
Abstract
Mutagenesis of in vitro propagated bananas is an efficient method to introduce novel alleles and broaden genetic diversity. The FAO/IAEA Plant Breeding and Genetics Laboratory previously established efficient methods for mutation induction of in vitro shoot tips in banana using physical and chemical mutagens as well as methods for the efficient discovery of ethyl methanesulphonate (EMS) induced single nucleotide mutations in targeted genes. Officially released mutant banana varieties have been created using gamma rays, a mutagen that can produce large genomic changes such as insertions and deletions (InDels). Such dosage mutations may be particularly important for generating observable phenotypes in polyploids such as banana. Here, we describe a Next Generation Sequencing (NGS) approach in Cavendish (AAA) bananas to identify large genomic InDels. The method is based on low coverage whole genome sequencing (LC-WGS) using an Illumina short-read sequencing platform. We provide details for sonication-mediated library preparation and the installation and use of freely available computer software to identify copy number variation in Cavendish banana. Alternative DNA library construction procedures and bioinformatics tools are briefly described. Example data is provided for the mutant variety Novaria and cv Grande Naine (AAA), but the methodology can be equally applied for triploid bananas with mixed genomes (A and B) and is useful for the characterization of putative Fusarium Wilt TR4 resistant mutant lines described elsewhere in this protocol book.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
New developments in the field of molecular biology enable fast and accurate identification of spontaneously occurring or induced changes of DNA sequence. This can allow a more precise use of induced mutations in crop improvement programmes. Mutagens produce various spectrum of changes. Chemical mutagens such as EMS predominantly induce point mutations, whereby physical mutagens such as gamma irradiation produce a broader spectrum of changes ranging from SNPs and small InDels to deletions greater than one million base pairs (Jankowicz-Cieslak and Till 2015; Till et al. 2018; Datta et al. 2018). While phenotypic consequences of large structural variants may be greater, datasets on the spectrum and density of such mutations are currently much smaller than that of EMS.
Mutation breeding may be especially useful in species with a narrow genetic base or those that are recalcitrant to traditional breeding methods such as obligate and facultative vegetatively propagated species. Additionally, mutagens that cause dominant or dosage-based phenotypes can increase the efficiency of generating novel traits in polyploids as the expression of phenotypes arising from recessive mutations requires the combination mutations from homologous sequences (Krasileva et al. 2017). Gamma irradiation has been used widely as a mutagenizing agent for breeding programmes for many crops. In poplar, treatment of pollen with gamma irradiation resulted in InDels varying between small fragments to whole chromosomes (Henry et al. 2015). This work further showed that large genomic InDels could be effectively recovered using low coverage whole genome sequencing (LC-WGS), making mutation discovery more cost-effective and data analyses more streamlined. Larger deletions range in size and may include loss of part of a chromosome (segmental aneuploidy) or loss of an entire chromosome (aneuploidy). Aneuploidy is better tolerated in polyploid plants and may be lethal for diploid plants and animals (Siegel and Amon 2012). These lead to changes in copy number of single genes to whole chromosomes, which have profound effects on phenotypes of the organism. Copy number variations especially affect haploinsufficient genes for which a single functional copy of a gene is not sufficient for normal function. Single copy mutations can potentially knock out the function of genes where only one functional copy is being maintained.
Inducing mutations in triploid banana provides an approach for generating novel variation that is heritable. The logistics of banana mutation breeding including tissue culture propagation, chimerism, polyploidy, heterozygosity, and field space required to find rare favourable mutations makes banana less tractable than seed propagated crops. However, these limitations can be overcome by tissue culture mutagenesis and genomic screening at earlier stages. Previously, we established a system for inducing and maintaining SNP mutations in clonally propagated banana plants. Treating shoot apical meristems of tissue cultured bananas with the chemical mutagen ethyl methanesulphonate (EMS) introduced a high density of GC-AT transitions mutations (Jankowicz-Cieslak et al. 2012). We further showed that mosaicism (chimerism) caused by accumulation of chemically induced mutations in different cells of the plant propagule could be rapidly removed via isolation of shoot apical meristems and subsequent longitudinal bisection. Further, induced mutations were maintained in mutant plants for more than six generations.
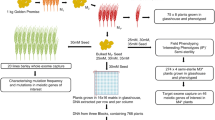
We sought to establish a similar system for inducing and maintaining insertions and deletions using physical irradiation. We aimed to develop an efficient pipeline for the generation and recovery of large copy number variations (CNVs) in gamma irradiated Cavendish banana cultivars, employing tissue culture, low coverage whole genome sequencing (LC-WGS) and chromosome dosage analysis (Fig. 8.1). We chose a chromosomal dosage analysis that was previously successful in detecting aneuploidy, insertions and deletions in Arabidopsis, rice and poplar (Tan et al. 2015, 2016). To establish a pipeline for banana, we first adapted sequencing and dosage analysis for the previously released mutant banana variety Novaria. Large genomic deletions of up to 3.8 Mbps were recovered. We next developed a newly mutagenized banana population and tested two different irradiation dosages to establish that new genetic variation can be induced and maintained in vitro (Datta et al. 2018). This work suggests that a large-scale mutagenesis pipeline can be created for routine production of mutant populations suitable for glasshouse and field evaluations. The efficacy of this approach is being further tested for Foc TR4 resistance (Fig. 8.1). We provide here the methodology for low-coverage DNA sequencing and data analysis to identify large indels in mutant populations of triploid (AAA) banana.

A pipeline for the generation and recovery of large copy number variations (CNVs) in gamma irradiated Cavendish banana cultivars, employing tissue culture, low coverage whole genome sequencing (LC-WGS) and chromosome dosage analysis. An in vitro mutant population was generated, and a subset was evaluated using the method described in this chapter. This ensures that mutagenesis was successful and mitotically heritable DNA lesions were produced during gamma irradiation and subsequent propagation. Genome sequencing can also be applied to plants showing improved resistance to disease such as Foc TR4 in order to identify mutations causative for the observed phenotype(s). (This figure is modified from Jankowicz-Cieslak et al. 2021)
2 Materials
2.1 Library Preparation and Sequencing
2.1.1 DNA Isolation and Quantification
-
1.
DNA isolation kit (e.g. DNeasy Plant Mini Kit, Qiagen, Cat Nr: 69104).
-
2.
Vortex mixer.
-
3.
Microcentrifuge.
-
4.
Micropipettes (1000 μl, 200 μl, 20 μl, 10 μl).
-
5.
Microcentrifuge tubes (1.5 ml, 2.0 ml).
-
6.
Metal beads (e.g. tungsten carbide beads, 3 mm, Qiagen, Cat Nr: 69997; see Note 1).
-
7.
Distilled or deionized water (dH2O).
-
8.
RNase A (10 μg/ml).
-
9.
Absolute ethanol.
-
10.
10× TE buffer (100 mM Tris-HCl, 10 mM EDTA, pH 8.0).
-
11.
Equipment for horizontal gel electrophoresis (combs, casting trays, gel tank, power supply).
-
12.
Agarose (for gel electrophoresis).
-
13.
0.5× TBE (Tris/Borate/EDTA) buffer (for gel electrophoresis).
-
14.
Ethidium bromide or equivalent double stranded DNA dye (see Note 2).
-
15.
Lambda DNA (e.g. Invitrogen, Cat Nr: 25250-010) for the preparation of concentration standards (see Note 3).
-
16.
DNA gel loading dye (containing Orange G or bromophenol blue) (see Note 4).
-
17.
Gel photography system (digital camera, light box).
2.1.2 Library Preparation and Sequencing
-
1.
Covaris M220 Ultrasonicator.
-
2.
microTUBE AFA Fiber Pre-Slit Snap-Cap (Cat Nr: 520077).
-
3.
TruSeq Kit (e.g. TruSeq®Nano DNA Library Prep kit; see Note 5).
-
4.
Fresh 70% EtOH.
-
5.
Magnetic stand.
-
6.
Qubit system (Thermo Fisher Scientific).
-
7.
Qubit dsDNA HS Assay Kit (Cat Nr: Q32854).
-
8.
PCR cycler.
2.2 DATA Analyses
-
1.
Computer with minimum processor requirement (see Note 6).
-
2.
Burrows-Wheeler Aligner (BWA) (http://bio-bwa.sourceforge.net/).
- 3.
-
4.
Samtools (http://www.htslib.org/).
-
5.
Bin-by-Sam-tool (see Note 7).
-
6.
Python version 2.7 (pre-installed with the Ubuntu operating system).
-
7.
Spreadsheet software (pre-installed with the Ubuntu operating system).
3 Methods
3.1 Library Preparation and Sequencing
3.1.1 DNA Isolation
-
1.
Collect 100 mg fresh weight tissue per sample and freeze at −80° C (see Note 8).
-
2.
Extract DNA using the DNeasy Plant Mini Kit (Qiagen, Cat Nr: 69106) or equivalent (see Note 9).
3.1.2 Assay DNA Quality and Quantity
-
1.
Measure concentration using a Qubit fluorometer (see Note 10).
-
2.
Prepare DNA concentration standards for gel electrophoresis (Huynh et al. 2017).
-
3.
Dilute lambda DNA to a set of DNA concentrations covering the expected range of the genomic DNA samples being assayed (see Note 11).
-
4.
Prepare a 1.5% agarose gel in 0.5× TBE buffer with 0.2 μg/ml ethidium bromide or alternative dye.
-
5.
Add 3 μl of DNA sample plus 2 μl DNA loading dye (see Note 12).
-
6.
Load samples and concentration standards on the gel.
-
7.
Run gel at 5–6 V/cm for 30–60 min (see Note 13).
3.1.3 Library Preparation for Sequencing
-
1.
Choose library preparation method, sequencing chemistry and read-length (see Note 14).
-
2.
If using low-DNA input library preparation with 550 bp fragments for 2×300PE (paired-end) sequencing, add 500 ng of genomic DNA to a Covaris Snap-Cap microTUBE.
-
3.
Shear the DNA in the M220 Covaris sonicator with the following settings: Peak Incident Power (W); 50, Duty Factor (%) 10; Cycles per Burst (cpb) 200; Treatment Time (sec) 50 (see Note 14).
-
4.
Proceed to library preparation using a TruSeq®Nano DNA Library Prep kit or equivalent, following manufacturers protocol. Use unique barcodes/indices for each sample.
-
5.
Quantify libraries using Qubit and determine molarity using the following equation where 660 is the molecular weight of a DNA base pair and median size is the average size of fragments in base pairs (see Note 15): ng/ul * 1 mol/(660)g * MEDIAN SIZE* 1 g/10ee9ng*10ee9nmol/mol*1ul/10ee-6 l = nmol/l
-
6.
Determine sample pooling based on sequencing throughput and genome size (see Note 16).
-
7.
Adjust each sample to 4 nM and pool samples together according to Note 16.
-
8.
Sequence pooled library (see Note 17)
3.1.4 Data Analysis
-
1.
Obtain Illumina raw sequence reads (see Note 18 and Fig. 8.2).
-
2.
Set up your computer for analysis (see Note 6).
-
3.
Install BWA following installation instructions on SourceForge. Alternatively, if running Ubuntu, the software is available from the repository and can be loaded by opening a terminal window and typing: sudo apt install bwa.
-
4.
Install samtools following instructions on htslib.org. Alternatively, if using Ubuntu, type the command sudo apt install samtools into the terminal window.
-
5.
Quality filter the sequence reads and trim to remove adapter sequences.
-
6.
Identify samples according to index and de-multiplex (see Note 19).
-
7.
Prepare the fastq data for mapping. In the example data provided (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA627139), the sequencing library was prepared using a kit that includes a post adapter-ligation amplification step. This can produce duplicated reads that affect data analysis. Duplicate reads can be removed using a tool in the BBmap suite. Install BBmap following the instructions found at https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/installation-guide/. Place the processed fastq.gz into the BBmap directory. Open a terminal window (found in the applications folder in Ubuntu 18.04, enter (cd) the BBmap directory and then execute the reformat software. The command to execute this from the BBmap directory is
-
./clumpify.sh in = Sample.R1.fq.gz in2 = Sample.R2.fq.gz out = Sample.R1.dedup.fastq.gz out2 = Sample.R2.dedup.fastq.gz dedupe;
-
Where Sample.R1 and Sample.R2 are the two paired-end reads from one of the samples. For example, G2.R1.fq.gz, and G2.R2.fq.gz in the example data for the cv Grande Naine. Repeat this for all samples.
-
-
8.
Prepare reference genome for mapping (see Note 20). The reference genome used for the banana example data can be found here https://www.ncbi.nlm.nih.gov/assembly/GCF_000313855.2. Click the download assembly button and select RefSeq and Genomic Fasta options.
-
9.
Create a new directory for your analysis. For the test data, make a folder titled BananaGamma in the home directory. In this directory, create a new folder titled Genome. Place the .fna file downloaded in step 8 into this folder.
-
10.
Index the genome for mapping. In the terminal window enter (cd) the Genome folder and execute the following command: bwa index *.fna. Five additional files will be produced.
-
11.
Map the data using BWA-mem. Move (mv) the dedup.fastq.gz files created in step 7 into the project directory (e.g. BananaGamma). Execute the following command: bwa mem -M -t 4 ./Genome/*.fna Sample.R1.dedup.fastq.gz Sample.R2.dedup.fastq.gz > Sample.dedup.sam
-
This step will take many hours on a personal computer. The -t option sets the number of threads. If using Ubuntu, it may be helpful to launch the System Monitor software and select the Resources tab. This will graphically show the CPU usage and allow to monitor your computer to ensure it has not crashed.
Fig. 8.2 
Diagram of bioinformatics steps to recover candidate mutations from gamma irradiated bananas
-
-
12.
Sort the sam file using samtools. In the terminal window, enter the following command:
-
samtools sort -O sam -T sample.sort -o G2_aln.sam G2.dedup.sam.
-
Where G2 is replaced with the sample name. Note that the output file name should end in _aln.sam for the bin-by-sam tool to work.
-
-
13.
Convert SAM files to BAM format and index it for visual analysis in step 2 of Sect. 3.1.5. In the terminal window, enter the following command: samtools view -b G2_aln.sam > G2.bam
-
Where G2 is replaced with the sample name. When complete, enter the following command:
-
samtools index G2.bam. This will create an index file titled G2.bam.bai. Replace G2 with sample name.
-
-
14.
Repeat steps 11–13 with all samples.
-
15.
Install and run the bin-by-sam_2.0.py script. Download the Bin-by-Sam-tool into the sample processing folder (e.g. BananaGamma). Samples can either be compared to a wild-type reference, or to each other. Move the _aln.sam files created in step 12 to the Bin-by-Sam-tool folder. Open a terminal window, enter (cd) the directory and execute the following command: python bin-by-sam_2.0.py -o N3_100kbin.txt -s 100000 -b -p 3 -c G2_aln.sam. Where -o sets the output file, -s the bin size in base pairs (in the example data, a 100 kb bin size is used) -b inserts empty lines in the results table, -p sets the ploidy (3 for banana), and -c sets the cultivar control sample (sample G2 in the example data provided). When complete you should see the output file (in the example it is N3_100kbin.txt, for Novaria with 100 kb binning, change this name for different samples and binning), This folder can be opened with e.g. a spreadsheet to view and graph data (see Note 21 and Table 8.1).

3.1.5 Data Visualisation
-
1.
Graph the data. The sample/control columns of the bin-by-sam output can be plotted as an Overlay Plot using a standard spreadsheet software such as Microsoft Excel or LibreOffice Calc, or alternatives such as JMP or R. If using LibreOffice Calc (which comes preinstalled in Ubuntu), open the .txt file created in Sect. 3.1.4.15 step 15, select data from column G (the ratio of mutant to reference in the example) for one chromosome (chromosome 5 in the example data is labelled HE813979.1). Select Insert Chart from the drop-down menu. Select “Line Points Only” to produce a coverage graph (Fig. 8.3).
-
2.
View data with IGV (optional). This tool provides a graphical view of mapped reads and can be a useful visual check of your mapping data. IGV can be used as a web app, which is preferred if the analysis computer has less than 16 Gb RAM. The genome file (.fna) from Sect. 3.1.4 step 8 needs to be renamed and indexed for IGV. Copy the .fna file to a new folder and change the extension from .fna to .fa. Next, open a terminal window, enter (cd) to the new folder and index by typing the following command: samtools faidx genome.fa. Where genome is the name of your genome file. Open a web browser and go to https://igv.org/app/. In the Genome pull down menu, go to the bottom (you may need to expand your browser to full screen in Ubuntu) and select Local File. Select both the .fa file and also the .fa.fai file that was created with samtools. Next, select Tracks, Local File to upload your bam files. This produces a graphical view of mapped reads (Fig. 8.4).

Dosage plot analysis of chromosome 5 of mutant variety Novaria. Each dot represents a bin that is the mean coverage for 100 kb. Relative coverage values less than 3.0 indicate a putative deletion of one or more copies of a chromosome fragment while higher values (>3.0) indicate potential insertional events. The previously identified ~3.8 Mbp single copy deletion is underlined (Datta et al. 2018)

Graphical view of mapped reads of cv Grande Naine (G2) and mutant variety Novaria (N3) example data using IGV
3.1.6 Validation of Predicted Variants
-
1.
Select variants for validation. Review the bin-by-sam output table to select candidates and the graphed data as in Fig. 8.3.
-
2.
Select regions falling within and outside the candidate regions from step 1.
-
3.
Design primers for each of the regions, using e.g. Primer3, suitable for quantitative PCR (Rozen and Skaletsky 2000).
-
4.
Perform quantitative PCR as outlined in Datta et al. (2018) for regions within and outside the candidate CNV. Variations in relative amplification abundance should correspond to the observed copy number change (Fig. 8.5).

(a) Stably inherited large deletion of 3.8 Mbp identified via LC-WGS in a ‘Novaria’ mutant. One hundred and eighty-nine genes are affected in the validated region by losing one copy. (b) qPCR verification of identified mutation (CL control left border, CR control right border, CNV Region(s) showing deletion). (Figure modified from Datta et al. 2018)
4 Notes
-
1.
While costly, tungsten carbide beads are durable and can be washed and re-used many times, making the actual cost per extraction extremely low. An alternative is to use 3-mm steel ball-bearings. Ball-bearings can be purchased in bulk at a low-cost allowing for disposal of bearings after a single use.
-
2.
Ethidium bromide is mutagenic. Wear gloves, lab coat and goggles. Dispose of gloves in toxic trash when through. Avoid contaminating other lab items (equipment, phones, door handles, light switches) with ethidium bromide. Consult Material Safety Data Sheet (MSDS) for proper handling and disposal procedures. Alternative DNA stains, e.g. SYBR Safe DNA gel stain can be used instead of ethidium bromide but detection may vary with different dyes.
-
3.
Lambda DNA is a high molecular weight DNA. Dilute this lambda stock in 1x TE to about 10 different concentrations in the range between 2.5 ng/μl and 150 ng/μl to serve as DNA concentration standards. The choice of the optimal DNA concentration standards depends on the concentrations of the sample DNAs. Calculate dilutions based on the information printed on the stock tube of the lambda DNA (note that the concentration of lambda DNA may vary from batch to batch). Store stocks at 4 °C.
-
4.
Prepare gel loading dye containing 30% glycerol and color dye. Avoid loading dyes containing bromophenol blue or other dyes that migrate in a molecular weight range where you expect to observe DNA fragments. The presence of loading dyes can reduce the intensity of bands.
-
5.
A variety of options exist for creating libraries compatible with Illumina sequencing-by-synthesis equipment. The per-sample library cost does not vary much at the time of writing this protocol, but cost savings can be achieved if library preparation will be routine and at a high scale as some suppliers sell library components individually. If using TruSeq, the kit comes with paramagnetic beads and all other components except for the adapter sequences (this may vary depending on the type of kit purchased). It is also common for sequencing service providers to provide library preparation services. Outsourcing the library preparation may be wise if you are new to the methods as the higher cost per library from a service provider will be balanced by the cost of new equipment and the time needed to gain expertise in your own laboratory.
-
6.
All steps can be performed on a 64-bit computer with a minimum of 8Gb RAM. Mapping with BWA is computationally intense and can be very slow on computers with limited RAM. If necessary, mapping can be done on the cloud using a free option such as galaxy (https://usegalaxy.org/). If you are setting up a new computer for this work, we suggest a minimum of 16Gb RAM, 4 cores, and a free Linux operating system (for example, https://ubuntu.com/download/desktop). Guidelines for installing and running software in this protocol are written for Ubuntu 18.04 LTS.
-
7.
Scripts have been developed to automate fastq processing and alignment (see http://comailab.genomecenter.ucdavis.edu/index.php/Bwa-doall) that are suitable for the bin-by-sam tool. Various tools have been updated since Bwa-doall was released and we have found that the python scripts require editing to properly work on Linux Ubuntu 18.04 LTS at the time of writing. Because of this, we provide alternative command line tools to remove duplicate reads and map data. Bash scripts can be written in Linux to automate the analysis steps if processing many samples. The bin-by-sam software version is no longer available at the original link. This link contains the version used in this protocol: (https://u.pcloud.link/publink/show?code=kZLR1xXZWNCbL3m6HK7wRt50OvDfe8tGb9Mk).
-
8.
The amount of tissue needed depends on the yield of DNA from the extraction procedure used and the method of sequencing library preparation. It is advised to test tissue collection and DNA extraction procedures in advance to ensure that the necessary quality and quantity parameters are met. It is advised to label collected samples with the line number, treatment number and generation. This is important in order to track the inheritance of induced mutations in tissue culture should material harbor chimeric sectors in the generation tested.
-
9.
It is advised to store extracted DNA in a buffered solution (e.g. 10 mM Tris pH 8). Many DNA extraction kits come with an elution buffer containing Tris buffer. DNA can also be eluted/suspended in Tris-EDTA buffer as a buffer exchange is typically carried out in the first steps of library preparation.
-
10.
Genomic DNA should be free of RNA and not degraded. In addition to fluorometry, it is recommended that DNA is evaluated by electrophoresis.
-
11.
See Huynh et al. (2017) for a detailed description on the use of lambda DNA standards and image analysis for DNA quantification.
-
12.
Use the Qubit concentration data to choose a volume and concentration of genomic DNA to produce a band with a pixel density within the range of the DNA standards used.
-
13.
The DNA sample (genomic DNA band) should be completely out of the well and into the gel at least 2 cm. Do not run the gel too long as the genomic DNA band will become diffuse and hard to quantify. You may be able to accurately quantify degraded samples by altering electrophoresis conditions.
-
14.
The required size range of DNA fragments and concentration of fragmented input DNA will vary depending on the library preparation method, sequencing chemistry and read length used. The original method to identify gamma-induced indels in banana was optimized for 2×300PE (paired-end) Illumina MiSeq sequencing with library preparation using a low-input Illumina “nano” kit. These kits enabled a low input of 200 ng by utilizing post-ligation PCR amplification to produce sufficient library for sequencing using Illumina sequencing-by-synthesis. A variety of kits available from different companies are suitable for discovery of gamma induced indels using this protocol. The 2×300PE sequencing chemistry supports 550 bp fragments. Note that newer sequencing platforms (e.g. NextSeq and NovaSeq) can produce higher throughputs at lower cost using shorter read lengths. If outsourcing sequencing, the lowest cost option (in terms of raw Gb per dollar) will be suitable and fragment size and library preparation parameters can be adjusted for this. For the purposes of this protocol, parameters for low input libraries with 2×300PE sequencing are described. Different methods are available for DNA fragmentation. It is best to optimize fragment size utilizing a high sensitivity DNA system (e.g. Fragment Analyzer), however fragment sizes can be estimated using gel electrophoresis. Illumina 2×300PE sequencing-by-synthesis were used to prepare this protocol. However, higher throughput, shorter read sequencing provides a lower-cost alternative if sequencing is being outsourced.
-
15.
Sequencing libraries are normalized using molarities so that the number of DNA molecules, independent of size or weight, can be adjusted for the sequencing run.
-
16.
Libraries are typically pooled prior to sequencing because a sequencing run (e.g. flow cell lane) produces much more data (in base pairs) than is needed for a single sample. Pool samples at an equal concentration such that enough coverage is produced for each. For example, for a 660 Mb genome, if 10× coverage is desired, 6.6 Gb of sequence is needed per sample. If a sequencing run will produce 100 Gb of raw data, then 15 (100/6.6) samples can be pooled provided that quantification and pooling are accurate so that each of the 15 samples are equally represented in the sequencing reaction.
-
17.
If using an Illumina MiSeq, follow manufacturer’s protocols for on-board cluster generation and sequencing. If sequencing is being outsourced, the sequencing service will provide library QC, concentration determination, any necessary dilution, cluster generation and sequencing according to the equipment used.
-
18.
Sequence reads are provided as compressed fastq format files that contain both reads that passed quality filters and those that did not.
-
19.
Demultiplexing and trimming is often carried out by the sequencing provider. Check with your provider to determine what is included and how your data has been processed.
-
20.
Data analysis involves (1) processing the raw data (fastq files), (2) mapping the raw (fastq) sequencing data to a reference genome, (3) calculating the average read depth over a set interval, or bin and (4) displaying the read depth bins to identify regions that deviate from expected values. Different tools have been described for these steps (e.g. BWA, Bowtie) and algorithms are under constant improvement. Users are encouraged to evaluate these scripts and modify algorithms as desired.
-
21.
The output file contains one row per selected bin size with one column showing the number of reads in each bin and another column with the calculated dosage relative to the control sample. It is advised to try different bin sizes as results will vary depending on quality of sequence and depth of coverage. A bin size of 100 kb was previously used to detect gamma induced indels in banana. Ploidy (p) should be set to 3 if analysing triploid banana. When evaluating data of triploids, a relative coverage value less than 3 indicates a deletion of one or more copies of a chromosomal region while values greater than three indicate potential insertional events. Different thresholds can be applied to filter potential false positive signals. In previous work, a threshold of three consecutive bins showing the same trend (below or above 3) was applied. This filter can be applied to the table of data independent of the visualization methods. Such variants were experimentally validated.
References
Datta S, Jankowicz-Cieslak J, Nielen S, Ingelbrecht I, Till BJ (2018) Induction and recovery of copy number variation in banana through gamma irradiation and low coverage whole genome sequencing. Plant Biotechnol J 16:1644–1653
Henry IM, Zinkgraf MS, Groover AT, Comai L (2015) A system for dosage-based functional genomics in poplar. The Plant Cell 27(9):2370–2383. Online, tpc.15.00349
Huynh OA, Jankowicz-Cieslak J, Saraye B, Hofinger B, Till BJ (2017) Low-cost methods for DNA extraction and quantification. In: Jankowicz-Cieslak J, Tai T, Kumlehn J, Till B (eds) Biotechnologies for plant mutation breeding. Springer, Cham
Jankowicz-Cieslak J, Till BJ (2015) Forward and reverse genetics in crop breeding. In: Al-Khayri JM, Jain SM, Johnson DV (eds) Advances in plant breeding strategies: breeding, biotechnology and molecular tools. Springer, Cham, pp 215–240
Jankowicz-Cieslak J, Huynh OA, Brozynska M, Nakitandwe J, Till BJ (2012) Induction, rapid fixation and retention of mutations in vegetatively propagated banana. Plant Biotechnol J 10:1056–1066
Jankowicz-Cieslak J, Goessnitzer F, Datta S, Viljoen A, Ingelbrecht I, Till JB (2021) Induced mutations for generating bananas resistant to Fusarium wilt tropical race 4 – symposium proceedings. In: Sivasankar S, Noel Ellis TH, Jankuloski L, Ingelbrecht I (eds) Mutation breeding, genetic diversity and crop adaptation to climate change. CABI, Oxfordshire
Krasileva KV, Vasquez-Gross HA, Howell T, Bailey P, Paraiso F, Clissold L et al (2017) Uncovering hidden variation in polyploid wheat. Proc Natl Acad Sci 114:E913–E921
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Methods in molecular biology bioinformatics methods and protocols. Humana Press, Totowa, pp 365–386
Siegel JJ, Amon A (2012) New insights into the troubles of aneuploidy. Annu Rev Cell Dev Biol 28:189–214
Tan EH, Henry IM, Ravi M, Bradnam KR, Mandakova T, Marimuthu MP et al (2015) Catastrophic chromosomal restructuring during genome elimination in plants. eLife Sci 4:e06516
Tan E, Comai L, Henry I (2016) Chromosome dosage analysis in plants using whole genome sequencing. Bio-Protocol 6. https://doi.org/10.21769/BioProtoc.1854
Till BJ, Datta S, Jankowicz-Cieslak J (2018) TILLING: the next generation. Adv Biochem Eng Biotechnol 164:139–160
Acknowledgments
Authors wish to thank Dr. Bernhard Hofinger and Dr. Prateek Gupta for support in setting up the bioinformatic pipelines. Funding for this work was provided by the Food and Agriculture Organization of the United Nations and the International Atomic Energy Agency through their Joint FAO/IAEA Centre of Nuclear Techniques in Food and Agriculture. This work is part of IAEA Coordinated Research Project D22005.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
The opinions expressed in this chapter are those of the author(s) and do not necessarily reflect the views of the [NameOfOrganization], its Board of Directors, or the countries they represent
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 3.0 IGO license (http://creativecommons.org/licenses/by/3.0/igo/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the [NameOfOrganization], provide a link to the Creative Commons license and indicate if changes were made.
Any dispute related to the use of the works of the [NameOfOrganization] that cannot be settled amicably shall be submitted to arbitration pursuant to the UNCITRAL rules. The use of the [NameOfOrganization]'s name for any purpose other than for attribution, and the use of the [NameOfOrganization]'s logo, shall be subject to a separate written license agreement between the [NameOfOrganization] and the user and is not authorized as part of this CC-IGO license. Note that the link provided above includes additional terms and conditions of the license.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Jankowicz-Cieslak, J., Ingelbrecht, I.L., Till, B.J. (2022). Mutation Detection in Gamma-Irradiated Banana Using Low Coverage Copy Number Variation. In: Jankowicz-Cieslak, J., Ingelbrecht, I.L. (eds) Efficient Screening Techniques to Identify Mutants with TR4 Resistance in Banana. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-64915-2_8
Download citation
DOI: https://doi.org/10.1007/978-3-662-64915-2_8
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-64914-5
Online ISBN: 978-3-662-64915-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)