
Abstract
When individual immune cells are triggered by pathogens or signalling molecules, a highly complex set of pathways regulate the response that involves a diverse range of intracellular components. Typically assessed through either fluorescent labelling or by invasive sequencing techniques, an alternative approach is becoming possible: the label-free measurement of the changes that occur on activation. Label-free analysis that can discriminate between different cell types or activation states uses contrast based on the endogenous molecular components of the cell, and can have sensitivity to both the molecular and morphological phenotype of the cells. Using optical scattering spectroscopy, which highlights molecular characteristics of the cell, along with quantitative phase imaging that provides morphology information, the elucidation of activation states can be achieved using only light. These techniques show that label-free optical methods can be an important part of the framework for future directions in single cell analysis.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


First I would like to acknowledge the support of the Uehara Foundation, who supported the research discussed here. Aside from financial support, which is crucial, members of the foundation also expressed their interest in developing new ways to visualize and measure cellular processes. Other recipients of Uehara support also were enthusiastic and provided useful feedback and support of this research. It has been an honor to be a part of this program. I would also like to thank the members of my lab, especially Dr. Nicolas Pavillon and Dr. Alison Hobro, without whom this work would not have been what it is. Also my colleagues and collaborators, particularly in Raman imaging at Osaka university, and in immunology at our institute have been very supportive.
Since joining the IFReC immunology institute, our lab research has been aimed at creating new optical tools to understand some of the less clear aspects of the immune response, especially on the single-cell level. Most of the tools used by researchers to study the cell response involve tagging specific target molecules and seeing how they are expressed in different cell types, and determining how that expression changes during dynamic processes such as the stimulation of different immune pathways. There are a large number of pathways, signaling ions, expression levels of different proteins, RNA, and more that can be targeted (Spiller et al. 2010) and this number continually grows as researchers unlock new information on how the cell is built, and how it responds. The growing number of known pathways means that at any given time, we have had, and continue to have, only a partial view of the basic mechanisms of the cell. This is where the developments in this project can play a role: using label-free optical methods to interrogate the cell, we are able to analyze changes in molecular content and morphology which can correlate with how the cell is responding to immune stimulation. This approach does not give the same level of specificity as using fluorescent markers, PCR, or other analytical chemistry-based techniques, but instead allows the generation of quantitative data which corresponds to the entire cellular composition and shape, which in turn, can be correlated with cell type, cell state, or other features of interest. It also allows mining the data to look for emergent patterns, correlations with other measurements, or to look for new features or indicators of cellular reactions which may not have been observed with other measurements.
A main technique we use is based on measuring how light is scattered from the sample, and by spectrally resolving the scattered light, we see the shifts, known as Raman shifts, in the light where the wavelength of scattered light is shifted due to the molecular vibrations in the sample. Rather than just elastically scattering the incident light, the sample molecules appropriate some of the photon energy, so that the scattered photons are red-shifted. The degree of energy absorbed, and therefore the wavelength shift, depends on vibrational modes in the sample molecules. Using this process, Raman imaging then gives us an image where the contrast, color, or both are determined by the molecules that make up the sample. This can then be exploited to visualize some of the basic activities of the cell such as cell division (Hamada et al. 2008), apoptosis (Okada et al. 2012), and can be expanded to take advantage of emerging high-resolution techniques (Watanabe et al. 2015). Taking the Raman spectra from a cell provides a signature of the cell’s chemical makeup. Not surprisingly, most cells have a roughly similar composition and therefore Raman spectra look similar between different cells. The slight differences, however, in Raman spectral features either within cells, between cells, or from different timepoints, can have enough statistical weight to discriminate cellular behavior or type that can be difficult to distinguish using any other methods, except by breaking the cell apart for analysis or by introducing a specific exogenous material to probe the cell states (Fig. 14.1).

(Left panel) Label-free Raman image of mouse embryonic fibroblast cells, cultured on a quartz substrate. Cells were excited by 532 nm laser, in a line-scan configuration, with spectrometer and cooled CCD detector. The green channel is a summation of Raman shifts between 2930–2940 cm−1 (shared by protein and lipid) and the red channel is between 2848–2856 cm−1 (predominantly lipid). Even with some overlap between the channels, nuclear features appear in saturated green, while lipid droplets appear as strong yellow. It is possible to further separate channels by processing if required. (Right panel) Average Raman spectra for different lymphocyte cell lines showing overall similarity, but statistically significant differences between ratios of peaks allows the discrimination of cell type by spectra. (See Hobro et al. 2016 for details)
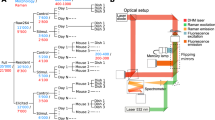
Raman imaging produces large amounts of highly useful data, and when performed with conservative laser excitation powers, does not harm or modify the cell. However, it is possible to simultaneously multiplex Raman imaging with other label-free modes (Pavillon et al. 2014) and thereby gain complementary data from the sample. The only downside to this approach is complexity of the optical setup, which once solved, allows simultaneous multimodal quantitative label-free imaging and analysis. To this end we also use quantitative phase imaging, which works by measuring the differences in optical path length across the cells, and provides a spatially resolved map of the sample morphology. From a collimated beam of laser light, a partial mirror splits the beam, sending half of the light through the sample, while the other beam acts as a reference and does not pass through the sample. Both beams are then combined and interfered on a detector. This gives a spatially resolved map of the optical path length and attenuation characteristics of the sample. The technique we use, known as digital holographic microscopy (DHM), provides quantitative morphological data that has been shown to be highly useful, for example demonstrating the ability to predict cell death at an early stage by detecting the changes in cellular ionic homeostasis (Pavillon et al. 2012)
Combining these two optical measurement approaches, see Fig. 14.2, allows us to use either or both sets of complementary information. Of particular note is the difference in imaging speed of these two modalities. Raman imaging can be performed on a time scale on the order of minutes, while the DHM phase imaging can be as fast as video rate. This allows the detection of relatively fast moving objects in the cell. Typical speeds of intracellular transport or bacterial motion are on the order of micrometers per second, which are easily resolved by the DHM mode, but too fast for the Raman imaging. The Raman mode, however, has a unique ability to give insight into the nature of the dynamic intracellular objects. In our data of moving objects we have utilized the DHM imaging mode to observe intracellular objects and then used the Raman measurements to classify the types of moving objects, such as vesicles, nuclear components, or invasive objects such as bacteria, nanoparticles or adjuvants.

Simultaneous label-free quantitative phase imaging (DHM) and Raman imaging of MH-S macrophages (a)–(f) and HeLa (g) and (h) cells. The image contrast is as labelled for each panel in (a)–(f), from (a) the C-H stretching band (2935–2955 cm−1), (b) the cytochrome C band (740–780 cm−1), (c) the lipid-dominated (2860–2880 cm−1), (d, g) phase, with (e, f, h) constructed as overlay images. Scale bars are 5 μm. (Image from Pavillon et al. 2014)
One example of this type of intracellular invasive object identification and tracking was done in our lab, where macrophages were exposed to hemozoin nanocrystals. Hemozoin is the byproduct of malaria infection, where the parasite biocrystallizes free heme to make a heme-based crystal. The uptake of the particles can produce an immune response, and can also act as an adjuvant in the response to other immune-triggering substances. Figure 14.3 shows Raman imaging of the macrophage hemozoin uptake, with the color channels determined automatically by PCA analysis. The images show the response can be visualized in terms of how endogenous compounds (see figure caption) move around in the cell in response to the presence of hemozoin. By comparison, the control cells look relatively homogenous. This in contrast to the cell imaging shown above, and is a result of the PCA analysis used here: the method determines the strongest contributions to the spectral contrast. When control data, 3 h, and 5 h post-incubation data is analyzed together, the differences within the cells at 3 and 5 h show the strongest contrast. If only control data is analyzed, then variance within the cell components, such as cytosolic components and nuclear material, is highlighted similar to the cell images shown above in Figs. 14.1 and 14.2.

Label-free Raman imaging of the uptake of malaria-related hemozoin by modified bone-marrow derived macrophages. The red channel shows hemozoin, appearing only at 3 and 5 h post-incubation, while the other channels show how the endogeneous cell components rearrange in response to the presence of the hemozoin. Color channels are assigned by principal components (PC1–PC4) determined from Raman spectral contrast. (See Hobro et al. 2015 for details)
We can also use the quantitative phase information to determine a number of different features in the cells that correlate with immune reactions or other processes in the cell. For macrophages exposed to LPS, toll-like receptors trigger a cascade of signals in the inflammatory response (Kawai and Akira 2010) the details of which are still an important topic of investigation. However, in a sample dish of relatively homogenous cultured cells, with tens of thousands of cells, each cell responds slightly differently. Using both Raman and quantitative phase data, it is possible to observe the effects of LPS stimulation on the composition and morphology of individual cells, with single-cell precision and relatively high throughput. The use of quantitative phase imaging for this application in particular has the potential to be high throughput since the imaging time for a field of view is in the order of milliseconds. Figure 14.4 shows the concept of imaging and processing macrophages using this approach.

Quantitative DHM phase images of J774 macrophages, in control conditions and when exposed to bacteria-related LPS. Morphological features as well as Raman spectral features can both be used to investigate, on a single-cell level, how individual cells respond to immune stimulation (Pavillon et al. 2018)
Interestingly, more and more reports are now emerging of how individual cells respond differently, and the expression of different markers can be highly variable. The approaches shown here provide a means to evaluate these single cell level responses, without changing or modifying the sample. While not having the specificity of more traditional techniques such as labelling or invasive PCR type methods, these label-free techniques allow the evaluation of different cellular functions before, during, and after they occur, and can then be used over time to evaluate the progression as well as heterogeneity of cell responses. We also use complementary techniques such as ELISA or other wetlab techniques to validate these methods, and can use the resulting data to form a set of independent measurements, covering both population level responses (from ELISA) and single cell variations in response.
I sincerely appreciate the Uehara foundation funding for supporting this fundamental research and we look forward to future progress building on these results.
References
Hamada K et al (2008) Raman microscopy for dynamic molecular imaging of living cells. J Biomed Opt 13:44027
Hobro AJ, Kumagai Y, Akira S, Smith NI (2016) Raman spectroscopy as a tool for label-free lymphocyte cell line discrimination. Analyst 141:3756–3764
Hobro AJ et al (2015) Label-free Raman imaging of the macrophage response to the malaria pigment hemozoin. Analyst 140:2350–2359
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384
Okada M et al (2012) Label-free Raman observation of cytochrome c dynamics during apoptosis. Proc Natl Acad Sci 109:28–32
Pavillon N, Fujita K, Smith NI (2014) Multimodal label-free microscopy. J Innov Opt Health Sci 7:1330009
Pavillon N et al (2012) Early cell death detection with digital holographic microscopy. PLoS One 7:e30912
Pavillon N, Hobro AJ, Akira S, Smith NI (2018) Noninvasive detection of macrophage activation with single-cell resolution through machine learning. Proc Natl Acad Sci U S A 115(12):E2676–E2685
Spiller DG, Wood CD, Rand DA, White MRH (2010) Measurement of single-cell dynamics. Nature 465:736–745
Watanabe K et al (2015) Structured line illumination Raman microscopy. Nat Commun 6:10095
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Smith, N.I. (2020). Multimodal Label-Free Imaging to Assess Compositional and Morphological Changes in Cells During Immune Activation. In: Toyama, Y., Miyawaki, A., Nakamura, M., Jinzaki, M. (eds) Make Life Visible. Springer, Singapore. https://doi.org/10.1007/978-981-13-7908-6_14
Download citation
DOI: https://doi.org/10.1007/978-981-13-7908-6_14
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-7907-9
Online ISBN: 978-981-13-7908-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)