
Abstract
Bone is a target tissue in many inflammatory diseases including rheumatic diseases such as rheumatoid arthritis (RA), ankylosing spondylitis (AS), systemic lupus erythematosus (SLE), and psoriatic arthritis.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others



1 Introduction
Bone is a target tissue in many inflammatory diseases including rheumatic diseases such as rheumatoid arthritis (RA), ankylosing spondylitis (AS), systemic lupus erythematosus (SLE), and psoriatic arthritis.
A relationship between inflammation and bone disease has been established in a variety of clinical settings and animal models of inflammatory disease [1,2,3,4]. It has been established that the nature of the inflammatory disease can influence on the extent and type of bone disease and that even a small rise in the level of systemic inflammation can impact on bone remodeling and increase fracture risk [5].
The inflammatory joint disorders, namely, rheumatic diseases, are usually accompanied with extra-articular side effects, mainly bone loss, or osteoporosis that would result in an increased risk of fractures and deformities, which are in turn associated with increased morbidity and mortality [6]. Therefore, such types of musculoskeletal diseases are considered as one of the major causes of disability around the world and can explain the enormous cost of the musculoskeletal conditions to the community.
In order to easily understand the underlying pathology and mediators that affect bones in rheumatic conditions, a brief overview on bone structure, biology, physiology, and essential molecular mechanism and signaling pathways needs to be explained clearly.
2 Objectives
-
1.
To explain the underlying bones pathology among patients with rheumatic diseases.
-
2.
To identify the common bone lesions occur with rheumatic diseases.
-
3.
To recognize the serious impact of developing secondary osteoporosis among patients with rheumatic diseases.
-
4.
To provide an updated approach for prevention and treatment of glucocorticoid-induced osteoporosis (GIOP) and bone fractures among patients with rheumatic diseases receiving glucocorticoids.
3 Bone Structures
Bone is a dynamic and highly specialized form of connective tissue, in which the extracellular components are mineralized, thus giving the property of marked rigidity and strength while retaining some degree of elasticity. Bone represents a store of calcium and other inorganic ions and actively contributes to the maintenance of calcium homeostasis.
Two types of bone can be identified macroscopically: compact or cortical bone and cancellous or trabecular bone. Microscopically both types of bone have the same histological structure. Like other supporting connective tissues, bone is composed of cells and extracellular matrix that is made up of 35% of organic component and 65% inorganic component [7]. The inorganic part consists of calcium and phosphorus in hydroxyapatite crystal form, while the organic component consists of type 1 collagen and ground substance containing proteoglycan aggregates and several specific structural glycoproteins [8] (Fig. 10.1).

Components of bone structure
4 Bone Remodeling and Bone Cells
Bone remodeling is the lifelong process whereby old bone is removed by bone resorbing cells and subsequently replaced by new bone via the action of bone-forming cells to maintain the bone structure. Bone remodeling occurs normally in all individuals, and in adults about 25% of trabecular and 3% of cortical bone is replaced by such process each year [9]. Bone remodeling also helps to maintain mineral homeostasis via the liberation of calcium and phosphorus into the circulation. The remodeling process occurs at discrete sites on cortical and cancellous bone surfaces and involves the integrated and sequential actions of osteoclasts (bone resorbing cells) and osteoblasts (bone-forming cells), comprising anatomic structures known as basic multicellular units (BMUs).
4.1 Bone Cells
Mesenchymal stem cells have the potential to differentiate into various cell types including osteoblasts, chondrocytes, adipocytes, myoblasts, and fibroblasts. Determination of the final fate of the differentiation process is determined by and depending on the signaling transcription pathways that are activated during the initial phase of differentiation of mesenchymal progenitor cells [10, 11].
Among the important signaling pathways that are responsible to direct the differentiation into osteoblast lineage are the mitogen-activated protein kinase (MAPK) and protein kinase A (PKA)-dependent pathway [12] and Wnt-signaling pathway with its related β-catenin protein [13, 14]. Moreover, of the transcriptional factors, at least two are shown to be absolutely essential for osteoblast differentiation from mesenchymal precursors including Runx2 [15,16,17].
The plasma membrane of activated osteoblast is rich in alkaline phosphatase and exhibits receptors for parathyroid hormone (PTH) [18], whereas the nuclei have receptors for estrogens [19], vitamin D3 [20], and glucocorticoids [ 21], which all are involved in the regulation of osteoblast differentiation and activity.
Osteoblasts contribute in the synthesis and secretion of new organic part of bone matrix (but not yet mineralized), called osteoid, between the secreting osteoblast layer and in contact with older bone matrix of previously formed bone. This process is referred to as bone apposition and is completed by further mineralization of the newly formed bone matrix (deposition of calcium salts into matrix), a process regulated by osteoblast too. At the end of the secreting period of osteoblasts, those osteoblasts are embedded within the bone and differentiated into osteocytes.
Osteoclasts originate from hematopoietic stem cells (HSCs), precisely from cells of the colony-forming unit of macrophage (CFU-M) that differentiate to multinucleated, giant, motile cells on stimulation with macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-kappa B “NFkB” ligand (RANKL) (Fig. 10.2).

Pathways regulating the development of (a) osteoblasts and (b) osteoclasts. Hormones, cytokines, and growth factors that control cell proliferation and differentiation are shown above the arrows. Transcription factors and other markers specific for various stages of development are depicted below the arrows. BMPs, bone morphogenetic proteins; Wnts, wingless-type mouse mammary tumor virus integration site; PTH, parathyroid hormone; Vit D, vitamin D; IGFs, insulin-like growth factors; Runx2, Runt-related transcription factor 2; M-CSF, macrophage colony-stimulating factor; PU-1, a monocyte- and B lymphocyte-specific ets family transcription factor; NFB, nuclear factor B; TRAF, tumor necrosis factor receptor-associated factors; RANK ligand, receptor activator of NFB ligand; IL-1, interleukin-1; IL-6, interleukin-6 [24]
Firstly, the osteoclast progenitors proliferate and differentiate into mononuclear preosteoclasts and then fuse with each other to form multinucleated cells. The terminal differentiation in this lineage is characterized by acquisition of mature phenotypic markers, such as the calcitonin receptor, tartrate resistant acid phosphatase (TRAP), and integrin αvβ3 [22]. The mature and active osteoclasts are characterized by a moderate rough endoplasmic reticulum, a well-developed Golgi apparatus, and abundant mitochondria, while the surface of their plasma membrane facing bone matrix is having ruffled border (clear or sealing zone), which is devoid of organelles but rich in actin microfilaments that form a ring of contractile protein serving to attach the cell to the bone surface via integrin receptors during the resorptive process [8]. The clear zone is a site of adhesion of the osteoclast to the bone matrix and creates a microenvironment where bone resorption takes place. From the ruffled border, osteoclasts secrete collagenase (and other proteolytic enzymes) and pump protons (low pH) into microenvironment and thus promoting the localized digestion of matrix and the dissolving of bone mineral (calcium salt crystal), respectively.
Several systemic and local factors have influenced osteoclasts and their bone resorption ability. In normal physiological conditions, the osteoclast activity is highly balanced by those factors. However, in pathological conditions, this balance becomes disturbed such as during excessive activation of the immune system, due to the secretion of additional pro-inflammatory cytokines, produced mainly by activated T cells [23].
4.2 The Remodeling Cycle
The remodeling cycle is comprised of four distinct phases including activation, resorption, reversal, and formation phase (Fig. 10.3). Bone remodeling starts with activation of the lining cells via increasing the surface expression of RANKL.

Process and phases of normal bone remodeling [26]
In the activation phase, RANKL interacts with its receptor RANK, thus triggering the recruitment of osteoclast progenitors to bone where they proliferate and differentiate into osteoclasts and attach tightly to the bone matrix.
Next is the resorption phase, when the activated osteoclasts possess ruffled borders under which the proteolytic enzymes are secreted and the hydrogen ions are pumped resulting in digestion of collagens and dissolving the mineralized matrix with the formation of a resorption cavity and allowing the release of several growth factors usually stored in the bone matrix. In addition, there is an accumulation of high concentration of calcium that directly controls osteoclasts activity resulting in cell retraction [25] and movement of osteoclasts across the bone surface to resorb a new area. At the end of this stage, osteoclasts undergo apoptosis after a life span of about 3 weeks, and thus the process of remodeling requires the continual production of osteoclast precursors.
In the reversal phase, the remnant debris of matrix degradation will be removed, while the released growth factors including bone morphogenetic proteins (BMPs), fibroblast growth factors (FGFs), and transforming growth factor-β (TGFβ) are likely to be responsible for recruitment of osteoblasts to cover the bottom of the resorption cavity, forming osteoid tissue until the cavity is filled.
In the final formation phase of bone remodeling, osteoblasts initially synthesize the organic matrix and then preside over its mineralization, thus completing the bone remodeling process. Toward the end of this process, some osteoblasts start to flatten and become quiescent lining cells; others become embedded in the matrix and differentiate into osteocytes, while the remaining of osteoblasts will undergo programmed cell death.
4.3 Factors Influencing Remodeling
The rate at which new osteoblasts and osteoclasts are supplied and the timing of apoptosis of these cells are crucial determinants of bone remodeling. The development of osteoclasts and osteoblasts is controlled by growth factors and cytokines produced in the bone marrow microenvironment and is modulated by systemic hormones and immunological mechanisms [27,28,29,30]. Certain signaling pathways, systemic hormones, pro-inflammatory cytokines, and growth factors are considered as fundamental regulators of bone remodeling.
Taken together, positive stimulator of osteoblast activity includes PTH, vitamin D3, IGFs, BMPs, and Wnt signaling, while those that promote osteoclast activation are monocyte-macrophage colony-stimulating factor M-CSF, RANKL, IL-1, and IL-6.
Eventually, the recent discovery of osteoprotegerin (OPG) and the subsequent identification of its cognate ligand, OPG ligand (OPGL or RANKL), have illuminated our understanding of the molecular basis that links between osteoblastogenesis and osteoclastogenesis and thereby the rate of bone remodeling upon which other inputs (hormonal, biomechanical, etc.) operate.
4.4 RANK/RANKL/OPG System
Despite that the principal function of the osteoblasts is to synthesize bone matrix proteins and to enhance bone mineralization, osteoblasts also play a crucial role in osteoclast biology that has been clearly demonstrated by the release of key molecules, which regulate osteoclastogenesis and bone resorption. Of these regulators are RANKL which is expressed on the surface of the osteoblast and interact with its receptor RANK [22] to mediate signals for osteoclast proliferation, differentiation, activation, and function [31] (Fig. 10.4), while OPG is acting as a decoy receptor for RANKL [32], noting that the OPG/RANK/RANKL system accounts only for signaling of osteoblasts to osteoclasts.

Mechanisms of action for OPG, RANKL, and RANK [34]
The human RANK is a polypeptide of 616 amino acids, related to the type 1 transmembrane protein class [33], and is expressed in various tissues such as the skeletal muscle, liver, and small and large intestines. Among bone cells RANK-mRNA is exclusively expressed in osteoclast precursor cells [22, 32]. On the other hand, RANKL is a TNF-related cytokine that exists in both transmembrane, the predominant form, and soluble (cleaved) forms [22]. The gene expression of RANKL can be found abundantly in the skeleton and lymphoid tissues and is produced by osteoblasts, bone marrow stromal cells, and other cells under the control of various pro-resorptive growth factors, hormones, and cytokines. Moreover, osteoblasts and stromal cells produce OPG, which binds to and thereby inactivates RANKL.
Collectively, RANKL is of great importance for the development and function of osteoclasts through binding to its transmembrane-signaling receptor RANK [35]. RANK-RANKL interactions lead to pre-osteoclast recruitment, fusion into multinucleated osteoclasts, osteoclast activation, and osteoclast survival. These effects are very selective to bone and can be inhibited by the natural, soluble, decoy receptor OPG [32].
OPG is considered as a humoral regulator of bone resorption. It blocks osteoclast maturation and differentiation, and subsequently it can protect the bone from both normal osteoclast remodeling and ovariectomy-associated bone loss [36].
Certain human adult tissues showed a high level of OPG mRNA expression, namely, the heart, the bone, the placenta, and the thyroid gland [37]. It has been demonstrated that OPG expression is upregulated in various human osteoblastic cell systems by 1,25-dihydroxyvitamin D3, bone morphogenetic protein-2 (BMP-2), pro-inflammatory cytokines such as interleukin-1 (IL-1) [38], estrogen [39], as well as transforming growth factor-β (TGF-β) [40]. However, some discrepancies were noticed in the effect of these modulators on the expression of the OPG mRNA and OPG protein levels depending on the species of the cells used and on the stage of osteoblastic differentiation. In contrast, it has been established that glucocorticoids downregulate the OPG transcript in human osteoblast and in human marrow stromal cells [41, 42], and they can suppress OPG production resulting in acceleration of osteoclastic bone resorption [43].
5 Mediators of Bone Loss in Rheumatic Diseases
Systemic bone loss in rheumatic diseases occurs as a result of several factors including direct effects of inflammation, poor nutrition, reduced lean body mass, immobility, and the effects of therapeutic agents, specifically glucocorticoids. These mechanisms are complex and interrelated but are eventually mediated through influencing on the bone remodeling cycle and may result in increasing bone resorption, decreasing bone formation, but most commonly affecting both of these processes.
Adding to the referred mechanisms that cause bone loss, there are background predisposing factors, which increase the risk of fractures due to bone loss, and they include age, gender, family history of osteoporosis, low body mass index (BMI), falling risk, and sedentary lifestyle (Fig. 10.5) [44].

Risk factors for osteoporosis and fractures in inflammatory rheumatic diseases [44]
The following sections will discuss the main underlying mechanisms that cause bone destruction in different rheumatic conditions, namely, the disease activity (inflammation), immobility, and treatment with glucocorticoids are considered. Noting that each rheumatic disease has a unique effect on articular bone or on other site on skeleton whether local or generalized bone loss, however, they remarkably share common pathways of skeletal remodeling (the RANKL/OPG pathway), which is involved in the regulation of bone resorption. In addition, most human and animal studies in the field of rheumatic arthritis have referred to the osteoclast as the principal cell type mediating bone loss in arthritis [45].
5.1 Effects of Systemic Inflammation
Inflammatory process in rheumatic diseases is usually associated with skeletal destruction. The effects of inflammation in induction bone loss involve two mechanisms, the role of pro-inflammatory cytokines and/or the role of inflammatory cells.
5.1.1 Role of pro-Inflammatory Cytokines
-
Many of the pro-inflammatory cytokines and growth factors (Fig. 10.6) [46] involved in the inflammatory processes in rheumatic diseases have been found to have a great impact on osteoclast differentiation and activation either directly, by acting on cells of the osteoclast lineage, or indirectly, by modulating the expression of the key osteoclastogenic factor (RANKL) and/or its inhibitor, OPG [47].
Fig. 10.6 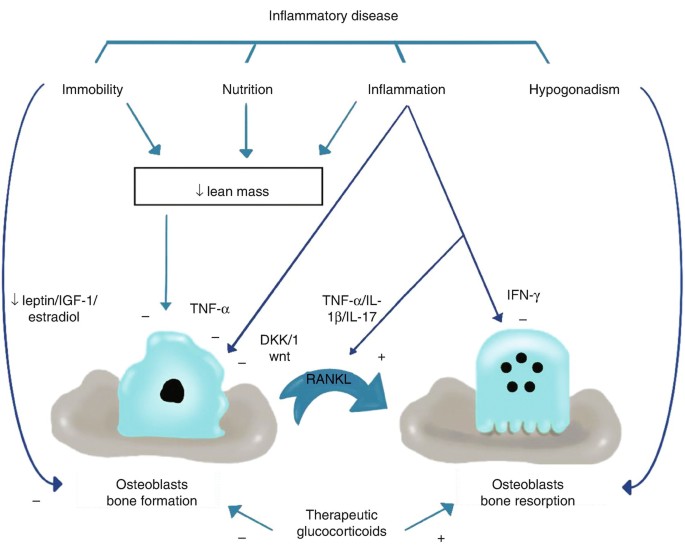
Illustration of the impacts of chronic inflammatory disease on bone formation and resorption. A stimulatory effect is indicated by C and an inhibitory effect by K [46]
-
Because a wide range of cytokines have positive and negative impact on OPG/RANKL system or directly on osteoclastogenesis, they are usually kept in balance in healthy subjects. However, imbalance of these cytokines occurs during inflammation but varies between disease states, and this variation would account for differences in predisposition to bone loss.
-
The cytokines that have positive (stimulatory) effects on osteoclastogenesis include TNF-α, IL-1b, IL-6, IL-11, and IL-17, whereas those that have negative (inhibitory) effects include interferon (IFN)-γ, IL-4, and transforming growth factor-β (TGF-β) [48].
-
For instance, tumor necrosis factor-alpha (TNF-α) can increase the expression of RANKL by osteoblasts and hence induce osteoclastogenesis and the bone-resorbing activity. However, TNF-α and interleukin-1 (IL-1) can synergize with RANKL to directly enhance bone resorption by osteoclasts.

5.1.2 Role of Inflammatory Cells
-
Under normal conditions, RANKL is derived from osteoblasts; however, during inflammation, a variety of inflammatory cells can also produce RANKL including lymphocytes and fibroblasts, which have been found in the inflamed synovium in various studies [49,50,51]. The expression or production of RANKL on/from non-osteoblastic cells causes a direct osteoclastogenic effect independent of osteoblasts.
-
An example of these cells is T lymphocytes that are derived from Th17 subset, which has been called so after the ability of these lymphocytes to secrete IL-17, and hence they are considered to have an osteoclastogenic cytokine profile [52]. The presence of this lymphocyte subset prominently in inflammatory arthritis could explain the tendency to local osteoclastogenesis and thus bone destruction in this condition [53].
-
A subsequent to the increased bone resorption, there should be also a stimulation of bone formation because the processes of bone resorption and formation are normally tightly coupled. However, during chronic inflammation, “uncoupling” of bone formation from resorption occurs with a suppressed or decreased bone formation relative to the high degree of resorption.
5.1.3 Causes of Uncoupling Process
5.1.3.1 The Wnt Signaling and its Antagonist, DKK1
Studying animal models of inflammatory arthritis could explain the uncoupling process via the implication of the Wnt-signaling pathway and precisely the Wnt antagonist dickkopf-1 (DKK1), in this process [54].
The canonical Wnt-signaling pathway is essential for bone development, directing differentiation of mesenchymal precursor cells into mature osteoblasts, as well as having a major role in the normal development of the skeleton in the embryo [55, 56]. The naturally occurring soluble Wnt antagonists such as DKK1, which suppress this process, are important during normal bone remodeling. This finding has been supported by that the DKK1 knockout mice develop an increased bone mass [57] and conversely myeloma cells with aberrant DKK1 expression are associated with purely lytic lesions with little evidence of bone formation [58].
The synovial fibroblast can secrete DKK1; however, in rheumatoid arthritis, the secretion is enhanced by TNF-α, and thus the circulating levels of DKK1 have been found much elevated in those patients [54]. Thus, the secreted DKK1 from the synovium would have a suppressive effect on osteoblast maturation and on OPG function leading to inhibition of local bone formation and increased bone resorption, respectively. Understanding the mechanism of Wnt signaling and its antagonist, DKK1 (Fig. 10.7), is very important, since administration of DKK1 antibody would be able to prevent bone erosions and reverse this block on osteoblast formation which resulted in a paradoxical excess of bone formation during inflammation as proved by the development of new osteophytes [54].

Schematic illustration of the possible role of DKK1 in the bone remodeling imbalance in inflammatory joint disease. Production of DKK1 in response to TNF-α production by inflammatory cells is proposed to inhibit bone formation but increase bone resorption by osteoclasts through a suppression of OPG production by osteoblasts [46]
5.1.3.2 Alteration of Glucocorticoid Signaling
The effects of glucocorticoids will be explained later in the following sections, but the current paragraph will discuss the influence of inflammation on glucocorticoid action in bone cells. Because of the intracellular metabolism of glucocorticoids by 11b-hydroxysteroid dehydrogenases (11β-HSDs) [59], it has become known that the levels of active glucocorticoids present within the circulation differ from that in the tissues. Specifically, 11β-HSD1 enzyme is expressed on osteoblast and can increase local glucocorticoid action in these cells by converting the inactive glucocorticoids such as cortisone and prednisone to their active counterparts’ cortisol and prednisolone, respectively.
Overexpression of the enzyme in osteoblasts reduces proliferation and the synthesis of bone-specific proteins such as osteocalcin when cells are exposed to inactive glucocorticoids [60, 61]. It was reported previously by [62] that pro-inflammatory cytokines such as TNF-α or IL-1b can effectively induce the expression and activity of this enzyme in osteoblasts. Thus, during inflammation, osteoblasts at the site of bone exposed to pro-inflammatory cytokines are likely to also be exposed to high doses of locally active glucocorticoid [62, 63]. This is potentially a major mechanism by which the uncoupling process of osteoblasts and osteoclasts occurs. Overall, a high glucocorticoid level in osteoblasts will decrease bone formation through direct effects on osteoblasts [64], but it can also induce osteoclastogenesis due to upregulation of RANKL and downregulation of OPG in osteoblast precursors [42].
Studying the correlation of locally generated glucocorticoids with other proposed mechanisms of uncoupling such as DKK1 induction is essentially needed for therapeutic purposes of rheumatic diseases.
5.2 Effects of Immobility
Immobility has consequences on all inflammatory diseases specifically neuromuscular and joint disease. The major impact on bone occurs due to uncoupling process that results in reduced bone formation and increased bone resorption [65] with overall bone loss. It has been found that osteocytes mediates mechanosensing, which means they can response to mechanical strain and maintain bony matrix via modulation of the major pathways such as the Wnt pathway that couple bone formation and resorption [66]. This effect may partly be dependent on estrogen receptor signaling, and thus hypogonadism would reduce the mechanosensing [67].
Regular exercises can maintain force on bone and thus control bone loss through mechanical stimulation. However, a more advanced approach is the administration of a vibration signal that could stimulate mechanosensing effects, which in turn will induce an anabolic response to bone [46].
5.3 Effects of Glucocorticoids
Glucocorticoids (GCs) are frequently prescribed for patients with variety of chronic inflammatory diseases such as rheumatic diseases. An excess of circulating GCs has a major negative effects on bone [64, 68, 69]. These adverse effects on bone are owing to reduced bone formation, characterized by a low mineral apposition rate that is explained by decreased numbers of osteoblasts, while bone resorption is unchanged or even elevated [70], leading to the development of glucocorticoid-induced osteoporosis (GIOP).
Overall negative effects of GCs on bone are either directly on bone cells or indirectly by affecting the bone metabolism. The underlying molecular mechanisms of GIOP include the increased apoptosis of osteoblasts and osteocytes and increased half-life time of osteoclasts, i.e., the direct effects on bone cells (Fig. 10.8) [71]. It has been reported that the increased osteoblast apoptosis results in a significant reduction in bone formation, while decreased osteocyte numbers result in a disturbed osteocyte-canalicular network and thus failure to respond to bone damage [72].

The direct effects of glucocorticoids on bone [71]
The process of apoptosis is induced by activating caspase-3 [73] and glycogen synthases kinase 3β (GSK3β), which suppresses the Wnt-signaling pathway by increasing the production of DKK-1, the Wnt pathway inhibitor [74, 75].
In addition to the increased apoptosis of osteoblasts, GCs impair osteoblast function by suppressing osteoblast differentiation [76] via interfering with both the bone morphogenetic protein (BMP) pathway and the Wnt-signaling pathway.
Moreover, recent studies proposed that high doses of GCs cause a shift of bone marrow stromal cells, the precursor cells of osteoblasts, to differentiate toward adipocytes instead of osteoblasts. This is mainly achieved either through an increased expression of the peroxisome proliferator-activated receptor-γ2 (PPR- γ2) and repression of the osteogenic transcription factor Runt-related protein 2 [77] or via suppression of AP-1, a process that not only mediates anti-inflammatory actions but also reduces bone strength [78].
In contrast to increased apoptosis of osteoblasts and osteocytes, GCs therapy would reduce the apoptosis of osteoclasts by extending their life span through upregulation of RANKL and suppression of OPG [42].
Likewise direct effects on osteoblasts, osteocytes, and osteoclasts, GCs have indirect effects on bone (Fig. 10.9). Previous studies asserted that GCs impair bone metabolism by inhibiting both the gastrointestinal absorption and the renal tubular reabsorption of calcium, leading to hypocalcaemia and the subsequent hyperparathyroidism [71]. Recent reports referred that GCs have influenced the bone mineralization by decreasing the production of important proteins for matrix formation, namely, osteocalcin and type 1 collagen [69]. Furthermore, GCs can cause steroid myopathy [79] [4] that may increase the risk of falling and thus indirectly increase the fracture risk.

The indirect effects of glucocorticoids on bone [71]
6 Common Bone Diseases Associated with Rheumatic Disease
Osteoporosis-related fragility fractures represent one of the most important complications that may occur in patients with rheumatic diseases; obviously, these fractures may contribute to an important decrease in quality of life, and hence osteoporosis becomes increasingly recognized as an eminent public health problem.
Osteoporosis is a metabolic bone disease characterized by both low bone density (mass) and low bone quality, which includes not only microarchitecture deterioration of bone tissue but also alterations in bone remodeling, damage accumulation (e.g., microfractures), and mineralization. These changes in bone density and quality enhance bone fragility with a consequent increase in fracture risk after minimal trauma. Osteoporosis is caused by an imbalance between bone formation and resorption with in favor of bone resorption over bone formation, leading to altered bone remodeling.
The reduction in bone mass can be quantified by measurement of bone mineral density (BMD) using dual-energy x-ray absorptiometry (DXA), which is the diagnostic method of osteoporosis [80]. Therefore, osteoporosis can be defined by DXA result when T score is ≤2.5 (i.e., bone density is 2.5 standard deviation below estimated peak BMD for the population), whereas osteopenia is defined when a T score is between −1 and −2.5.
6.1 Rheumatoid Arthritis and Bone Loss
Rheumatoid arthritis (RA) is characterized by three types of bone lesions: periarticular osteopenia, bone erosions, and osteoporosis:
-
Periarticular osteopenia is one of the first radiographic signs of RA. It appears markedly in early disease and is mainly associated with disease activity.
-
Bone erosions develop within the first months of the disease onset and account as the radiographic sign of RA and reflect undesirable prognosis of RA. Hence, the extent and severity of the erosions reflect the increasing disease activity and indicate the disability of the disease.
Within 6 months of disease onset, less than 50% of patients showed radiographic erosions, while almost 70% of the patients have erosions detected by MRI [81,82,83] and may be accompanied by bone edema, where CD34+ cells and potential osteoclast precursors [84] can be found during joint aspiration.
-
Osteoporosis in RA is mainly characterized by marked loss of bone in the hip and the radius, while the axial bone is scarce, a pattern not similar to that of postmenopausal osteoporosis. In addition, several cross-sectional studies reported a lower bone mineral density (BMD) in patients with RA, with a twofold increase in osteoporosis compared to age- and sex-matched controls.
6.1.1 Predisposing Factor of Osteoporosis in RA
In addition to the risk factors of osteoporosis (Fig. 10.5), other factors may also contribute in RA, such as muscle wasting, glucocorticoids therapy, and disease duration. Interaction between several factors should be considered, for example, additional muscle wasting contributes to increased immobilization [85].
6.1.2 Pathological Process
-
Several evidences suggested the presence of osteoclasts at the site of bone erosions, indicating the increased of bone resorption [86, 87].
-
In RA, the local and generalized bone loss share common pathways: the RANKL/OPG pathway. The main inflammatory cytokines that have been found in RA and involve in upregulating RANKL, with subsequent activation of osteoclastogenesis, include TNF-α, IL-1, IL-6, and IL-17 [88, 89].
-
The Wnt-signaling pathway is another pathway that regulates osteoblast activity, and thus the activation of the Wnt/β-catenin pathway is crucial for osteoblastic differentiation [90, 91]. There are two blockers of the Wnt-signaling pathway, dickkopf-1 (Dkk-1) and sclerostin, both of which play an important role in the pathogenesis of RA. TNF-α can induce both sclerostin and Dkk-1 [89], leading to inhibition of osteoblastic differentiation.
-
Further studies in RA patients confirmed these pathological processes and revealed that OPG/RANKL ratio was lower than in healthy controls, while Dkk-1 and sclerostin were higher. After treatment with anti-IL-6, OPG/RANKL increased, Dkk-1 decreased, and sclerostin increased [92].
6.1.3 Management of Bone Loss in RA
-
Recent treatments with biological agents were introduced in patients with rheumatoid arthritis. All available TNF-alpha blocking agents are quite successful in the prevention of erosion formation.
-
However, progression of structural damage in RA patients treated with methotrexate can be avoided by denosumab, a fully human monoclonal IgG2 antibody that binds RANKL [93].
-
It has also been found that in patients with RA treated with infliximab, the bone loss was abolished in the spine and hip, but not in the metacarpal cortical hand [94].
-
Moreover, preventing the loss of vertebral strength in patients with RA can be principally achieved by treatment with alendronate [95].
-
After this extensive review, here comes the value of early diagnosis of RA and early and aggressive intervention with disease-modifying anti-rheumatic drugs (DMARDs) to prevent bone destruction, osteoporosis, and erosions.
6.2 Systemic Lupus Erythematosus and Bone Loss
6.2.1 Predisposing Factors of Bone Loss in SLE
-
In addition to the traditional background factors, there are also disease-related factors (Fig. 10.5) such as inflammation, metabolic factors, hormonal factors, serologic factors, and medication-induced adverse effects [96].
-
Another factor that may contribute in decreased BMD in SLE is the associated high frequency of vitamin D deficiency [97,98,99], a metabolic condition that induces bone loss. Vitamin D deficiency might induce bone loss in SLE via several factors including (a) photosensitivity (so patients avoid exposure to the sun and use sunscreens), (b) dark skin pigment, (c) renal failure, and (d) treatment with GC (has a dual action, it can induce bone loss, but also it has a beneficial effect on bone mass by suppressing inflammation) and possibly hydroxychloroquine (HCQ) (via inhibiting hydroxylase α1 that form active vitamin D), which showed a controversial results [98, 100]. Due to these inconsistent results of HCQ, further studies in large groups of SLE patients and patients with other diseases treated with HCQ are needed to clarify the relationship between HCQ uses and bone metabolism.
-
Changes in hormonal pattern may also negatively influence the BMD in patients with SLE, where a relatively high estrogenic and low androgenic state and a decrease in dehydroepiandrosterone (DHEA) have been demonstrated and associated with low BMD [101].
-
Collectively, the factors that may adversely affect bone mass, resulting in osteoporosis and possible fracture risk in SLE, have been summarized in Table 10.1.
Table 10.1 Summary of risk factors for osteoporosis in patients with SLE [101]
6.2.2 Pathological Process
-
Chronic systemic inflammation is a cause of bone loss in SLE, where the activated inflammatory cells at sites of inflammation produce a wide spectrum of cytokines that stimulate local and generalized bone resorption.
-
Researchers have revealed increased serum levels of tumor necrosis factor (TNF) [103] and oxidized low-density lipoprotein (LDL) [104] in patients with active lupus. Oxidized LDL stimulates the activation of T cells, which in turn can increase the production of RANKL and TNF. Consequently, TNF and RANKL will induce the maturation and activation of osteoclasts [103]. In addition, oxidized LDL has the ability to inhibit osteoblast maturation, and hence it can negatively influence bone formation [105].
-
Moreover, high levels of homocysteine (caused by inflammation) have been reported in patients with SLE, and this might be attributed to the accelerated bone loss [106, 107] via enhancing the bone resorption and reduction of bone formation.
-
Until recently, the previous clinical studies have not been able to demonstrate the association between bone loss in SLE and the disease activity score [108,109,110]. However, several studies showed an association between organ damage and reduced BMD [111], and because prolonged active SLE usually causes organ damage in the patients, this finding suggests that disease activity contributes to reduced BMD in SLE. Moreover, the Hopkins Lupus Cohort study [112] has established that low complement C4 levels (a measure of active disease) were a predictor of low spine BMD among patients with SLE.
6.2.3 Management of Bone Loss in SLE
-
To approach bone health in SLE patients, the underlying risk factors for bone loss should be evaluated. For instance, evaluation of calcium and vitamin D levels and homocysteine status is recommended. Although there is not enough data relating the low levels of vitamin D and SLE activity, the possible association would suggest that replacement of vitamin D may have benefits beyond bone health for those patients [113].
-
Supplementation with vitamin D should aim to keep the serum 25-hydroxyvitamin D [25(OH)D] level above 25 ng/mL, and calcium supplementation should be at the recommended daily allowance for the age of the patient (Table 10.2).
Table 10.2 ACR 2010 Recommendations on counseling for lifestyle modification and assessment of patients starting glucocorticoids at any dose of >3 months duration -
Bear in mind that it takes approximately 3 months to achieve a steady state of 25(OH)D level once vitamin D supplementation is started, so rechecking a 25(OH)D should not be done earlier than 3 months [114, 115].
-
Moreover, if homocysteine levels are elevated, folic acid should be initiated at 1 mg daily [116].
-
Patients with SLE are at increased risk of bone loss due to the synergistic effect of the inflammatory process and its treatment with corticosteroids; therefore adequate management is essential to prevent osteoporotic fractures and maintain BMD. However, all preventative measures and pharmacological therapy will be mentioned later on under the section of “Glucocorticoid-Induced Osteoporosis” according to ACR 2010 recommendations.
6.3 Ankylosing Spondylitis and Bone Loss
Inflammation in ankylosing spondylitis (AS) is characterized by subchondral bone marrow edema with subchondral bone erosive lesions and eventually to subchondral new bone formation through the articular cartilage and ossification of the periarticular ligaments [85].
Bone edema is accounted as a sign of inflammatory activity and may affect limited or extensive parts of vertebrae (Fig. 10.10). Recent studies suggested a possible sequence of events of new bone formation in AS, as follows: first erosions at the site of inflammation, followed by repair reaction, and subsequently ended by new bone formation (10).
-
For instance, at the corners of the vertebral bodies, there might be marginal erosive lesions with adjacent subchondral edema and sclerosis (Romanus lesion). Also, a new periosteal intraosseous bone formation was found and provided the typical picture of squaring of the vertebrae [85].

Sites of bone edema, bone loss, and bone erosion in AS [85]
6.3.1 Fracture Risk in AS
Subsequent results to bone changes in AS lead to an increase in bone loss (osteoporosis) and bone fragility and therefore increased the risk of bone fractures.
-
AS is associated with an elevated risk of vertebral fractures, which are six to seven times higher than in healthy population [117, 118], and these fractures are often accompanied by neurological signs and symptoms [119]. However, the increased in morphometric and clinical vertebral fractures [120] but not in peripheral (forearm or hip) fractures indicates a more local effect of AS on bone, unlike RA, where the inflammatory effects are more systemic. Furthermore, despite sharing similar pathogenesis of osteoporosis but with different clinical phenotypes, bone loss in AS is accompanied by new bone formation contrasting to RA and postmenopausal conditions,
-
Of most important types of spinal fractures in AS includes wedging fracture, which contributes to spine rigidity and hyperkyphosis of upper part of the spine and impaired physical function [119, 121, 122]. In addition to wedging fracture, structural damage of the spine and the disease activity are other significant contributors to hyperkyphosis [123].
6.3.2 Management of Bone Loss in AS
-
Because of the concomitant bone loss and the new abnormal bone formation and the presence of syndesmophytes, the reliability of BMD measurement is affected, and there would be a large variation in the prevalence of osteoporosis in patients with AS [124, 125].
-
Taken together, AS is characterized by bone and cartilage degradation. The bone destruction reflects the systemic inflammatory effects on bone density and can be inhibited by TNF-α blocking agent. However, the cartilage damage might be related to syndesmophyte formation, which is not influenced by anti-inflammatory therapy [120]. This highlights the suggestion that bone degradation and new bone formation are uncoupled mechanisms in AS, the reason that might make their therapeutic intervention basically different.
-
A remarkable but yet not confirmed finding has shown that the risk of clinical fracture decreased in AS patients taking NSAIDs, which could relieve the inflammatory back pain and stiffness and thus improving the physical activity that helped in maintaining bone mass and reducing the risk of falling and subsequent fracture [126, 127]. In addition, it has recently been suggested that NSAIDs may also inhibit the formation and growth of syndesmophytes of AS in the spine via interfering with the prostaglandin metabolism. Therefore, if the divergent inhibitory effects of NSAIDs on osteoporotic fractures (bone loss) and progression of syndesmophytes (bone formation) can be confirmed, this would be an important clue in further explaining pathophysiological mechanisms in AS.
-
In contrast to the treatment of osteoporosis in patients with RA, treatment of osteoporosis in patients with AS is not yet common. Data supporting the efficacy of this treatment in AS are rare. Of all bisphosphonates, alendronate and risedronate are found to be effective in increasing BMD in men. Alendronate and risedronate significantly increase BMD in both vertebrae and femur, with a significant reduction of vertebral fractures [128, 129]. More recently teriparatide was tested with the same aims, but only a positive effect on BMD could be shown [130]. It is clear that there is a need for evidence-based knowledge in these fields in the near future. Our studies highlight the need to develop strategies to identify high-risk patients with AS. Research on the treatment of osteoporosis to prevent vertebral fractures in these patients is urgently needed.
6.4 Glucocorticoid-Induced Osteoporosis (GIOP)
Steroids are widely used in the medical practice to treat various diseases such as asthma, systemic connective tissue diseases, and other autoimmune diseases and in addition to rheumatic diseases. Treatment with GCs results in bone loss within 1 month after initiation of the therapy but primarily occurs in the trabecular bone, so that it mainly increases the risk of vertebral fracture rather than non-vertebral fractures [79]. Fractures are considered the most clinically relevant risk of prolonged steroid therapy.
GIOP is a common type of secondary osteoporosis which occurs at any age and in both men and women. It has been known that one loss in GIOP is biphasic, with a rapid reduction in BMD of 6–12%* which occurs followed by a slower annual loss of about 3%* for as long as the glucocorticoids are administered [131, 132].
6.4.1 Impact of GIOP
-
As a consequent to the bone loss during GCs therapy, it has been reported that the relative fracture risk within the first 3 months after initiation of the therapy increases by 75% even before any BMD changes occur [133].
-
Although the increase of fracture risk has appeared to be dose dependent [134], it was found to be partially reversible so that the fracture risk would gradually return to baseline [135].
6.4.2 Approaching Managements of Patients with GIOP
-
American College of Rheumatology (ACR) have developed and updated recommendations to provide guidance for prevention and treatment of GIOP in order to be applied by the physicians in light of each patient’s circumstances.
-
ACR recommendation 2001 [136] has been updated and replaced by ACR recommendation 2010 [137], which had expanded the recommendations for counseling (Table 10.2) and monitoring updated pharmacological guidelines and used patient’s overall clinical risk instead of T score alone.
-
Afterward, ACR 2017 recommendations have been released for GIOP prevention and treatment, based on the balance of relative benefits and harms of the treatment options and highly considering the quality of the evidence and patients’ values and preferences [138]. Therefore, due to limited evidence on the benefits and harms of interventions in GC users, most recommendations in ACR 2017 guidelines are conditional or of good clinical practice. The strength of the recommendations is based on the fracture risk categories in GC-treated patients [138].
-
The ACR 2017 recommendations for GIOP prevention and treatment have addressed, in addition to all adults’ categories (< 40 years and > 40 years of age), special populations categories, namely children, people with organ transplants, women of childbearing potential, and people receiving very high-dose GC treatment.
-
The initial approach of patients with GIOP begins with clinical assessment of fracture risk by interpreting detailed clinical and biochemical data, together with identifying the diagnostic criteria for assessment of bone mineral density (BMD) results, as follows:
-
-
1.
Clinical Assessment: This is concerned with having detailed medical history to identify the cumulative risk factors for bone loss (Fig. 10.11) and performing proper physical examination to detect any underlying medical conditions or evidence of osteoporosis such as fracture, kyphosis, and loss of height or determine muscle strength and size.
Fig. 10.11 
Risk factors that may shift an individual to a greater risk category for GIOP (ACR 2010) [137]
-
2.
Biochemical Assessment: The baseline levels of the following parameters are needed to be obtained in order to rule out any underlying medical diseases that may affect the outcome of GIOP such as low levels of calcium or vitamin D; those would affect the bone formation and metabolism [137, 138]. These parameters include:
-
Complete blood cell count.
-
Serum calcium and phosphorus.
-
Serum 25-hydroxyvitamin D.
-
Serum-free testosterone in males.
-
Estradiol in premenopausal women.
-
Renal Function Tests specifically 24-hour urinary calcium and sodium.
-
Liver function test, because healthy liver is important for synthesis of sex hormones.
-
-
3.
Assessment of Bone Mineral Density (BMD): Measuring the BMD is one of the salient determinants of bone strength. It can be measured at different sites in the body by distinct methods. For instance, dual-energy x-ray absorptiometry (DXA) measures BMD mainly at lumbar spine and proximal femur, while quantitative computed tomography (QCT) is used mostly to estimate bone density at the forearm, tibia, or lumbar spine. The World Health Organization (WHO) has defined the diagnostic criteria for assessment of BMD results (Table 10.3) [139].
Table 10.3 WHO criteria for assessment of BMD [139] -
4.
Assessment and Classification of Fracture Risk: Identifying patients with increased fracture risk solely using BMD assessment has some limitations due to its age dependency and its inaccuracy in measuring bone quality. Therefore, it has been recommended that fracture risk should be assessed using tools that calculate the absolute fracture risk for a given patient. One of the available tools proposed by the World Health Organization (WHO) is called Fracture Risk Assessment (FRAX) tool [140].
FRAX is a unique model that is considered in calculating the risk of the following factors, age, sex, race, family history, the BMD, and the usage of BMD, but excludes the dosage and the evaluation of the risk factors of falls and the presence or absence of prevalent vertebral deformities, although they are known as risk factors for fractures. The output of FRAX calculation is a 10-year probability of hip fracture and the 10-year probability of a major osteoporotic fracture (clinical spine, forearm, hip, or shoulder fracture) [140].
-
Based on the risk factors shown in Fig. 10.11 as well as the FRAX results, adult patients receiving GC can be classified into low-, moderate-, and high-risk categories accordingly (Fig. 10.12). The ACR 2017 recommendations for GIOP prevention and treatment have addressed, in addition to all adults’ categories (< 40 years and > 40 years of age), special populations, namely children, people with organ transplants, women of childbearing potential, and people receiving very high-dose GC treatment.
Fig. 10.12 
Fracture risk categories in GC-treated patients [138]
-
Therefore, the primary implication of ACR 2017 recommendation is to clarify that all clinicians treating patients with GCs have to be aware of the GIOP risk, identify patients at high fracture risk (Fig. 10.12), and be able to provide the appropriate treatment [138].
-
Moreover, the assessment of fracture risk may not only be useful in treatment decisions, but also in improving patients’ treatment compliance that would provide the patients a better insight into their future fracture risk.
-


6.4.3 Recommendations for Fracture Risk Assessment and Reassessment of Patient with GIOP
These recommendations are considered as good practice recommendations.
-
Initial fracture risk assessment:
-
For all adults and children, an initial clinical fracture assessment should be performed as soon within six months of the initiation of long-term GC treatment. This clinical assessment should include the following:
-
A detailed clinical history of GC use (dose, duration, mode, and pattern of use),
-
An evaluation of underlying risk factors for fracture including history of falls, fractures, frailty, others such as (malnutrition, significant weight loss or low body weight, hypogonadism, secondary hyperparathyroidism, thyroid disease, family history of hip fracture, history of alcohol use [at > 3 units/day] or smoking), and other clinical comorbidities.
-
A physical examination including measurement of weight and height, detailed examination of musculoskeletal system, and other clinical findings of undiagnosed fracture (e.g., spinal tenderness, deformity, and reduced space between lower ribs and upper pelvis).
-
-
For adults >40 years old, the initial absolute fracture risk should be evaluated using FRAX with correction of GC dose and BMD (if available) as prompt as possible but within at least six months of starting the GC therapy (Fig. 10.13) [138].
-
For adults <40 years old, BMD testing should be done as promptly as possible but at least within 6 months of starting the GC treatment if the patient has a history of previous OP fracture(s) (high risk) or if the patient has other significant OP risk factors (Fig. 10.13) [138].
Fig. 10.13 
Initial fracture risk assessment [138]
-
-
Reassessment of fracture risk:
-
For all adults and children, if GC therapy is used continuously, a clinical fracture risk reassessment (as referred earlier) should be performed every 12 months. For detailed pathways of reassessment of clinical fracture risk in adults <40 and ≥40 years of age, refer to Fig. 10.14 [138].
Fig. 10.14 
Reassessment of fracture risk [138]
-


6.4.4 Recommendations for Initial Treatment and Prevention of GIOP
-
In addition to adjusting the pharmacologic treatment of GIOP, optimizing the dose of calcium and vitamin D uptake and counseling lifestyle modification are included within both the ACR 2010 and the ACR 2017 recommendations for treating patients with GIOP.
-
A conditional recommendation is reported, generally for all adults on GC at a dose of ≥2.5 mg/day for ≥3 months, to optimize calcium intake (1000–1200 mg/day) and vitamin D intake (600–800 IU/day; serum level ≥20 ng/mL) [138, 141] alongside lifestyle modification with regard to weight, nutrition, smoking, and alcohol intake (Table 10.2).
-
For children 4–17 years of age receiving GC therapy, a calcium intake of 1000 mg/day and vitamin D of 600 IU/day is recommended.
-
The ACR 2017 recommendations of initial pharmacologic treatment are categorized according to the following groups and are highlighted in Fig. 10.15 and Table 10.4.
-
All adults >40 years of age, they are divided into women >40 years old but not of childbearing potential, and men >40 years old, who are at moderate to high risk of fracture (Fig. 10.15).
-
Adults <40 years of age, includes (women not of childbearing potential and men) with a history of OP fracture, or those continuing GC treatment (>6 months at a dose of >7.5 mg/day), who have either a hip or spine BMD with Z score <−3 or DXA result reveals bone loss of >10%/year at the hip or spine (Fig. 10.15).
Fig. 10.15 
Initial pharmacologic treatment for adults [138]. Recommended doses of calcium and vitamin D are 1000–1200 mg/day and 600–800 IU/day (serum level ≥20 ng/mL), respectively. Lifestyle modifications include a balanced diet, maintaining weight in the recommended range, smoking cessation, regular weight-bearing and resistance training exercise, and limiting alcohol intake to 1–2 alcoholic beverages/day. Very high-dose glucocorticoid (GC) treatment was defined as treatment with prednisone ≥30 mg/day and a cumulative dose of >5 gm in the past year. The risk of major osteoporotic (OP) fracture calculated with the FRAX tool should be increased by 1.15, and the risk of hip fracture by 1.2, if the prednisone dose is .7.5 mg/day (e.g., if the calculated hip fracture risk is 2.0%, increase to 2.4%)
-
Special populations that have further subgroups including (Table 10.4):
-
Women who meet criteria for moderate-to-high risk of fracture and are of childbearing potential but do not plan to become pregnant within the period of OP treatment and are using effective birth control or are not sexually active.
-
Adults >30 years of age who are receiving very high-dose GC treatment (initial prednisone dose of >30 mg/day [or equivalent GC exposure] and a cumulative annual dose of >5 gm) (Table 3 of main reference).
-
Adults who have received an organ transplant and who are continuing treatment with GCs.
-
GC-treated children at 4–17 years of age are further subdivided into two groups (Table 10.4).
-
-

6.4.5 Rationale of Pharmacotherapy of GIOP
-
GIOP can be partially prevented by using bisphosphonates (alendronate and zoledronic acid) [142]. However, oral bisphosphonates are limited by low adherence rates, and therefore zoledronic acid provides the intravenous form of this medication and can be prescribed rather than the patient receiving no additional therapy beyond calcium and vitamin D.
-
On the other hand, PTH 1-34 (teriparatide) therapy seems to be superior to oral bisphosphonates but is more expensive [143] and can be used if bisphosphonate is not appropriate.
-
If neither oral nor IV bisphosphonates nor teriparatide treatment is appropriate, denosumab should be used rather than the patient receiving no additional treatment beyond calcium and vitamin D. Denosumab is a humanized monoclonal antibody to RANKL and is useful for GC-treated patients with renal insult but with stable serum Ca+2 levels and are not candidates for bisphosphonates or teriparatide. Denosumab has been approved for the prevention of vertebral and non-vertebral fractures, in women with postmenopausal osteoporosis [144]. Moreover, it was revealed that denosumab therapy increased spine and hip BMD and reduced bone turnover markers for 12 months in patients received GC [145]. A recent randomized, doubleblind, comparative study of denosumab and risedronate in patients ≥19 years of age taking prednisolone ≥7.5 mg/day for ≥3 months reported that denosumab significantly increased spine and femoral BMD compared to risedronate [146].
-
If none of these medications is appropriate for postmenopausal women, raloxifene [selective estrogen receptor modulator (SERM)] should be used rather than the patient receiving no additional treatment beyond calcium and vitamin D. The order of the preferred treatments was established according to a comparison of efficacy (fracture reduction), toxicity, and cost.
6.4.6 Follow-up Treatment Recommendations
Initial treatment failure is defined if the osteoporotic fracture occurs after 18 months of treatment initiation with oral bisphosphonate or if there is significant BMD reduction (≥10%/year) at follow-up. Various categories of treatment failure of GIOP are explained in Table 10.5 with appropriate recommendations according to the reassessment of fracture risk status.
7 Summary
Chronic inflammatory diseases, namely, rheumatic diseases such as rheumatoid arthritis, systemic lupus erythematosus, and ankylosing spondylitis, are commonly associated with extra-articular side effects, including bone loss and fractures. Osteoporosis-related fragility fractures represent one of the most important adverse outcomes that may occur in patients with rheumatic diseases. These fractures may contribute to a significant decrease in quality of life and thus would have a great impact on the economic status of the society.
The concept of osteoimmunology elucidates in depth the links between the immune system and bone physiology. The predisposing factors that cause the underlying pathology of bone loss in rheumatic patients are multifactorial. In addition to the traditional background fracture risks, such as age, BMI, and gender, there are potential bone loss mediators that substantially increase fracture risk in these patients. Of these common mediators are inflammation (high disease activity), immobility, and treatment with glucocorticoids. Other mediators would contribute in bone loss in rheumatic patients and may include poor nutrition, the increase of catabolic state, and the decrease in reproductive hormones (hypogonadism) in both men and women.
These effector mediators appear to interact in a complex and synergistic way to reinforce each other through various mechanisms that act on a shared common pathway, the bone remodeling cycle. The net result of these mediators is the production of a wide spectrum of cytokines that stimulate local and/or generalized bone resorption and that inhibit (as in RA) or stimulate (as in AS) bone formation. For instance, during the inflammatory process, the Wnt-signaling antagonist (DKK-1) is secreted from the synovial fibroblast and inhibits osteoblast maturation and OPG function leading to suppression of local bone formation. Therefore, administration of anti DKK-1 would be useful to prevent bone erosions and reverse the inhibition on bone formation. However, immobility will suppress the mechanosensing process of osteocytes leading to uncoupling of bone formation and bone resorption through the Wnt-signaling pathway.
Although GCs are frequently prescribed for the rheumatic patients, they have a great adverse impact on bone quality leading to GIOP. The overall effects of GCs on bone are either directly on bone cells or indirectly by affecting the bone metabolism, both of which result in enhancing bone resorption and decreasing bone formation. Inhibition of bone formation by GCs occurs by increasing the osteoblast and osteocyte apoptosis and/or impairing osteoblast function via suppressing the BMP pathway and the Wnt-signaling pathway. On the other hand, GCs can stimulate bone resorption by reducing osteoclast apoptosis via upregulation of RANKL and inhibition of OPG.
GIOP is a significant clinical complication that occurs as a result of adverse effects of the prescribed GCs for patients with rheumatic disease. ACR 2010 had set several recommendations updated that of ACR 2001 for evaluating and monitoring patients, who has just initiated or received GCs for/or more than 3 months duration. However later, ACR 2017 recommendations have been released and aimed to standardize the classification of patients at risk of GC induced fracture (Fig. 10.12). So that the appropriate recommendations can be applied on each category, while reducing the risk and burden of radiological testing and the anti-fracture therapy. Therefore, all clinicians treating patients with GCs should be aware of these fracture risks and identify the patient’s level of fracture risk according to the ACR 2017 guidelines for assessment and reassessment of fracture risks. The recommendations of anti-fracture pharmacotherapy in order of preference, for prevention and treatment of GIOP, were based on their efficacy, potential harms, and cost. Hence, oral bisphosphonates were recommended as preferred first-line therapy over other recommended anti-fracture therapies.
Taking together, the salient approach for early diagnosis of rheumatic diseases would be very crucial and of great help in diminishing the magnitude of bone destruction that occur during the pathogenesis of these diseases and therefore preventing further bone erosion and osteoporosis. Finally, GIOP is a medical problem that patients should be aware of its fracture risk and clinicians should consider evaluating the fracture risks for all GC-treated patients and actively prevent reduction of bone mass.
References
Spector TD, Hall GM, McCloskey EV, Kanis JA. Risk of vertebral fracture in women with rheumatoid arthritis. BMJ. 1993;306:558.
Gough AK, Lilley J, Eyre S, Holder RL, Emery P. Generalised bone loss in patients with early rheumatoid arthritis. Lancet. 1994;344:23–7.
Bernstein CN, Blanchard JF, Leslie W, Wajda A, Yu BN. The incidence of fracture among patients with inflammatory bowel disease. A population based cohort study. Ann Intern Med. 2000;133:795–9.
Bultink IE, Lems WF, Kostense PJ, Dijkmans BA, Voskuyl AE. Prevalence of and risk factors for low bone mineral density and vertebral fractures in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52:2044–50.
Schett G, Kiechl S, Weger S, et al. High sensitivity C-reactive protein and risk of nontraumatic fractures in the Bruneck study. Arch Intern Med. 2006;166:2495–501.
Sinigaglia L, Varenna M, Girasole G, Bianchi G. Epidemiology of osteoporosis in rheumatic diseases. Rheum Dis Clin N Am. 2006;32:631–58.
Glimcher MJ. Composition, structure and organization of bone and other mineralized tissues and the mechanism of calcification. In: Aurbach GD, editor. Handbook of Physiology-Endocrinology, (Vol. 7/sec. 7). DC: American Physiological Society. Washington; 1976. p. 25–116.
Junqueira LC, Carneiro J, Kelley RO., “Bone”, in basic histology (9th edition)., Appleton & Lange, (1998).
Manolagas SC, Jilka RL. Bone marrow, cytokines, and bone remodeling: emerging insights into the pathophysiology of osteoporosis. N Engl J Med. 1995;332(5):305–11.
Caplan AI. Mesenchymal stem cells. J Orthop Res. 1991;9:641–50.
Owen M. Marrow stromal stem cells. J Cell Sci Suppl. 1988;10:63–76.
Danen EH, Lafrenie RM, Miyamoto S, Yamada KM. Integrin signaling: cytoskeletal complexes, MAP kinase activation, and regulation of gene expression. Cell Adhes Commun. 1998;6:217–24.
Komori T. Regulation of osteoblast differentiation by transcription factors. J Cell Biochem. 2006;99(5):1233–9.
Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–50.
Otto F, Thornell AP, Crompton T, Denzel A, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–71.
Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–64.
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29.
Dempster DW, Cosman F, Parisien M, Shen V, Lindsay R. Anabolic actions of parathyroid hormone on bone. Endocr Rev. 1993;14:690–709.
Boyce BF, Hughes DE, Wright KR, Xing L, Dai A. Recent advances in bone biology provide insight into the pathogenesis of bone diseases. Lab Investig. 1999;79:83–94.
Lian JB, Stein GS, Stein JL, van Wijnen AJ. Regulated expression of the bone specific osteocalcin gene by vitamins and hormones. Vitam Horm. 1999;55:443–509.
Ishida Y, Heersche JH. Glucocorticoid-induced osteoporosis: both in vivo and in vitro concentrations of glucocorticoids higher than physiological levels attenuate osteoblast differentiation. J Bone Miner Res. 1998;13:1822–6.
Lacey DL, Timms E, Tan H-L, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–76.
Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, et al. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117:122–32.
Fauci AS, Kasper DL, Braunwald E, et al., Harrison’s Principles of Internal Medicine, 17th Edition: http://www.accessmedicine.com, [Copyright of The McGray-Hill Companies, Inc, . (Modified from T Suda et al: Endocr Rev 20:345, 1999, with permission.)].
Zaidi M. Calcium receptors on eukaryotic cells with special reference to the osteoclast. Biosci Rep. 1990;10(6):493–507.
Teti A, Rucci N. The unexpected links between bone and the immune system. Medicographia. 2010;32(4):341–8.
Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, Suda T, Matsuo K. Bidirectional ephrinB2–EphB4 signaling controls bone homeostasis. Cell Metab. 2006;4:111–21.
Goldring SR, Goldring MB. Eating bone or adding it: the Wnt pathway decides. Nat Med. 2007;13:133–4.
Matsuo K, Irie N. Osteoclast–osteoblast communication. Arch Biochem Biophys. 2008;473:201–9.
Sims NA, Gooi JH. Bone remodeling: multiple cellular interactions required for coupling of bone formation and resorption. Semin Cell Dev Biol. 2008;19:444–51.
Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y. The TRAF family of signal transducers mediates NF-kappa B activation by the TRANCE receptor. J Biol Chem. 1998s;273(43):28355–9.
Burgess TL, Qian Y-X, Kaufman S, et al. The ligand for Osteoprotegerin (OPGL) directly activates mature osteoclasts. J Cell Biol. 1999;145(3):527–38.
Anderson MA, Maraskovsky E, Billingsley WL, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–9.
Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappa B ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev. 2008;29(2):155–92. Epub 2007 Dec 5. Review
Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20(3):345–57. Review
Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–19.
Yasuda H, Shima N, Nakagawa N, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139(3):1329–37.
Hofbauer LC, Dunstan CR, Spelsberg TC, Riggs BL, Khosla S. Osteoprotegerin production by human osteoblast lineage cells is stimulated by vitamin D, bone morphogenetic protein-2, and cytokines. Biochem Biophys Res Commun. 1998;250(3):776–81.
Saika M, Inoue D, Kido S, Matsumoto T. 17beta-estradiol stimulates expression of osteoprotegerin by a mouse stromal cell line, ST-2, via estrogen receptor-alpha. Endocrinology. 2001;142(6):2205–12.
Takai H, Kanematsu M, Yano K, et al. Transforming growth factor-beta stimulates the production of osteoprotegerin/osteoclastogenesis inhibitory factor by bone marrow stromal cells. J Biol Chem. 1998;273(42):27091–6.
Vidal NO, Brandstrom H, Jonsson KB, Ohlsson C. Osteoprotegerin mRNA is expressed in primary human osteoblast-like cells: down-regulation by glucocorticoids. J Endocrinol. 1998;159(1):191–5.
Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC, Khosla S. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology. 1999;140(10):4382–9.
Sasaki N, Kusano E, Ando Y, Yano K, Tsuda E, Asano Y. Glucocorticoid decreases circulating osteoprotegerin (OPG): possible mechanism for glucocorticoid induced osteoporosis. Nephrol Dial Transplant. 2001;16(3):479–82.
Bultink IE, Vis M, van der Horst-Bruinsma IE, Lems WF. Inflammatory rheumatic disorders and bone. Curr Rheumatol Rep. 2012;14(3):224–30. Review
Gravallese EM, Harada Y, Wang JT, et al. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol. 1998;152:943–51.
Hardy R, Cooper MS. Bone loss in inflammatory disorders. J Endocrinol. 2009;201(3):309–20. https://doi.org/10.1677/JOE-08-0568. Review
Walsh NC, Crotti TN, Goldring SR, Gravallese EM. Rheumatic diseases: the effects of inflammation on bone. Immunol Rev. 2005;208:228–51.
Lorenzo J, Horowitz M, Choi Y. Osteoimmunology: interactions of the bone and immune system. Endocr Rev. 2008;29(4):403–40.
Kong YY, Feige U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–9.
Gravallese EM, Manning C, Tsay A, et al. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43:250–8.
Kotake S, Udagawa N, Hakoda M, et al. 1 activated human T cells directly induce osteoclastogenesis from human monocytes: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum. 2004;4:1003–12.
Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–82.
Lundy SK, Sarkar S, Tesmer LA, Fox DA. Cells of the synovium in rheumatoid arthritis. T lymphocytes. Arthritis Res Ther. 2007;9(1):202.
Diarra D, Stolina M, Polzer K, et al. Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007;13:156–63.
Johnson ML, Harnish K, Nusse R, Van HW. LRP5 and Wnt signaling: a union made for bone. J Bone Miner Res. 2004;19:1749–57.
Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene. 2004;341:19–39.
Morvan F, Boulukos K, Clement-Lacroix P, et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21:934–45.
Tian E, Zhan F, Walker R, Rasmussen E, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–94.
Tomlinson JW, Walker EA, Bujalska IJ, Draper N, et al. 11b-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev. 2004;25:831–66.
Cooper MS, Walker EA, Bland R, Fraser WD, et al. Expression and functional consequences of 11b-hydroxysteroid dehydrogenase activity in human bone. Bone. 2000;27:375–81.
Rabbitt E, Lavery GG, Walker EA, Cooper MS, et al. Pre-receptor regulation of glucocorticoid action by 11b-hydroxysteroid dehydrogenase: a novel determinant of cell proliferation. FASEB J. 2002;16:36–44.
Cooper MS, Bujalska I, Rabbitt E, Walker EA, et al. Modulation of 11b-hydroxysteroid dehydrogenase isozymes by proinflammatory cytokines in osteoblasts: an autocrine switch from glucocorticoid inactivation to activation. J Bone Miner Res. 2001;16:1037–44.
Canalis E, Delany AM. 11b-hydroxysteroid dehydrogenase, an amplifier of glucocorticoid action in osteoblasts. J Bone Miner Res. 2002;17:987–90.
Cooper MS. Sensitivity of bone to glucocorticoids. Clin Sci. 2004;107:111–23.
Carmeliet G, Bouillon R. The effect of microgravity on morphology and gene expression of osteoblasts in vitro. FASEB J. 1999:13 S129–34.
Bonewald LF, Johnson ML. Osteocytes, Mechanosensing and Wnt signaling. Bone. 2008;42:606–15.
Armstrong VJ, Muzylak M, Sunters A, Zaman G, et al. Wnt/beta-catenin signaling is a component of osteoblastic bone cell early responses to load-bearing and requires estrogen receptor alpha. J Biol Chem. 2007;282:20715–27.
Manolagas SC, Weinstein RS, Bellido T, Bodenner DL. Opposite effects of estrogen on the life span of osteoblasts/osteocytes versus osteoclasts in vivo and in vitro An explanation of the imbalance between formation and resorption in estrogen deficiency. Journal Of Bone & Mineral Research. 1999;14(suppl. 1):S169.
Canalis E, Mazziotti G, Giustina A, Bilezikian JP. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int. 2007;18(10):1319–28. Epub 2007 Jun 14. Review
Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–82.
den Uyl D, Bultink IE, Lems WF. Advances in glucocorticoid-induced osteoporosis. Curr Rheumatol Rep. 2011;13(3):233–40. https://doi.org/10.1007/s11926-011-0173-y. Review
O’Brien CA, Jia D, Plotkin LI, et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–41.
Liu Y, Porta A, Peng X, et al. Prevention of glucocorticoid-induced apoptosis in osteocytes and osteoblasts by calbindin-D28k. J Bone Miner Res. 2004;19:479–90.
Ohnaka K, Tanabe M, Kawate H, Nawata H, et al. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem Biophys Res Commun. 2005;329:177–81.
Wang FS, Ko JY, Yeh DW, Ke HC, et al. Modulation of Dickkopf-1 attenuates glucocorticoid induction of osteoblast apoptosis, adipocytic differentiation, and bone mass loss. Endocrinology. 2008;149:1793–801.
Pereira RC, Delany AM, Canalis E. Effects of cortisol and bone morphogenetic protein-2 on stromal cell differentiation: correlation with CCAAT-enhancer binding protein expression. Bone. 2002;30:685–91.
Shi XM, Blair HC, Yang X, McDonald JM, et al. Tandem repeat of C/EBP binding sites mediates PPARgamma2 gene transcription in glucocorticoid-induced adipocyte differentiation. J Cell Biochem. 2000;76:518–27.
Carcamo-Orive I, aztelumendi A, Delgado J, et al. Regulation of human bone marrow stromal cell proliferation and differentiation capacity by glucocorticoid receptor and AP-1 crosstalk. J Bone Miner Res. 2010;25:2115–25.
Natsui K, Tanaka K, Suda M, et al. High-dose glucocorticoid treatment induces rapid loss of trabecular bone mineral density and lean body mass. Osteoporos Int. 2006;17:105–8.
Kanis JA. Diagnosis of osteoporosis and assessment of fracture risk. Lancet. 2002;359:1929–36.
Van Der Heijde DMFM. Joint erosions and patients with early rheumatoid arthritis. Br J Rheumatol. 1995;34(Suppl. 2):74–8.
McQueen FM, Stewart N, Crabbe J, et al. Magnetic resonance imaging of the wrist in early rheumatoid arthritis reveals a high prevalence of erosions at four months after symptom onset. Ann Rheum Dis. 1998;57:350–6.
McGonagle D, Conaghan PG, O’Connor P, et al. The relationship between synovitis and bone changes in early untreated rheumatoid arthritis: a controlled magnetic resonance imaging study. Arthritis Rheum. 1999;42:1706–11.
Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42.
Vosse D, de Vlam K. Osteoporosis in rheumatoid arthritis and ankylosing spondylitis. Clin Exp Rheumatol. 2009;27(4 Suppl 55):S62–7.
Romas E, Bakharevski O, Hards DK, Kartsogiannis V, et al. Expression of osteoclast differentiation factor at sites of bone erosion in collagen-induced arthritis. Arthritis Rheum. 2000;43:821–6.
Leisen JCC, Duncan H, Riddle JM, Pitchford WC. The erosive front: a topographic study of the junction between the pannus and the subchondral plate in the macerated rheumatoid metacarpal head. J Rheumatol. 1988;15:17–22.
Geusens PP, Lems WF. Osteoimmunology and osteoporosis. Arthritis Res Ther. 2011;13(5):242. Epub. This is a large overview on osteoimmunology in rheumatic diseases
Schett G, Saag KG, Bijlsma JWJ. From bone biology to clinical outcome: state of the art and future perspectives. Ann Rheum Dis. 2010;69:1415–9. This is an outstanding state-of-the-art article on bone biology
Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. 2011;377:1276–87.
Lories RJ, Luyten FP“O. Wnt antagonists: for better or worse? Nat Rev Rheumatol. 2009;5:420–1.
Terpos E, Fragidaki K, Konsta M, et al. Early effects of Il-6 receptor inhibition on bone homeostasis. Clin Exp Rheumatol. 2011;29:921–5.
Cohen SB, Dore RK, Lane NE, et al. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelvemonth, multicenter, randomized, double blind, placebo-controlled, phase II clinical trial. Arthritis Rheum. 2008;58:1299–309.
Vis M, Havaardsholm EA, Haugeberg G, et al. Evaluation of bone mineral density, bone metabolism, osteoprotegerin and receptor activator of the NF kappa B ligand serum levels during treatment with infliximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65:1495–9.
Mawatari T, Miura H, Hamai S, et al. Vertebral strength changes in rheumatoid arthritis patients treated with alendronate, as assessed by finite element analysis of clinical computed tomography scans: a prospective randomized clinical trial. Arthritis Rheum. 2008;58:3340–9.
Bultink IE. Osteoporosis and fractures in systemic lupus erythematosus. Arthritis Care Res. 2012;1:2–8. This is an outstanding overview on the multifactorial pathogenesis of osteoporosis and fractures in SLE
Borba VZ, Vieira JG, Kasamatsu T, et al. Vitamin D deficiency in patients with active systemic lupus erythematosus. Osteoporos Int. 2009;20:427–33.
Huisman AM, White KP, Algra A, et al. Vitamin D levels in women with systemic lupus erythematosus and fibromyalgia. J Rheumatol. 2001;28:2535–9.
Toloza SM, Cole DE, Gladman DD, et al. Vitamin D insufficiency in a large female SLE cohort. Lupus. 2010;19:13–9.
Ruiz-Irastorza G, Egurbide MV, Olivares N, et al. Vitamin D deficiency in systemic lupus erythematosus: prevalence, predictors and clinical consequences. Rheumatology (Oxford). 2008;47:920–3.
Formiga F, Moga I, Nolla JM, et al. The association of dehydroepiandrosterone sulphate levels with bone mineral density in systemic lupus erythematosus. Clin Exp Rheumatol. 1997;15:387–92.
Lee C, Ramsey-Goldman R. Bone health and systemic lupus erythematosus. Curr Rheumatol Rep. 2005;7:482–9.
Svenungsson E, Fei GZ, Jensen-Urstad K, et al. TNF-α: a link between hypertriglyceridaemia and inflammation in SLE patients with cardiovascular disease. Lupus. 2003;12:454–61.
Frostegard J, Svenungsson E, Wu R, et al. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum. 2005;52:192–200.
Moerman EJ, Teng K, Lipschitz DA, et al. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-γ2 transcription factor and TGF-β/BMP signaling pathways. Aging Cell. 2004;3:379–89.
McLean RR, et al. Homocysteine as a predictive factor for hip fracture in older persons. N Engl J Med. 2004;350:2042–9.
Yesilova Z, et al. Hyperhomocysteinemia in patients with Behcet’s disease: is it due to inflammation or therapy? Rheumatol Int. 2005;25:423–8.
Lakshminarayanan S, Walsh S, Mohanraj M, et al. Factors associated with low bone mineral density in female patients with systemic lupus erythematosus. J Rheumatol. 2001;28:102–8.
Almehed K, Forsblad DH, Kvist G, et al. Prevalence and risk factors of osteoporosis in female SLE patients: extended report. Rheumatology (Oxford). 2007;46:1185–90.
Mendoza-Pinto C, Garcia-Carrasco M, Sandoval-Cruz H, et al. Risk factors of vertebral fractures in women with systemic lupus erythematosus. Clin Rheumatol. 2009;28:579–85.
Lee C, Almagor O, Dunlop DD, et al. Disease damage and low bone mineral density: an analysis of women with systemic lupus erythematosus ever and never receiving corticosteroids. Rheumatology (Oxford). 2006;45:53–60.
Petri M. Musculoskeletal complications of systemic lupus erythematosus in the Hopkins lupus cohort: an update. Arthritis Care Res. 1995;8:137–45.
Kamen D, Aranow C. Vitamin D in systemic lupus erythematosus. Curr Opin Rheumatol. 2008;20:532–7.
Hollis BW, Wagner CL. Assessment of dietary vitamin D requirements during pregnancy and lactation. Am J Clin Nutr. 2004;79(5):717–26.
Kamen DL. Vitamin D in lupus - new kid on the block? Bull NYU Hosp Jt Dis. 2010;68(3):218–22. Review
Lane NE. Therapy Insight: Osteoporosis and osteonecrosis in systemic lupus erythematosus. Nat Clin Pract Rheumatol. 2006;2(10):562–9. Review
Cooper C, Carbone L, Michet CJ, et al. Fracture risk in patients with AS: a population based study. J Rheumatol. 1994;10:1887–2.
Vosse D, Landewé R, van der Heijde D, et al. AS and the risk of fracture: results from a large primary care based nested case–control study. Ann Rheum Dis. 2009;68:1839–42.
Vosse D, Feldtkeller E, Erlendsson, et al. Clinical vertebral fractures in patients with Ankylosing spondylitis. J Rheumatol. 2004;10:1981–5.
Ralston SH, Urquhart GDK, Brzeski M, Sturrock RD. Prevalence of vertebral compression fractures due to osteoporosis in ankylosing spondylitis. BMJ. 1990;300:563–5.
Mitra D, Elvins DM, Speden DJ, Collins AJ. The prevalence of vertebral fractures in mild ankylosing spondylitis and their relationship to bone mineral density. Rheumatology. 2000;39:85–9.
Nguyen HV, Ludwig S, Gelb D. Osteoporotic vertebral burst fractures with neurologic compromise. J Spin Dis Techn. 2003;16:10–9.
Voss D, Van Der Heijde DM, Landewe R, et al. Determinants of hyperkyphosis in patients with Ankylosing spondylitis. Ann Rheum Dis. 2006;65:770–4.
Bessant R, Keat A. How should clinicians manage osteoporosis in ankylosing spondylitis? J Rheumatol. 2002;29:1511–9.
Gran JT, Husby G. Clinical, epidemiological, and therapeutic aspects of ankylosing spondylitis. Curr Opin Rheumatol. 1998;10:292–8.
Gillespie LD, Gillespie WJ, Robertson MC, Lamb SE, et al. Interventions for preventing falls in elderly people. Cochrane Database Syst Rev. 2003;4:CD000340.
Wolf SL, Sattin RW Kutner M, O’Grady M, Greenspan AI, et al. Intense tai chi exercise training and fall occurrences in older, transitionally frail adults: a randomized, controlled trial. J Am Geriatr Soc. 2003;51:1693–701.
Orwoll E, Ettinger M, Weiss S, Miller P, et al. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604–10.
Ringe JD, Faber H, Farahmand P, Dorst A. Efficacy of risedronate in men with primary and secondary osteoporosis\: results of a 1-year study. Rheumatol Int. 2006;26:427–31.
Kaufman JM, Orwoll E, Goemaere S, San Martin S, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005;16:510–6.
LoCascio V, Bonucci E, Imbimbo B, et al. Bone loss in response to long-term glucocorticoid therapy. Bone Miner. 1990;8:39–51.
Reid DM, Devogelaer JP, Saag K, et al. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet. 2009;373:1253–63.
van Staa TP, Laan RF, Barton IP, et al. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arth Rheum. 2003;48:3224–9.
van Staa TP, Geusens P, Pols HA, de Laet C, et al. A simple score for estimating the long-term risk of fracture in patients using oral glucocorticoids. QJM. 2005;98:191–8.
De Vries F, Bracke M, Leufkens HG, et al. Fracture risk with intermittent high-dose oral glucocorticoid therapy. Arthritis Rheum. 2007;56:208–14.
American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum 2001;44:1496–1503.
Grossman JM, Gordon R, Ranganath VK, Deal C, et al. American College of rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken). 2010;62(11):1515–26. https://doi.org/10.1002/acr.20295. Epub 2010 Jul 26. Review. Erratum in: Arthritis Care Res (Hoboken). 2012 Mar;64(3):464. PubMed PMID: 20662044].
Buckley L, Guyatt G, Fink HA, et al. American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis. Arthritis Rheumatol. 2017;69(8):1521–1537. https://doi.org/10.1002/art.40137. Epub 2017 Jun 6. Erratum in: Arthritis Rheumatol. 2017;69(11):2246. PMID: 28585373.
Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. Geneva, World Health Organization, 1994 (WHO Technical Report Series, No. 843).
FRAX: (https://www.shef.ac.uk/FRAX/tool.jsp).
Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011;96:53–8.
Weinstein RS. “Clinical Practice: Glucocorticoid-induced bone disease”. N Engl J Med. 2011;365:62–70. [PubMed: 21732837].
Weinstein RS, Jilka RJ, Roberson PK, et al. “Intermittent parathyroid hormone administration prevents glucocorticoid-induced osteoblast and osteocyte apoptosis, decreased bone formation, and reduced bone strength in mice”. Endocrinol. 2010;151:2641–649.
Cummings SR, San Martin J, McClung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–65. [PubMed: 19671655].
Dore RK, Cohen SB, Lane NE, et al. Effects of denosumab on bone mineral density and bone turnover in patients with rheumatoid arthritis receiving concurrent glucocorticoids or bisphosphonates. Ann Rheum Dis. 2010;69:872–5.
Saag KG, Wagman RB, Geusens P, et al. Denosumab versus risedronate in glucocorticoid-induced osteoporosis: a multicentre, randomised, double-blind, active-controlled, double-dummy, non-inferiority study. Lancet Diabetes Endocrinol. 2018;6:445–54.
Acknowledgments
The author of this chapter is grateful to the medical student Somayya Khan for her efforts in drawing some of this chapter’s figures. The authors also would like to thank Dr. Waleed Hafiz for his assistance in the development of this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Abdulkhaliq, A. (2021). Bones and Rheumatology. In: Almoallim, H., Cheikh, M. (eds) Skills in Rheumatology . Springer, Singapore. https://doi.org/10.1007/978-981-15-8323-0_10
Download citation
DOI: https://doi.org/10.1007/978-981-15-8323-0_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-8322-3
Online ISBN: 978-981-15-8323-0
eBook Packages: MedicineMedicine (R0)