered patients in 187 countries/regions [2].
Cutaneous manifestations reports published in periodicals indexed in PubMed are occasionally rising, but often clinical images and/or histopathological findings of these lesions are not included. Using MeSH (Medical Subject Headings) in PubMed, authors searched for “cutaneous and COVID-19”, as well as, “skin and COVID-19”, making it possible to retrieve more than 160 articles. Moreover, these papers have published many aspects from patients’ cutaneous manifestations with COVID-19 [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38] to economic impact [40] and protective measures for the cutaneous system during COVID-19 exposure [41,42,43,44,45,46,47,48,49,50,51,52]. Another aspect are skin damages in healthcare workers [42, 49, 53,54,55,56,57,58], medical education and telemedicine during the pandemic [59,60,61,62], the use of immunomodulators [63, 64]; immunosuppressors and immunobiological agents in dermatology [65], as well as, in rheumatology skin conditions [63, 66, 67].
What was reported about cutaneous lesions in COVID-19 patients?
Concerning integumentary clinical manifestations, unfortunately, we cannot access clinical images or histopathological registers of part of such cases reported until now. Some authors described these cutaneous lesions under distinct dermatological terms: erythematous rash [4], urticarial eruptions [18], varicella-like vesicles [4], chilblain-like lesions [7], acrocyanosis [8], retiform purpura [11], livedo [13], among others.
Probably due to lack of adequate personal protective equipment (PPE) for frontline health care workers, including respirators, face shields, gloves, ocular glasses, gowns, and hand sanitizers, dermatologists have not adequately registered the cutaneous findings in COVID-19 patients [18]. Viral infections can produce specific clinical and non-specific manifestations, due to the direct action in infected human cells or as a phenomenon of immune system hyperactivity. Since some of the associations are considered to be either causal or probably causal whereas others are not, it is useful to consider, through specific case studies, what clinical evidence is well-accepted to establish a causal relation, and which factors may be dispensable [68].
Regarding dermatological manifestations reported until May of 2020, related to COVID-19, we summarized the case studies described in Table 1.
In addition to cases mentioned above, the prospective study conducted by Galván Casas et al. [39] in Spain reported 375 cases of skin lesions associated with COVID-19, classifying them in 5 different patterns. It is important to state that, for the first time, a temporal relationship between skin lesions and other systemic symptoms, and moreover, a severity of the disease was created. The study showed that vesicular eruptions appeared in the early course of the disease, even before other symptoms. Also, that pseudo-chilblain pattern appeared at a later time during the evolution of the disease and in most cases after other symptoms. When associating severity, the authors described a gradient, from less to most severe disease, illustrating pseudo-chilblain with less severe pulmonary disease in contrast to livedoid presentations which were associated with worse pulmonary diagnosis once these patients required intensive care treatment [39].
It is remarkably interesting that PubMed indexed literature, up to now has retrieved not over 600 cutaneous manifestations in COVID-19 cases reported, despite over 4 million COVID-19 patients [1]. Many correlated possibilities are as follows: under cutaneous manifestation notifications, the presence of severe pulmonary COVID-19, and physicians do not take into account that the skin also needs careful observation, lack of dermatologists in hospitals due to social isolation, patients with mild COVID-19 who have not referred cutaneous manifestations to their general practitioners, among others. However, dermatologists should conduct more photograph registrations and cutaneous biopsies for better scientific documentation, as well as, adequate differential diagnosis when assisting such patients.
Recently, Jones et al. [69] described a case of a 6-month-old infant diagnosed with classic Kawasaki disease, at the same time screened positive for COVID-19 with fever and minimal respiratory symptoms believing there may be a potential association between both diseases. Kawasaki disease has previously been associated with other pathogens, including coronaviruses [70,71,72,73]. In an article by Principi et al. [70], there was histological evidence suggested that Kawasaki disease has an infectious origin, with persistent intracytoplasmic inclusion bodies and RNA staining which may be a clue of a viral infection that shall persist indefinitely. They also reported that the first site of infection was the respiratory tract, and later dissemination occurred through macrophages to all body sites, including the medium-sized arteries. This can occur by the direct action of SARS-COV-2 on the complement system and generating MAC (C5b-9), ending with endothelial and coronary arteries damage, as well as, thrombosis by the procoagulant surface [11].
Possible actions of SARS-CoV-2 on human skin and the resulting potential dermatological manifestations
We highlight some interesting aspects of SARS-CoV-2 infections for dermatologists and their possible correlation with the skin:
-
1.
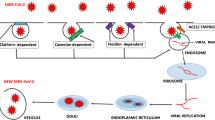
SARS-Cov-2 is a single-stranded RNA virus composed of 16 non-structural proteins (named as NSP 1-16) with specific roles in the replication of coronaviruses (CoVs) [74]. One of them, NSP3, has the property to block host’s innate immune response and to promote cytokine expression. The other two, NSP5 can inhibit interferon (IFN) signaling and NSP16 avoids MAD5 (melanoma differentiation-associated gene 5) recognition, depressing innate immunity [74]. Four proteins are structural and essential for viral assembly and infection: homotrimers of S proteins (spikes on the viral surface), M protein (three transmembrane domains), E protein (involved in viral pathogenesis), and N protein (2 domains, both of which can bind virus RNA genome) [74].
-
2.
Aerosolized uptake of SARS-Cov-2 leads to infection of angiotensin-converting enzyme (ACE) type II (ACE2) expressing target cells such as alveolar type 2 (that produce lung surfactant) or other unknown target cells [75].
-
3.
Dendritic cells, monocytes, and macrophages are the first cellular lineage to fight viral infections. The interferons type I (α and β) are the danger signal for the human body during this clinical setting. Protective immune responses to viral infection are initiated by innate immune sensors that survey extracellular and intracellular space for foreign nucleic acids [76]. Enzymes that metabolize or modify endogenous nucleic acids are critical for preventing an upregulation activation of the innate antiviral response [76]. Humans and their primate ancestors have been under intensive selective pressure due to viral infections for tens of millions of years [76]. These events occurred due to viruses like influenza, polio, measles, and smallpox in the recent past of our history [75]. Antiviral proteins that are at the front lines of immune defense are the main targets of virus-encoded antagonists.
Protein suppressors and/or others involved in upregulation of the human immune innate responses can emerge from mutant genes, which appear randomly in each generation, restore the antiviral response and provide an advantage to the host immune defense to a specific viral infection [76]. Inadequate negative or positive regulation of innate immune receptors, may lead to signals that stimulate nucleic acid and subsequent protein transcription, which can occur as monogenic genetic disorders, with gain in function (GOF) or loss of function (LOF). These are known as type I interferonopathies or other autoinflammatory diseases [76,77,78,79,80,81].
Some studies have shown direct T cell viral infection by the detection SARS-like viral particles and SARS-CoV RNA in T lymphocytes from peripheral blood sample, spleen, lymph nodes, and lymphoid tissue of various organs [82]. It is questioned whether the alveolar macrophages can internalize the viral particles and then transfer to lymphocytes [82]. However, the direct attack on other organs by disseminated SARS-CoV-2, the immune pathogenesis caused by the systemic cytokine storm, and the microcirculation dysfunctions together lead to viral sepsis [82].
-
4.
In COVID-19, like MERS-CoV, the pathologies are not yet fully understood, moreover viral and host factors play a key role in these infections. However, it should be noted that immunopathogenesis is associated with an out-of-control immune response, which may result in pulmonary tissue damage, functional impairment, and reduced lung capacity [83]. Chemotactic factors are essential to the immune responses against the virus infections, given their regulatory effect on dilations and positions of leukocytes in the host lungs [83]. Therefore, spectral changes in chemotactic factors may lead to severely maladjusted immune responses [83]. Immune insufficiency or misdirection may increase viral replication and cause tissue damages [83]. In a subset of patients, by the end of the first week, the disease can progress to pneumonia, respiratory failure, and death [1]. This progression is related to an extreme rise in inflammatory cytokines including interleukin (IL)2, IL7, IL10, GCSF, IP10, MCP1, MIPI A, and TNFα [1]. The increase in the pro-inflammatory cytokines, in particular, IL6 is associated with severe pneumonia and it can have deleterious effects on the adaptive immune system [8, 84]. In these subsets of patients, overactive immune responses may induce immunopathological conditions, named as “cytokine storm” and in some individuals leads to macrophage activation syndrome (MAS)-like, often causing a fatal outcome [74, 83, 85, 86]. Cytokines could reach the skin and stimulate dermal dendritic cells, macrophages, mast cells and lymphocytes, in addition to polymorphonuclear cells and promote eruptions such as erythema, urticarial lesions, vesicles and others. These have already been reported in situations with cytokine release during similar situations in patients with systemic lupus erythematosus, antiphospholipid syndrome, adult Still’s disease [81]. Magro et al. [11] found complement deposition (C5b-9 and C4d) using immunohistochemistry in dermal capillaries of patients with retiform purpura. This may play a role in pathogenicity, as the data present co-localization of products linked to complement activation with SARS-CoV-2 spike glycoproteins. Llamas-Velasco et al. [87] observed endothelial damage not only in kidney, small bowel and lung, but also in the dermo-hypodermal junction. These biological self-aggressive host behaviors run in the same pattern of the monogenic type I interferonpathies, but in COVID-19 is an acquired viral disease, acute and has a faster clinical evolution. An IL6 blockage, with JAK inhibitors, is one of the possible targeted treatments currently in clinical trial studies [88, 89].
-
5.
Hammining et al. [90] identified in 2004 the metallopeptidase named angiotensin-converting enzyme 2 (ACE2) as the functional receptor for SARS-CoV responsible for an epidemic outbreak during 2003–2004. Using IHC methods, these authors demonstrated that the most remarkable finding was the surface expression of ACE2 protein on lung alveolar epithelial cells (pneumocytes), macrophages, and enterocytes of the small intestine. Furthermore, ACE2 was present in arterial and venous endothelial cells and arterial smooth muscle cells in all organs studied, including the skin in the basal layer of the epidermis, endothelial cells of dermal blood vessels and eccrine adnexal tissue [90,91,92,93].
With electron microscopy, Varga et al. [91] revealed viral inclusion structures in endothelial cells across vascular beds of different organs in some patients with COVID-19. This showed direct viral infection of the endothelial cell and diffuse endothelial inflammation [91]. The ACE2 receptor is also widely expressed on endothelial cells in multiple organs, suggesting that endotheliitis could occur in several sites as a direct consequence of viral involvement and host inflammatory response [91]. COVID-19-endotheliitis could explain the systemic impaired microcirculatory function in different vascular beds and their clinical sequelae in patients with COVID-19 [91].
-
6.
A transmembrane protease, serine 2 (TMPRSS2), a type II transmembrane serine protease (TTPS), plays a critical role in SARS and MERS coronavirus (CoV) and in 2003 Asian H7N9 influenza virus and several H1N1 subtype influenza A viruses infections, indicating that TMPRSS2 could be a novel antiviral strategy to treat coronavirus and some low pathogenic influenza virus infections [94]. SARS-Cov-2 (viral agent of the COVID-19) and SARS-CoV bind to ACE2 by the protein S (Spike) and allows the virus to enter and infect cells [95]. In order for the virus to complete entry into the cell following this initial process, the spike protein has to be primed by a protease (TMPRSS2) to complete this process [95]. In order to attach virus receptor (spike protein, S) to host cellular ligand (ACE2), activation by TMPRSS2 as a protease is needed [95]. Bjerregaard et al. [93] studied the expression of ACE2 in various human tissues and it was found on the skin; therefore, for the cleavage protease action of TMPRSS2, it could occur on the skin by the enzyme itself, which has yet to be demonstrated by immunohistochemistry studies, or by other proteases such as human tryptase, that could be released by resident cells of the dermis, such as perivascular mast cells. This may open a window of opportunity for future studies.
-
(i)
TMPRSS2 gene is located on human chromosome 21, and a significant feature of the TMPRSS2 gene is that several androgen receptor elements (AREs) are located upstream of the transcription start site and the first intron [94]. TMPRSS2 gene encodes a predicted protein of 492 amino acids which anchors to the plasma membrane, and after autocatalytic cleavage, a noticeable portion of them can gain blood circulation [94]. TMPRSS2 is predominantly expressed in the prostate, with a relatively lower level of expression in lungs, colon, liver, kidneys, and pancreas [94].
-
(ii)
Recently, Wambier and Goren [96] proposed that COVID-19 could affect the male gender more due to this relation between TMPRSS2 and androgen levels, since androgenic hormones stimulate this transmembrane growth. Goren et al. [97] demonstrated a greater prevalence of COVID-19 in male gender among patients in Spain proposing a potential clue to the role of androgens in COVID-19 severity due to the higher prevalence of androgenetic alopecia among patients with COVID-19, both in men and women hospitalized with the disease.
-
(iii)
Several members of the subfamily of Coronavirinae use peptidases as receptors for host cell entry: most alpha coronaviruses use CD13, and among betacoronavirus’ species, the novel coronavirus MERS binds to CD26, and both SARS-CoV and the human coronavirus NL63 engage the carboxypeptidase ACE2. ACE2 expression in human membrane cell protects against acute respiratory distress syndrome [98]. Cleavage of the SARS-CoV S protein (SARS-S) by host cell proteases is essential for viral infectivity, and the responsible enzymes constitute potential targets for intervention [99]. The SARS-CoV can hijack two cellular proteolytic systems to ensure the adequate processing of its S protein [99]. Cleavage of SARS-S can be facilitated by cathepsin L, a pH-dependent endo-/lysosomal host cell protease, upon uptake of virions into target cell endosomes [99]. Alternatively, TMPRSS2 and human airway trypsin-like protease (HAT) can activate SARS-S, presumably by cleavage of SARS-S at or close to the cell surface, and activation of SARS-S by TMPRSS2 allows for cathepsin L-independent cellular entry [99]. Both TMPRSS2 and HAT are expressed in ACE2-positive cells in the human lung, and results obtained with surrogate cell culture systems suggest that TMPRSS2 might play a significant role in SARS-CoV spread in the human respiratory tract [99]. Heurich et al. [99] suggested that TMPRSS2 and HAT impact SARS-S-driven entry via two independent mechanisms: ACE2 cleavage by these proteases increases entry efficiency, while SARS-S cleavage by TMPRSS2 activates the S protein for cathepsin L-independent host cell entry. Meanwhile, human airway trypsin-like protease (HAT), and TMPRSS2 (transmembrane protease, serine 2) are known to cleave the glycoprotein hemagglutinin (HA) of influenza A viruses, a prerequisite for the fusion between viral and host cell membranes and viral cell entry [100]. Cleavage of HA is critical for viral infection, with the tissue distribution of proteases determining cell tropism of virus strains [101].
Until the present day, the cardinal points in severe COVID-19 are upregulated innate immune human response; hypercoagulable state; polymorphous clinical manifestations, due to pulmonary tissue damage, neurological and/or gastrointestinal tract involvement; and fatal outcome in severe cases of macrophage activation syndrome-like (MAS) [102].
Additional considerations
Other possibilities in developing ischemic/coagulopathy lesions as livedo racemosa, retiform purpura, and acro-ischemia in COVID-19 patients, besides activation of coagulation system [11] due to viral load, it is the presence in some subjects in selected populations with a background of thrombophilia factors. Coagulopathy is shown to be associated with higher mortality and the use of anticoagulants has been reported with variable outcomes [103]. However, some reports have shown more promising results with the use of soluble thrombomodulin and heparin [104].
Afro-Americans have genetically and increased serum levels of lipoprotein a [Lp(a)] than Caucasians and Asians subjects. Lp(a) is an isolated risk factor for cardiovascular, peripheral arterial and cerebrovascular diseases, and impaired fibrinolysis, because it competes with plasminogen, and has prothrombogenic properties. There are ethnic differences in Lp(a) levels because subjects of African descent have about twice as high levels as Caucasians, Hispanics, and many Asian populations, while intermediate levels are reported for South Asians [105]. This observed interethnic difference could also be due to the apo(a) allele distribution in the subset of the population that presumably left Africa and subsequently gave rise to other population groups [105, 106]. We might make a parallel clinical setting with multi-organ inflammation and hypercoagulability in severe COVID-19, besides complement system activation, and endothelial dysfunction demonstrated by Magro et al. [11]. Enkhmaa et al. [105] suggested a significant association between elevated serum amyloid A, an HDL-associated systemic inflammatory biomarker, and a higher allele-specific Lp(a) level for smaller apo(a) size was found in African and Afro-Americans subjects.
Taken together, these findings suggest a potential for an additive effect between molecular properties of Lp(a), in particular, small size apo(a) (with elevated serum levels), and inflammation in promoting Lp(a)-associated CVD risk [105]. Furthermore, fibrinogen was positively correlated with Lp(a) levels in Japanese and Caucasians and independently predicted levels [105,106,107,108]. Consistent with this finding, fibrinogen was also significantly associated with Lp(a) levels in older Italian subjects [105, 109]. An inflammatory score summarizing the intensity of the pro-inflammatory state based on four different biomarkers (CRP, fibrinogen, IL-6, and IL-1 receptor antagonist) was significantly correlated with Lp(a) concentration in this study [109]. Among Spanish Caucasian subjects with metabolic syndrome, the CRP concentration was twofold greater in subjects with high Lp(a) concentrations (≥ 30 mg/dl) [110].
Otherwise, Caucasian patients have more prevalence of minor allele frequency for Factor V (Leiden) mutation (rs6025) (0.6–7.3%) among different regions of Europe, than other ethnic groups, as Hispanics (0.4–1.4%), Asians (0.0–3.8%), and Afro-Americans (0.6–0.7%), but almost similar to African subjects (1.0–10.2%) [111]. Thus, in the presence of genetic thrombophilia in a patient with COVID-19, there is theoretically a greater possibility of thrombotic events occurring, both in the macro and microcirculation.
ABO gene polymorphism rs8176719, the insG allele which produces non-O blood group phenotype confers a 1.5-fold risk of developing venous thromboembolism (VTE), presumably because of a 25% increase levels of von Willebrand factors and factor VIII of coagulation in subjects of non-O blood groups, then in those with O blood group [111,112,113].
Taking all data together, these thrombophilic genetic conditions might begin to explain the elevated number of fatalities among American, Afro-American, and European populations when compared to Asian subjects in the clinical setting of severe COVID-19.
When discussing possible actions of SARS-CoV-2 on human skin that could result in potential dermatological manifestations, there is still room for more investigation. Excessive activation of inflammatory mediators creating a “cytokine storm”, leading to damage to the endothelium; formation of multiple thromboses in the microvasculature of the skin; changes in the cellular component of immunity with activation of the complement system, as well as, the possibility of direct entry of SARS-CoV-2 entry via receptor ACE2 and protease TMPRSS2 in the human endothelial cell in dermal blood vessels cannot be excluded such as possible mechanisms if the possibility of virus circulation in the blood is proved. In any case, the problem of skin manifestations pathogenesis in patients with COVID-19 needs further investigation.
Conclusions
Several viral infections are related to eliciting innate and adaptive human immune responses. In some circumstances, as previous SARS-Cov, MERS-Cov, and SARS-CoV-2 the monocytic–macrophage system may produce an upregulated immune response (sHLH/MAS-like), severe inflammatory systemic state and damage in lungs and other internal organs, often hematological system, gastrointestinal tract, and kidneys. The activation of mast cells and basophils, by direct and/or indirect viral effect, is a possible event and it is important to be alert to skin manifestations such as the onset of urticaria, atopic dermatitis, or the exacerbation of these conditions, rashes, neutrophilic dermatoses and skin manifestations of hypercoagulable states, such as acral ischemia. Considering the cutaneous manifestations described, some aspects of SARS-CoV-2 infection may be caused by cytopathic effects on the endothelium dermal vessels or even stimulated by cytokines in arterioles and capillaries. The direct cytopathic effect of SARS-CoV-2 can occur in the described vesicular or papular–vesicular lesions, which are very similar to those caused by the Herpesviridae family. Rashes may be para-viral due to cytokines or due to drug exposure during treatment of the disease. Photography and anatomopathological studies are critical for the correct diagnosis and for establishing differential diagnosis with other cutaneous or systemic conditions, which may or may not be chronologically and causally related to COVID-19 or be casual events.
References
Singhal T. A review of coronavirus disease-2019 (COVID-19). Indian J Pediatr. 2020;87(4):281–6.
Coronavirus Resource Center of the John Hopkins University. Disposable on https://coronavirus.jhu.edu/map.html, 3.
Recalcati S. Cutaneous manifestations in COVID-19: a first perspective. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16387.
Henry D, Ackerman M, Sancelme E, Finon A, Esteve E. Urticarial eruption in COVID-19 infection. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16472.
Kamali Aghdam M, Jafari N, Eftekhari K. Novel coronavirus in a 15-day-old neonate with clinical signs of sepsis, a case report. Infect Dis (Lond). 2020. https://doi.org/10.1080/23744235.2020.1747634.
Joob B, Wiwanitkit V. COVID-19 can present with a rash and be mistaken for dengue. J Am Acad Dermatol. 2020;82(5):e177. https://doi.org/10.1016/j.jaad.2020.03.036.
Alramthan A, Aldaraji W. A case of COVID-19 presenting in clinical picture resembling chilblains disease. First report from the Middle East. Clin Exp Dermatol. 2020. https://doi.org/10.1111/ced.14243.
Zhang Y, Cao W, Xiao M, Li YJ, Yang Y, Zhao J, Zhou X, Jiang W, Zhao YQ, Zhang SY, Li TS. Clinical and coagulation characteristics of 7 patients with critical COVID-2019 pneumonia and acro-ischemia. Zhonghua Xue Ye Xue Za Zhi. 2020;41:E006. https://doi.org/10.3760/cma.j.issn.0253-2727.2020.0006.
Li Taisheng, Hongzhou Lu, Zhang Wenhong. Clinical observation and management of COVID-19 patients. Emerg Microbes Infect. 2020;9(1):687–90. https://doi.org/10.1080/22221751.2020.1741327.
Mazzotta F, Toccoli T. Acute acro-ischemia in the child at the time of COVID-19. Eur J Ped Dermatol. https://www.ejpd.com/en/Mondayscase. Accessed 9 Apr 2020.
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, Baxter-Stoltzfus A, Laurence J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020. https://doi.org/10.1016/j.trsl.2020.04.007.
Marzano AV, Genovese G, Fabbrocini G, Pigatto P, Monfrecola G, Piraccini BM, Veraldi S, Rubegni P, Cusini M, Caputo V, Rongioletti F, Berti E, Calzavara-Pinton P. Varicella-like exanthem as a specific COVID-19-associated skin manifestation: multicenter case series of 22 patients. J Am Acad Dermatol. 2020. https://doi.org/10.1016/j.jaad.2020.04.044.
Manalo IF, Smith MK, Cheeley J, Jacobs R. A dermatologic manifestation of COVID-19: transient livedo reticularis. J Am Acad Dermatol. 2020. https://doi.org/10.1016/j.jaad.2020.04.018.
Estébanez A, Pérez-Santiago L, Silva E, Guillen-Climent S, García-Vázquez A, Ramón MD. Cutaneous manifestations in COVID-19: a new contribution. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16474.
Mahé A, Birckel E, Krieger S, Merklen C, Bottlaender L. A distinctive skin rash associated with coronavirus disease 2019? J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16471.
Jimenez-Cauhe J, Ortega-Quijano D, Prieto-Barrios M, Moreno-Arrones OM, Fernandez-Nieto D. Reply to “COVID-19 can present with a rash and be mistaken for Dengue”: petechial rash in a patient with COVID-19 infection. J Am Acad Dermatol. 2020. https://doi.org/10.1016/j.jaad.2020.04.016.
Ehsani AH, Nasimi M, Bigdelo Z. Pityriasis rosea as a cutaneous manifestation of COVID-19 infection. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16579.
Lu S, Lin J, Zhang Z, Xiao L, Jiang Z, Chen J, Hu C, Luo S. Alert for nonrespiratory symptoms of Coronavirus Disease (COVID-19) patients in epidemic period: a case report of familial cluster with three asymptomatic COVID-19 patients. J Med Virol. 2020. https://doi.org/10.1002/jmv.25776.
Fernandez-Nieto D, Ortega-Quijano D, Segurado-Miravalles G, Pindado-Ortega C, Prieto-Barrios M, Jimenez-Cauhe J. Comment on: Cutaneous manifestations in COVID-19: a first perspective. Safety concerns of clinical images and skin biopsies. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16470.
Dong X, Cao YY, Lu XX, Zhang JJ, Du H, Yan YQ, Akdis CA, Gao YD. Eleven faces of coronavirus disease 2019. Allergy. 2020. https://doi.org/10.1111/all.14289.
van Damme C, Berlingin E, Saussez S, Accaputo O. Acute urticaria with pyrexia as the first manifestations of a COVID-19 infection. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16523.
Morey-Olivé M, Espiau M, Mercadal-Hally M, Lera-Carballo E, García-Patos V. Cutaneous manifestations in the current pandemic of coronavirus infection disease (COVID 2019). An Pediatr (Engl Ed). 2020. https://doi.org/10.1016/j.anpede.2020.04.002.
Paolino G, Canti V, Raffaele Mercuri S, Rovere Querini P, Candiani M, Pasi F. Diffuse cutaneous manifestation in a new mother with COVID-19 (SARS-Cov-2). Int J Dermatol. 2020. https://doi.org/10.1111/ijd.14919.
Tosti G, Barisani A, Queirolo P, et al. Skin signs resembling vascular acrosyndromes during the COVID-19 outbreak in Italy. Clin Exp Dermatol. 2020. https://doi.org/10.1111/ced.14267.
Bouaziz JD, Duong T, Jachiet M, et al. Vascular skin symptoms in COVID-19: a French observational study. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16544.
Amatore F, Macagno N, Mailhe M, et al. SARS-CoV-2 infection presenting as a febrile rash. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16528.
Diaz-Guimaraens B, Dominguez-Santas M, Suarez-Valle A, et al. Petechial skin rash associated with severe acute respiratory syndrome coronavirus 2 infection. JAMA Dermatol. 2020. https://doi.org/10.1001/jamadermatol.2020.1741.
Tammaro A, Adebanjo GAR, Parisella FR, Pezzuto A, Rello J. Cutaneous manifestations in COVID-19: the experiences of Barcelona and Rome. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16530.
Avellana Moreno R, Villa E, Avellana Moreno V, Estela Villa C, Aparicio M, Fontanella A. Cutaneous manifestation of COVID-19 in images: a case report. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16531.
Recalcati S, Barbagallo T, Frasin LA, Prestinari F, Cogliardi A, Provero MC, Dainese E, Vanzati A, Fantini F. Acral cutaneous lesions in the time of COVID-19. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16533.
Zengarini C, Orioni G, Cascavilla A, Horna Solera C, Fulgaro C, Misciali C, Patrizi A, Gaspari V. Histological pattern in Covid-19 induced viral rash. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16569.
Gianotti R, Veraldi S, Recalcati S, Cusini M, Ghislanzoni M, Boggio F, Fox LP. Cutaneous clinico-pathological findings in three COVID-19-positive patients observed in the metropolitan area of Milan, Italy. Acta Derm Venereol. 2020. https://doi.org/10.2340/00015555-3490.
Ahouach B, Harant S, Ullmer A, Martres P, Bégon E, Blum L, Tess O, Bachmeyer C. Cutaneous lesions in a patient with COVID-19: are they related? Br J Dermatol. 2020. https://doi.org/10.1111/bjd.19168.
Piccolo V, Neri I, Filippeschi C, Oranges T, Argenziano G, Battarra VC, Berti S, Manunza F, Belloni Fortina A, Di Lernia V, Boccaletti V, De Bernardis G, Brunetti B, Mazzatenta C, Bassi A. Chilblain-like lesions during COVID-19 epidemic: a preliminary study on 63 patients. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16526.
Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2002032.
Zhang Y, Xiao M, Zhang S, et al. Coagulopathy and antiphospholipid antibodies in patients with COVID-19. N Engl J Med. 2020. https://doi.org/10.1056/NEJMc2007575.
Hoehl S, Rabenau H, Berger A, et al. Evidence of SARS-CoV-2 infection in returning travelers from Wuhan, China. N Engl J Med. 2020;382(13):1278–80.
Ma J, Xia P, Zhou Y, et al. Potential effect of blood purification therapy in reducing cytokine storm as a late complication of critically ill COVID-19. Clin Immunol. 2020;214:108408. https://doi.org/10.1016/j.clim.2020.108408.
Galván Casas C, et al. Classification of the cutaneous manifestations of COVID-19: a rapid prospective nationwide consensus study in Spain with 375 cases. Br J Dermatol. 2020. https://doi.org/10.1111/bjd.19163.
Gupta M, Abdelmaksoud A, Jafferany M, Lotti T, Sadoughifar R, Goldust M. COVID-19 and economy. Dermatol Ther. 2020. https://doi.org/10.1111/dth.13329.
Yan Y, Chen H, Chen L, et al. Consensus of Chinese experts on protection of skin and mucous membrane barrier for health-care workers fighting against coronavirus disease 2019. Dermatol Ther. 2020. https://doi.org/10.1111/dth.13310.
Elston DM. Occupational skin disease among health care workers during the coronavirus (COVID-19) epidemic. J Am Acad Dermatol. 2020;82(5):1085–6. https://doi.org/10.1016/j.jaad.2020.03.012.
Tao J, Song Z, Yang L, Huang C, Feng A, Man X. Emergency management for preventing and controlling nosocomial infection of 2019 novel coronavirus: implications for the dermatology department. Br J Dermatol. 2020. https://doi.org/10.1111/bjd.19011.
Jakhar D, Kaur I, Kaul S. Art of performing dermoscopy during the times of coronavirus disease (COVID-19): simple change in approach can save the day! J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16412.
Cavanagh G, Wambier C. Rational hand hygiene during COVID-19 pandemic. J Am Acad Dermatol. 2020. https://doi.org/10.1016/j.jaad.2020.03.090.
Chandrasekharan P, Vento M, Trevisanuto D, Partridge E, Underwood MA, Wiedeman J, Katheria A, Lakshminrusimha S. Neonatal resuscitation and post-resuscitation care of infants born to mothers with suspected or confirmed SARS-CoV-2 infection. Am J Perinatol. 2020. https://doi.org/10.1055/s-0040-1709688.
Der Sarkissian SA, Kim L, Veness M, Yiasemides E, Sebaratnam DF. Recommendations on dermatologic surgery during the COVID-19 pandemic. J Am Acad Dermatol. 2020. https://doi.org/10.1016/j.jaad.2020.04.034.
Rudnicka L, Gupta M, Kassir M, Jafferany M, Lotti T, Sadoughifar R, Goldust M. Priorities for global health community in COVID-19 pandemic. Dermatol Ther. 2020. https://doi.org/10.1111/dth.13361.
Lan J, Song Z, Miao X, et al. Skin damage among health care workers managing coronavirus disease-2019. J Am Acad Dermatol. 2020;82(5):1215–6. https://doi.org/10.1016/j.jaad.2020.03.014.
Yu N, Wu L, Su J, Huang K, Zhao S, Chen X. One more paper towel, longer protection. J Am Acad Dermatol. 2020. https://doi.org/10.1016/j.jaad.2020.03.064.
Türsen Ü, Türsen B, Lotti T. Coronavirus-days in dermatology. Dermatol Ther. 2020. https://doi.org/10.1111/dth.13438.
Gupta S, Jangra RS, Gujrathi AV, Mahendra A, Singla R, Sharma A, Gupta S. A novel clinical set up for examining healthy dermatology outpatients in time of Covid-19. Clin Exp Dermatol. 2020. https://doi.org/10.1111/ced.14246.
Lee SW, Cheong SH, Byun JY, Choi YW, Choi HY. Occupational hand eczema among nursing staffs in Korea: self-reported hand eczema and contact sensitization of hospital nursing staffs. J Dermatol. 2013;40:182–7.
Hamnerius N, Svedman C, Bergendorff O, Bjork J, Bruze M, Ponten A. Wet work exposure and hand eczema among healthcare workers: a cross-sectional study. Br J Dermatol. 2018;178:452–61.
Wollina U. Challenges of COVID-19 pandemic for dermatology. Dermatol Ther. 2020. https://doi.org/10.1111/dth.13430.
Balato A, Ayala F, Bruze M, et al. European Task Force on Contact Dermatitis statement on coronavirus 19 disease (COVID-19) outbreak and the risk of adverse cutaneous reactions. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.1655.
Lin P, Zhu S, Huang Y, Li L, Tao J, Lei T, Song J, Liu D, Chen L, Shi Y, Jiang S, Liu Q, Xie J, Chen H, Duan Y, Xia Y, Zhou Y, Mei Y, Zhou X, Wu J, Fang M, Meng Z, Li H. Adverse skin reactions among healthcare workers during the coronavirus disease 2019 outbreak: a survey in Wuhan and its surrounding regions. Br J Dermatol. 2020. https://doi.org/10.1111/bjd.19089.
Gheisari M, Araghi F, Moravvej H, Tabary M, Dadkhahfar S. Skin reactions to non-glove personal protective equipment: an emerging issue in the COVID-19 pandemic. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16492.
Reinholz M, French LE. Medical education and care in dermatology during the SARS-CoV2 pandemia: challenges and chances. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16391.
McDowell L, Goode S, Sundaresan P. Adapting to a global pandemic through live virtual delivery of a cancer collaborative trial group conference: the TROG 2020 experience. J Med Imaging Radiat Oncol. 2020. https://doi.org/10.1111/1754-9485.13047.
Wollenberg A, et al. European Task Force on Atopic Dermatitis (ETFAD) statement on severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2)-infection and atopic dermatitis. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16411.
Marasca C, Ruggiero A, Fontanella G, Ferrillo M, Fabbrocini G, Villani A. Telemedicine and support groups in order to improve the adherence to treatment and health related quality of life in patients affected by inflammatory skin conditions during COVID-19 emergency. Clin Exp Dermatol. 2020. https://doi.org/10.1111/ced.14245.
Wang C, Rademaker M, Baker C, Foley P. COVID-19 and the use of immunomodulatory and biologic agents for severe cutaneous disease: an Australia/New Zealand consensus statement. Australas J Dermatol. 2020. https://doi.org/10.1111/ajd.13313.
Rademaker M, Baker C, Foley P, Sullivan J, Wang C. Advice regarding COVID-19 and use of immunomodulators, in patients with severe dermatological diseases. Australas J Dermatol. 2020. https://doi.org/10.1111/ajd.13295.
Price KN, Frew JW, Hsiao JL, Shi VY. COVID-19 and immunomodulator/immunosuppressant use in dermatology. J Am Acad Dermatol. 2020. https://doi.org/10.1016/j.jaad.2020.03.046.
Arora G, Kassir M, Jafferany M, Galadari H, Lotti T, Satolli F, Sadoughifar R, Sitkowska Z, Goldust M. The COVID-19 outbreak and rheumatologic skin diseases. Dermatol Ther. 2020. https://doi.org/10.1111/dth.13357.
Atzori L, Mugheddu C, Addis G, Sanna S, Satta R, Ferreli C, Atzori MG, Montesu MA, Rongioletti F. Psoriasis health care in the time of the coronavirus pandemic: insights from dedicated centers in Sardinia (Italy). J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16473.
Ranney ML, Griffeth V, Jha AK. Critical supply shortages—the need for ventilators and personal protective equipment during the Covid-19 Pandemic. N Engl J Med. 2020. https://doi.org/10.1056/NEJMp2006141.
Jones VG, Mills M, Suarez D, et al. COVID-19 and Kawasaki disease: novel virus and novel case. Hosp Pediat. 2020. https://doi.org/10.1542/hpeds.2020-0123.
Principi N, Rigante D, Esposito S. The role of infection in Kawasaki syndrome. J Infect. 2013;67(1):1–10. https://doi.org/10.1016/j.jinf.2013.04.004.
Kim GB, Park S, Kwon BS, Han JW, Park YW, Hong YM. Evaluation of the temporal association between Kawasaki disease and viral infections in South Korea. Korean Circ J. 2014;44(4):250–4. https://doi.org/10.4070/kcj.2014.44.4.250.
Shirato K, Imada Y, Kawase M, Nakagaki K, Matsuyama S, Taguchi F. Possible involvement of infection with human coronavirus 229E, but not NL63, in Kawasaki disease. J Med Virol. 2014;86(12):2146–53. https://doi.org/10.1002/jmv.23950.
Chang LY, Lu CY, Shao PL, et al. Viral infections associated with Kawasaki disease. J Formos Med Assoc. 2014;113(3):148–54. https://doi.org/10.1016/j.jfma.2013.12.008.
Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92(4):418–23. https://doi.org/10.1002/jmv.25681.
Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38(1):1–9. https://doi.org/10.12932/AP-200220-0772.
Crowl JT, Gray EE, Pestal K, Volkman HE, Stetson DB. Intracellular nucleic acid detection in autoimmunity. Annu Rev Immunol. 2017;35:313–36. https://doi.org/10.1146/annurev-immunol-051116-05233.
Wirestam L, Arve S, Linge P, Bengtsson AA. Neutrophils-important communicators in systemic lupus erythematosus and antiphospholipid syndrome. Front Immunol. 2019;10:2734. https://doi.org/10.3389/fimmu.2019.02734.
Marzano AV, Ortega-Loayza AG, Heath M, Morse D, Genovese G, Cugno M. Mechanisms of inflammation in neutrophil-mediated skin diseases. Front Immunol. 2019;10:1059. https://doi.org/10.3389/fimmu.2019.01059.
Martorana D, Bonatti F, Mozzoni P, Vaglio A, Percesepe A. Monogenic autoinflammatory diseases with Mendelian inheritance: genes, mutations, and genotype/phenotype correlations. Front Immunol. 2017;8:344. https://doi.org/10.3389/fimmu.2017.00344.
Sun NZ, Brezinski EA, Berliner J, Haemel A, Connolly MK, Gensler L, McCalmont TH, Shinkai K. Updates in adult-onset Still disease: atypical cutaneous manifestations and associations with delayed malignancy. J Am Acad Dermatol. 2015;73(2):294–303. https://doi.org/10.1016/j.jaad.2015.04.063.
Criado PR, de Carvalho JF, Ayabe LA, Brandt HR, Romiti R, Maruta CW. Urticaria and dermographism in patients with adult-onset still’s disease. Rheumatol Int. 2012;32(8):2551–5. https://doi.org/10.1007/s00296-011-2025-5.
Li H, Liu L, Zhang D, et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 2020;395(10235):1517–20. https://doi.org/10.1016/S0140-6736(20)30920-X.
Li G, Fan Y, Lai Y, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92(4):424–32. https://doi.org/10.1002/jmv.25685.
Ferro F, Elefante E, Baldini C, Bartoloni E, Puxeddu I, Talarico R, Mosca M, Bombardieri S. COVID-19: the new challenge for rheumatologists. Clin Exp Rheumatol. 2020;38(2):175–80.
Chamseddin B, Marks E, Dominguez A, Wysocki C, Vandergriff T. Refractory macrophage activation syndrome in the setting of adult-onset Still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutan Pathol. 2019;46(7):528–31. https://doi.org/10.1111/cup.13466.
Gansner JM, Berliner N. The rheumatology/hematology interface: CAPS and MAS diagnosis and management. Hematology Am Soc Hematol Educ Program. 2018;2018(1):313–7. https://doi.org/10.1182/asheducation-2018.1.313.
Llamas-Velasco M, Muñoz-Hernández P, Lázaro-González J, et al. Thrombotic occlusive vasculopathy in skin biopsy from a livedoid lesion of a COVID-19 patient. Br J Dermatol. 2020. https://doi.org/10.1111/bjd.19222.
Sarzi-Putting P, Giorgi V, Sirotti S, Marotto D, Ardizzone S, Rizzardini G, Antinori S, Galli M. COVID-19, cytokines and immunosuppression: what can we learn from severe acute respiratory syndrome? Clin Exp Rheumatol. 2020;38(2):337–42.
Peterson D, Damsky W, King B. Calm before the storm: understanding the role of JAK inhibitors in COVID-19. J Am Acad Dermatol. 2020. https://doi.org/10.1016/j.jaad.2020.04.097.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–7. https://doi.org/10.1002/path.1570.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–8. https://doi.org/10.1016/S0140-6736(20)30937-5.
Soler MJ, Batlle M, Riera M, Campos B, Ortiz-Perez JT, Anguiano L, Roca-Ho H, Farrero M, Mont L, Pascual J, Perez-Villa F. ACE2 and ACE in acute and chronic rejection after human heart transplantation. Int J Cardiol. 2019;275:59–64. https://doi.org/10.1016/j.ijcard.2018.10.002.
Bjerregaard B, Larsen OB. The Danish pathology register. Scand J Public Health. 2011;39(7 Suppl):72–4.
Shen LW, Mao HJ, Wu YL, Tanaka Y, Zhang W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie. 2017;142:1–10. https://doi.org/10.1016/j.biochi.2017.07.016.
Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: their roles in pathogenesis. J Microbiol Immunol Infect. 2020. https://doi.org/10.1016/j.jmii.2020.03.022.
Wambier CG, Goren A. SARS-COV-2 infection is likely to be androgen mediated. J Am Acad Dermatol. 2020. https://doi.org/10.1016/j.jaad.2020.04.032.
Goren A, Vano-Galvan S, Wambier CG, McCoy J, Gomez-Zubiaur A, Moreno-Arrones OM, Shapiro J, Sinclair R, Gold MH, Kovacevic M, Mesinkovska NA, Goldust M, Washeni K. A preliminary observation: male pattern hair loss among hospitalized COVID-19 patients in Spain—a potential clue to the role of androgens in COVID-19 severity. J Cosmet Dermatol. 2020. https://doi.org/10.1111/jocd.13443.
Lukassen S, Lorenz Chua R, Trefzer T, Kahn NC, Schneider MA, Muley T, Winter H, Meister M, Veith C, Boots AW, Hennig BP, Kreuter M, Conrad C, Eils R. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020. https://doi.org/10.15252/embj.20105114.
Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pöhlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88(2):1293–307. https://doi.org/10.1128/JVI.02202-13.
Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol. 2010;84(24):12658–64. https://doi.org/10.1128/JVI.01542-10.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020. https://doi.org/10.1016/j.cell.2020.02.052.
McGonagle D, Sharif K, O’Regan A, Bridgewood C. The role of cytokines including Interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. 2020. https://doi.org/10.1016/j.autrev.2020.102537.
Thachil J. The versatile heparin in COVID-19. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14821.
Lillicrap D. Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J Thromb Haemost. 2020;18(4):786–7. https://doi.org/10.1111/jth.14781.
Enkhmaa B, Anuurad E, Berglund L. Lipoprotein (a): impact by ethnicity and environmental and medical conditions. J Lipid Res. 2016;57(7):1111–25. https://doi.org/10.1194/jlr.R051904.
Caforio ALP, Adler Y, Agostini C, Allanore Y, Anastasakis A, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J. 2017;38(35):2649–62. https://doi.org/10.1093/eurheartj/ehx321.
Slunga L, Asplund K, Johnson O, Dahlen GH. Lipoprotein (a) in a randomly selected 25–64 year-old population: the Northern Sweden Monica Study. J Clin Epidemiol. 1993;46:617–24.
Nago N, Kayaba K, Hiraoka J, Matsuo H, Goto T, Kario K, Tsutsumi A, Nakamura Y, Igarashi M. Lipoprotein(a) levels in the Japanese population: influence of age and sex, and relation to atherosclerotic risk factors. The Jichi Medical School Cohort Study. Am J Epidemiol. 1995;141(9):815–21. https://doi.org/10.1093/oxfordjournals.aje.a117516.81.
Volpato S, Vigna GB, McDermott MM, Cavalieri M, Maraldi C, Lauretani F, Bandinelli S, Zuliani G, Guralnik JM, Fellin R, Ferrucci L. Lipoprotein(a), inflammation, and peripheral arterial disease in a community-based sample of older men and women (the InCHIANTI study). Am J Cardiol. 2010;105(12):1825–30. https://doi.org/10.1016/j.amjcard.2010.01.370.
Muñoz-Torrero JF, Rivas D, Alonso R, Crespo L, Costo A, Roman M, Martín C, Zamorano J. Influence of lipoprotein (a) on inflammatory biomarkers in metabolic syndrome. South Med J. 2012;105(7):339–43. https://doi.org/10.1097/SMJ.0b013e31825b5fb2.
Tang L, Hu Y. Ethnic diversity in the genetics of venous thromboembolism. Thromb Haemost. 2015;114(5):901–9. https://doi.org/10.1160/TH15-04-0330.
Ji HL, Zhao R, Matalon S, Matthay MA. Elevated plasmin(ogen) as a common risk factor for COVID-19 susceptibility. Physiol Rev. 2020;100(3):1065–75. https://doi.org/10.1152/physrev.00013.2020.
Wenzhong L, Hualan L. COVID-19: attacks the 1-beta chain of hemoglobin and captures the porphyrin to inhibit human heme metabolism. ChemRxiv. 2020. https://doi.org/10.26434/chemrxiv.11938173.v7 (Preprint).
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Additional information
Responsible Editor: Anatoliy Kubyshkin.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Criado, P.R., Abdalla, B.M.Z., de Assis, I.C. et al. Are the cutaneous manifestations during or due to SARS-CoV-2 infection/COVID-19 frequent or not? Revision of possible pathophysiologic mechanisms. Inflamm. Res. 69, 745–756 (2020). https://doi.org/10.1007/s00011-020-01370-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-020-01370-w