
Abstract
Mutagenesis of in vitro propagated bananas is an efficient method to introduce novel alleles and broaden genetic diversity. Mutations can be induced by treatment of plant cells with chemical mutagens or ionizing radiation. The FAO/IAEA Plant Breeding and Genetics Laboratory established efficient methods for mutation induction of in vitro shoot tips in banana using physical and chemical mutagens as well as methods for the efficient discovery of EMS-induced single nucleotide mutations in targeted genes or amplicons and identification of large genomic changes, including deletions and insertions. Mutagenesis of in vitro propagated tissues requires large populations serving as starting material, and a long process to dissolve genetic mosaics (chimeras) resulting from the mutagenesis of multicellular tissues. However, treating shoot apical meristems of tissue cultured bananas with a mutagen is a commonly used practice for banana mutation breeding programmes, and still the most effective. In our previous studies, we showed that chimeras, unique mutations accumulated in different cells of the plant propagule, could be rapidly removed via isolation of shoot apical meristems and subsequent longitudinal bisection. Further, induced mutations were maintained in mutant plants for several generations. We established such systems for inducing and maintaining both point mutations caused via EMS mutagenesis as well as insertions and deletions caused by gamma irradiation and describe hereafter methods for dose selection, gamma irradiation and chimera dissolution.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Bananas and plantains are among the most important staple food crops for people living in tropical and subtropical countries. They are herbaceous monocots belonging to the genus Musa; most are seedless, polyploid, sterile and clonally propagated. The majority of banana and plantains are consumed locally.
Vegetatively propagated crops (VPCs) such as banana pose unique problems compared to cereals because they have a reduced genetic diversity as they can’t be cross pollinated to enhance variation. Furthermore, because of its triploid parthenocarpic nature, bananas do not produce seeds and conventional breeding is thus a long process. Advances in biotechnology for crop improvement have had a great impact on vegetatively propagated crops (Gosal et al. 2010). Biotechnology based on tissue culture is complementary to conventional breeding technology.
One advantage of vegetatively propagated crops is that methods have been developed for rapid clonal propagation. Micropropagation is currently used in many countries for rapid propagation of disease-free planting material for distribution on a large scale. Such tissue culture techniques that ensure genetic stability (e.g. using shoot tip/nodal cultures for propagation) are particularly useful for in vitro mutation induction and maintenance of mutant plant populations. This technique is also of particular interest to breeders since the multiplication of the new lines for field trials and evaluation could be hastened, thereby shortening the time required for the release of new cultivars. In vitro techniques also offer possibilities to use induced mutation for further manipulation aimed at improvement. New genetic variation conferring a desirable trait can be fixed, and identical material rapidly deployed.
The structure of meristematic regions and the development of new meristems from differentiated tissue are particularly important when investigating radiation-induced mutation of vegetatively propagated crops (VPCs). In most cases, the new shoots originate from a single epidermal cell from a tissue, and this could directly lead to homohistant mutant plants whose genetics may be investigated further (Spencer-Lopes et al. 2018).
One of most critical prerequisites for successful mutation breeding is the determination of the optimal mutagen dose. The dose required for a particular experiment depends on the desired effects but may be restricted by undesirable effects of the mutagenic treatment, which could lead to lethality. There is a strong correlation between the genotype and the sensitivity of the plant material to the mutagenic treatments in plants (Jankowicz-Cieslak et al. 2012). The dose increase causes severe mutations, such as chromosomal aberrations, and can cause cell damage and subsequently death. While little is known for VPCs, the data from seed propagated plants suggests that fine-tuning of dose applied may be needed. Therefore, radiation sensitivity tests should be carried out to determine the mutagen dose that results in a 50 percent reduction in e.g., plant height or plant weight. In practice, a breeder applying irradiation treatment on vegetatively propagated crops may decide to settle for a growth reduction of 30–50 percent (GR30–50) for M1V1 plants or a survival rate of 40–60 percent depending on the sensitivity of the plant material. An equal number of control materials for the comparison should be planted at the same time. Measurement of growth reduction of in vitro treated material should be recorded 30 days after the treatments.
For VPCs, various methods have been developed which involve tissue isolation and dissection during post-mutagenesis aiming at reducing the genotypic complexity of the resulting plants.
The following procedures are routinely used in the Plant Breeding and Genetics Laboratory to micropropagate banana and plantain by shoot tip culture and perform mutagenesis (Fig. 1.1).

Mutation induction pipeline using banana in vitro shoot tip cultures. The first step of the process consists of establishment of banana cultures, either via sucker dissection from the field or alternatively obtaining accessions of interest from germplasm repository. Upon sufficient multiplication of banana shoot tips, the bulk mutagenesis can be performed or, if the knowledge on radio-sensitivities of this particular genotype doesn’t exist, a radiosensitivity test should be carried out. After mutagenic treatment of a bulk number of shoot tips, chimeras need to be dissolved and at the M1V3 stage plantlets can be rooted and hardened for field or screenhouse selection
2 Materials
2.1 In Vitro Culture Media
-
1.
Stock solutions prepared under sterile conditions and vacuum filtering after chemicals are completely dissolved in the given amount of distilled water.
(a) | Thiamine hydrochloride Stock: | 100 mg | in 100 ml dest. Water |
(b) | 6-benzylaminopurine (BAP): | 112.6 mg | in 0.5 l dest. Water |
(c) | L-Cysteine HCl: | 2 g | in 0.5 l dest. Water |
(d) | Indole-3-butyric acid (IBA): | 20.3 mg | in 100 ml dest. Water |
(e) | Isopentenyl adenine (2iP): | 20.32 mg | in 100 ml dest. Water |
-
2.
Initiation medium / Maintenance medium composition (1 l).
(a) | Thiamine hydrochloride | 1 ml |
(b) | 6-benzylaminopurine (BAP) | 20 ml |
(c) | L-Cysteine HCl | 10 ml |
(d) | Sucrose | 40 g |
-
3.
Rooting medium composition (1 l).
(a) | B5 Vitamin | 1 ml |
(b) | 2iP | 5 ml |
(c) | IBA | 0.1 ml |
(d) | L-Cysteine HCl | 1 ml |
(e) | Sucrose | 40 g |
-
4.
Sucrose (household grade).
-
5.
Murashige and Skoog basal salt with minimal organics (MS) (e.g. Sigma Cat Nr.: M-6899).
-
6.
Tissue culture grade water.
-
7.
Gelling agent (e.g. Gelrite).
-
8.
0.22 μm Cellulose Acetate (CA) sterilising, low binding filters.
-
9.
Vacuum pump.
-
10.
Sterile tubes (50 ml).
-
11.
Analytical balance.
-
12.
Weighing trays.
-
13.
Spatula.
-
14.
Magnetic stir bar.
-
15.
Hot plate.
-
16.
pH meter.
-
17.
NaOH.
-
18.
KOH.
-
19.
Medium dispenser.
-
20.
Culture tubes.
-
21.
Closures for culture tubes.
-
22.
Erlenmeyer flasks (250 ml).
-
23.
Aluminium foil.
-
24.
Autoclave.
-
25.
Cold room for media storage.
2.2 Gamma-Ray Mutagenesis
-
1.
Physical mutagenesis source.
-
2.
Tissue culture laboratory equipped with flow benches (fitted with gas).
-
3.
Sterile S-27 liquid culture medium.
-
4.
High quality, disease-free in vitro cuttings (e.g. 1000 propagules per treatment).
-
5.
Petri plates (94 mm and 145 mm diameter).
-
6.
Whatman filter papers for 90 mm Petri dishes.
-
7.
Sterile distilled water.
-
8.
Sterile forceps.
-
9.
Sterile scalpel.
-
10.
Parafilm.
-
11.
Orbital shaker.
-
12.
Growth rooms with light and temperature control (light regime 65 μmol/m2/s; e.g. Cool White fluorescent tubes, Philips TLP 36/86; temperature regime of 22° +/− 2 °C).
-
13.
Personal protective equipment (laboratory coat, shoe protection, nitril gloves).
2.3 Calculation of Lethality and Growth Reduction
-
1.
Tissue culture laboratory equipped with sterile flow benches (fitted with gas).
-
2.
Analytical balance.
-
3.
Sterile Petri plates for weighing in vitro material (94 mm).
-
4.
Sterile Whatman filter papers for 90 mm Petri dishes.
-
5.
Forceps.
-
6.
Sterile S-27 liquid culture medium.
-
7.
Standard spreadsheet software e.g. Microsoft Excel.
2.4 Chimera Dissolution
-
1.
Tissue culture laboratory equipped with flow benches (fitted with gas).
-
2.
Growth rooms with light and temperature control (light regime 65 μmol/m2/s; e.g. Cool White fluorescent tubes, Philips TLP 36/86; temperature regime of 22° +/− 1 °C).
-
3.
Sterile S-27 liquid culture medium.
-
4.
Sterile S-27 solid culture medium.
-
5.
Ethanol for surface and tools sterilisation.
-
6.
Forceps.
-
7.
Scalpels.
-
8.
Scalpel blades.
-
9.
Petri plates (94 mm and 145 mm diameter).
-
10.
Orbital shaker.
2.5 Hardening
-
1.
Facility with light, temperature and humidity control or plastic bags to cover pots.
-
2.
Peat.
-
3.
Sand.
-
4.
Soil mixer (for large amounts).
-
5.
Running tap water.
-
6.
5 l Container with room temperature tap water.
-
7.
Small trays or jiffy pots.
-
8.
Forceps.
-
9.
Plastic labels.
-
10.
Permanent pen or pencil.
3 Methods
3.1 Preparation of Liquid Maintenance Medium S-27
-
1.
Prepare stock solutions of thiamine (1 mg/ml), BAP (0.23 mg/ml) and L-Cysteine (4 g/l).
-
2.
Filter sterilise stock solutions.
-
3.
Dispense into 50 ml batches and freeze for future use. The working solution should be stored at 4 °C.
-
4.
For 1 litre of the liquid culture media, use the following: 4.4 g of Murashige and Skoog basal salt with minimal organics, 40 g sucrose, 10 ml L-Cysteine, 20 ml BAP and 1 ml of thiamine stock solutions. Use double distilled water.
-
5.
Place the media on the mixer and let it mix for 30 min.
-
6.
Calibrate the pH meter as per manufacturer instruction.
-
7.
While stirring, adjust medium to pH 5.8 using NaOH and HCl.
-
8.
Dispense 12 ml of the culture medium per Erlenmeyer flask.
-
9.
Close each Erlenmeyer flask tightly over the top with an aluminium foil.
-
10.
Autoclave for 20 min at 121 °C at 1 bar.
-
11.
Allow the medium to cool to room temperature.
-
12.
Store the medium in a cold room (see Note 1).
3.2 Preparation of Solid Culture Medium
-
1.
For 1 litre of solid medium cook 1.8 g of Gelrite and 40 g sucrose in 400 ml of tissue culture grade water.
-
2.
In a separate beaker containing 400 ml water mix:
-
(a)
For initiation/maintenance medium: 4.4 g of Murashige and Skoog basal salt with minimal organics, 10 ml L-Cysteine, 20 ml BAP and 1 ml of thiamine stock solutions.
-
(b)
For rooting medium: 4.4 g of Murashige and Skoog basal salt with minimal organics, 1 ml B5 Vitamin, 5 ml 2iP, 0.1 mL IBA and 1 ml of L-Cysteine HCl stock solution.
-
(a)
-
3.
Heat the mixture while stirring.
-
4.
When completely dissolved and hot enough add the Gelrite/sucrose mixture.
-
5.
Heat while stirring until the solution is homogenous and clear, add tissue culture grade water to 1 litre.
-
6.
While stirring adjust the pH to 5.8 using NaOH and HCl.
-
7.
Dispense 8 ml of the medium into culture tubes.
-
8.
Sterilise the medium for 20 min at 121 °C.
-
9.
Allow medium to cool to room temperature prior to use.
3.3 Initiation and Maintenance of Banana Cultures
-
1.
Select disease free vegetative buds or peepers or juvenile sword suckers (see Note 2).
-
2.
Cut the explants to ca. of 5 mm in size consisting of the meristematic dome with two to four leaf primordia.
-
3.
Wash explants under running tap water to remove dirt and keep in covered Petri dishes. From this point on all procedures need to be carried out in the sterile airflow cabinet.
-
4.
Rinse explants with 70% ethanol for surface sterilization.
-
5.
Transfer them in 40% commercial Chlorox with a drop of tween 20 (any available brand of bleach in a final concentration of 0.5–1% NaOCI can be used) for 20 min.
-
6.
Agitate explants several times while in Chlorox solution.
-
7.
Wash the explants with sterilized water three times.
-
8.
Remove two or three leave sheaths, not damaging the meristem, and transfer to a solid initiation medium.
-
9.
Cultures are kept at 28 °C for 16 h light/day (65 μmol/m2/s; Cool White fluorescent tubes, Philips TLP 36/86).
-
10.
Monitor cultures for contamination on a daily basis (see Note 3).
-
11.
Once plantlets are fully developed, increase the number of plantlets by micropropagation of shoot tip meristems.
-
12.
Isolate meristematic tips from each plantlet and propagate through longitudinal division into two propagules using a scalpel.
-
13.
Transfer isolated meristematic shoot tips into culture flasks containing fresh liquid media.
-
14.
Place the flasks on a horizontal gyratory shaker at 60 rpm and allow the explants to grow for approx. 30 days (light 65 μmol/m2/s; Cool White fluorescent tubes, Philips TLP 36/86 and the temperature of 26° +/− 1 °C).
-
15.
Repeat the process in order to increase the population size for mutagenesis purposes (see Note 4).
3.4 Establishment of the Radiosensitivity Curve and Calculation of Growth Reduction (GR)
3.4.1 Tissue Irradiation
-
1.
Autoclave all non-disposable materials (e.g. sieves, forceps).
-
2.
Prepare fresh liquid S-27 culture medium and autoclave.
-
3.
Prepare meristematic cuttings per each treatment chosen (see Note 5).
-
4.
Place cuttings into a Petri plate with a drop of water or on a moist filter paper (see Note 6).
-
5.
Calculate the exposure time (sec., min.) based on the dose rate of gamma cell irradiator that will be used (Gy/s or Gy/min) (see Note 7).
-
6.
Label Petri plates according to the dose required and genotype when handling more than one genotype. Include an untreated Petri plate as control.
-
7.
Wrap all Petri plates with Parafilm in order to avoid surface contamination.
-
8.
Transfer the material for irradiation to the physical mutagen source laboratory (see Note 8).
-
9.
Apply the required dose (Table 1.1) by placing Petri plates into irradiator chamber for the exposure time to produce the dosage (see Note 9).
-
10.
Irradiate each Petri plate with the chosen dose.
-
11.
Transfer irradiated samples back to the tissue culture laboratory.
-
12.
Surface sterilize each Petri plate with 70% ethanol before placing them in the laminar flow bench.
-
13.
Remove the Parafilm under sterile environment.
-
14.
Transfer the irradiated explants (5 explants per flask) to sterile, labelled conical flasks (Erlenmeyer flasks) containing 12 ml liquid S-27 medium (see Note 10).
-
15.
With the non-irradiated control sample, place the flasks on a horizontal gyratory shaker at 60 rpm (light 65 μmol/m2/s; Cool White fluorescent tubes, Philips TLP 36/86 and the temperature of 26° +/− 1 °C).
-
16.
If necessary, transfer cultures weekly into fresh liquid media to reduce possible accumulation of phenolic compounds due to the stress caused by mutagenesis (see Note 11).
-
17.
Thirty days after the treatment, take measurements of the chosen parameters (e.g. weight of the explants) and record survival rates of the mutagenized population (see Note 12).
3.4.2 Data Collection
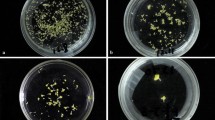
-
1.
After 30 days of incubation, assess the viability of plants by counting the surviving plantlets and measuring the fresh weight of each plant (see Note 13 and Fig. 1.2).
-
2.
Place a balance in the laminar bench and weigh each mutated plant separately in a sterile Petri plate.
-
3.
Record the data for each treatment and enter it into a spreadsheet (e.g. Microsoft Excel) (see Note 14).

Effect of irradiation on propagules weight of six different banana accessions expressed as percentage of non-irradiated (control) and percentage reduction in mean weight. Blue, orange, green and red lines indicate measurements taken with weekly intervals (1 week – blue, 2 weeks – orange, 3 weeks – green and 4 weeks – red post mutagenesis). A clear trend in response stabilisation takes place already after 2 (orange) to 3 (green) weeks
3.4.3 Data Analysis
-
1.
For every parameter measured, calculate the average per treatment for each replication.
-
2.
Repeat point 1 for control material.
-
3.
Express the reduction/increase of the radiated materials as a percentage of the non-irradiated control (see Note 15).
-
4.
Plot the calculated values as a dose-response curve using the dose as x-axis and the percentages on the y-axis. The control treatment is set as 100 percent (see Fig. 1.2).
-
5.
Repeat these steps with all the measured parameters (reduction in fresh weight percentage, plantlet height, root length, survival) (see Note 16).
-
6.
Compare the graphs from the different parameters (see Note 17).
-
7.
Estimate the mutagen dose required to obtain e.g. 50% of the control (see Fig. 1.2, red line).
-
8.
Identify the doses suitable for bulk mutagenesis of your material (see Note 18 and Table 1.2).
3.5 Bulk Mutagenesis by Physical Agents
-
1.
Autoclave all non-disposable materials (e.g. sieves, forceps).
-
2.
Prepare fresh liquid S-27 culture medium and autoclave.
-
3.
Prepare meristematic cuttings (e.g. 1000) per treatment chosen (see Note 20).
-
4.
Distribute cuttings evenly into Petri plates with a drop of water or on a moist filter paper.
-
5.
Calculate the exposure time (sec., min.) based on the dose rate of the gamma cell irradiator that will be used (Gy/s or Gy/min).
-
6.
Label Petri plates according to the dose required and genotype when handling more than one genotype. An untreated Petri plate (control) with the same number of propagules will be held in the same conditions as the treated ones.
-
7.
Transfer the Petri dishes to physical mutagen source laboratory.
-
8.
Apply the required dose by placing Petri plates into irradiator chamber for the exposure time to produce the dosage.
-
9.
Transfer irradiated samples to the tissue culture laboratory.
-
10.
Surface sterilize the Petri dish with 70% ethanol before removing the Parafilm.
-
11.
Transfer the irradiated explants along with a non-irradiated control into sterile labelled conical flasks (Erlenmeyer flasks) containing 12 ml liquid S-27 medium (5 explants per flask).
-
12.
Place flasks on a horizontal gyratory shaker at 60 rpm (light 65 μmol/m2/s; Cool White fluorescent tubes, Philips TLP 36/86 and the temperature of 26° +/− 1 °C).
-
13.
If necessary, transfer cultures weekly into fresh liquid media to reduce possible accumulation of phenolic compounds due to the stress caused by mutagenesis. Monitor cultures for the presence of any contamination.
3.6 Post-Mutagenesis Handling
3.6.1 Chimera Dissolution
-
1.
Grow explants in liquid culture media under a constant horizontal rotation at 60 rpm with a continuous light (65 μmol/m2/s; Cool White fluorescent tubes, Philips TLP 36/86) at 22° +/− 2 °C.
-
2.
Each shoot tip is the starting material for subsequent cultures and is referred to as a line as previously described (Jankowicz-Cieslak et al. 2012; Jankowicz-Cieslak and Till 2016) (see Note 21).
-
3.
In order to remove potential chimeric sectors, propagate shoot tip meristems through longitudinal division into two propagules using a scalpel (Fig. 1.3).
-
4.
Define each mutated meristem as a source of an individual line and assign to it a number or a barcode, depending on the system you have (see Note 22). This number corresponds to the mutant line that will be generated from this individual meristem.
-
5.
Isolate meristematic tips from each of mutated plants.
-
6.
Transfer every single isolated meristem into a separate culture tube or flask containing fresh liquid or solid media.
-
7.
Incubate cultures for 4–5 weeks.
-
8.
Repeat the process in order to make the population of M1V3 individuals (see Fig. 1.3 and see Note 23).
-
9.
At each meristematic division transfer the material into fresh culture media. Remember to follow the nomenclature assigned for each line.
-
10.
Transfer plants into the solid media for a long-term storage (culture media supplemented by 1.8% Gelrite) (see Note 24).
-
11.
Maintain cultures at 22° +/− 2° under stationary incubation and 12 h light cycle for the duration of the study.

Dissolution of chimeras in bananas. Plants after the mutagenic treatment are allowed to grow for 30 days in a liquid culture media at a constant rotary shaking (60 rpm). Each mutated plant is given a unique line number and assigned a population stage starting with M1V1. Chimeras may exist after 30 days recovery period if surviving meristematic cells harbour different mutations. In order to dissolve potential chimeras, meristems are isolated and divided into two parts through a longitudinal cut which results in most cases in generation of 2 daughter plants. These are allowed to recover and grow for another 30 days. The procedure is repeated 3 times. At the M1V3 stage, plants are being transferred into the solid culture media for long-term storage
3.6.2 Shoot Elongation and Rooting
-
1.
Transfer regenerants on to a rooting medium (containing half-strength MS, 1 mg/l thiamine, 5 μmol/l IBA, 20 g/l sucrose, and 7 g/l agar).
-
2.
Maintain cultures at 22° +/− 2 °C under stationary incubation and 12 h light cycle.
-
3.
After 3–5 weeks, transfer well-rooted plantlets to non-aseptic conditions or the greenhouse (see Note 25).
3.6.3 Hardening of In Vitro Mutant Population
-
1.
Use facility with light, temperature and humidity control or plastic bags to cover pots with sufficient water in the soil to maintain high ambient humidity.
-
2.
Prepare soil mixture of 50:50 (m/m) peat and sand by hand or soil mixture until both components are evenly mixed.
-
3.
Fill pots with soil before starting the procedure.
-
4.
Gently remove well rooted plantlets from culture vessels.
-
5.
Wash remaining medium off through running room-temperature tap water.
-
6.
Collect cleaned plantlets in a container with room-temperature water.
-
7.
Transplant plantlets in previously prepared pots.
-
8.
Water plantlets immediately after transfer to soil.
-
9.
Keep surface of plants moistened until transferred to humidity chamber.
-
10.
Decrease relative humidity weekly by 10% until 60% relative humidity in the chamber is reached (see Note 26).
-
11.
After 3–4 weeks move plants to the greenhouse.
-
12.
Allow plantlets to grow for about 4 weeks or until reaching sufficient size to be transferred to the greenhouse, nursery or field in preparation for screening and selection (see Note 27 and Chap. 7).
3.7 Phenotypic and Genotypic Selection
4 Notes
-
1.
It is recommended to prepare fresh media in the amount that is needed. If a cold room or refrigerator is available, media can be stored at 4–8 °C up to 6 weeks.
-
2.
The mutation breeding process usually starts with in vitro propagation of the plant material using shoot tips, corms, and also embryonic cells suspensions. Shoot tips are found to be the most commonly used hence, the protocol described is for mutation induction of in vitro shoot tips.
-
3.
In case of contamination, repeat the process of surface sterilisation.
-
4.
Excessive blackening in some genotypes, (e.g., AAB, BB) is normally overcome by rinsing the explants in an antioxidant solution made from 50 mg/l citric acid, 50 mg/l ascorbic acid and 5 ml of commercial Chlorox.
-
5.
Estimate how many explants can fit into a Petri plate. Usually, each treatment replicate should be placed in a separate Petri plate (e.g. 30 cuttings/plate), however if the number of explants is high, split them equally. When the applied dose for the genotype is unknown, a radiation test must be conducted. It is advisable that even if a dosage is known and was determined in a different laboratory, that a radiosensitivity test is performed as laboratory-to-laboratory differences can affect growth reduction measurements. To perform a radiosensitivity test on vegetative tissue, use 30 cuttings per dose with a wide range from 0 to 100 Gray (Gy). The range of 0, 10, 20, 30, 40, 50 and 60 Gy of gamma may be sufficient to establish the optimal dose due to the high moisture content of banana in vitro tissue in comparison to seeds. The same number of non-irradiated explants should be used as control.
-
6.
The amount of water has an important impact on the irradiation efficiency. Be consistent with the water used for irradiation by using a device to transfer it (e.g. a pasteur pipette or micro pipette). In the case of a long-distance transfer of samples for mutagenesis/radiosensitivity, consider placing explants on a solid media (e.g. Petri plates, magenta boxes).
-
7.
Exposure time is equal to the required dose divided by the dose rate of the day. The Gy is the unit used to quantify the absorbed dose of radiation (1 Gray = 1 J/kg).
-
8.
Physical mutagens comprise all nuclear radiations and sources of radioactivity including ultraviolet light (a non-ionising radiation), several types of ionizing radiations, namely X- and gamma-rays, alpha and beta particles, protons and neutrons. This protocol describes gamma ray induced mutagenesis. Gamma ray mutagenesis may be performed using different facilities, such as gamma cell irradiator, gamma phytotron, gamma house, or gamma field (Spencer-Lopes et al. 2018). Gamma cells are the most commonly used emitters for plant mutation induction. The isotopes Cobalt-60 (60Co) and Caesium-137 (137Cs) are the main sources of gamma rays. The International Basic Safety Standards published by the IAEA provides details for the safe handling of these sources (IAEA 2014).
-
9.
Radioactivity is mutagenic and carcinogenic. The safety precautions for exposing plant material to a gamma irradiation source must be strictly observed. Radioactive sources should therefore be operated only by trained and authorized personnel. Local regulations are usually explicit.
-
10.
The gamma irradiated samples are safe to handle.
-
11.
Blackening of liquid cultures can be the result of accumulation of phenolic compounds in the culture. Observe cultures for such media discoloration or for an eventual contamination. Remove contaminated flasks and decontaminate by autoclaving.
-
12.
The dose of a mutagen that achieves the optimum mutation frequency with the least possible unintended damage, is regarded as the optimal dose for induced mutagenesis. For physical mutagens, this is estimated by carrying out tests of radiosensitivity. These estimates of the percentage in plant growth reduction are good parameters for estimating the damage due to mutagenic treatment. By reading off the dose corresponding to 50% reduction, the so-called lethal dose 50 (LD50), is obtained. The LD50 is an appropriate dose for irradiation but, in practice, a range of doses around this value should be used. In case of VPCs and seed propagated plants, also a growth reduction (GR) is being taken into count. Additional tests can be carried out such as genome sequencing to identify the spectrum and density of induced mutations (see Chap. 8 and 9).
-
13.
Previous studies indicated that the radiosensitivity reading can be taken only 30 days post-irradiation. Results presented in Fig. 1.2 clearly demonstrate that for some genotypes the response stabilises already in the second week post-mutagenesis and for all in the third week.
-
14.
Collect data on the plantlet height, fresh weight, survival, root length, number of shoots. For optimal results all these measurements should be performed on the same day for all treatments and replications to reduce bias. In the case of counting survival rate, count the number of healthy plantlets.
-
15.
This calculation is made by dividing the weight of the mutated material (numerator) by the weight of the control material measured at the same time (denominator) multiplied by 100. For example, the weight of material treated is 1.46 g. The control material is 1.6 g. The percentage is then 1.46/1.60 x 100 = 91.25%.
-
16.
At low doses fresh weight of irradiated material may exceed control values.
-
17.
They may show different percent reductions at the same dose, but an overall trend can be derived by combining the data.
-
18.
Keep in mind when planning later field trials that emergence and survival obtained under in vitro testing conditions may differ considerably from that under field conditions due to environmental stresses.
-
19.
Jain (2010) published recommended doses for various banana cultivars: 10–20 Gy of gamma irradiation for diploid clones ‘Calcutta 4’ (AA genome) and ‘Tani’ (BB genome); 30–40 Gy of gamma irradiation for the triploids ‘Three Hand Planty’ (AAB genome), ‘Grande Naine’ (AAA genome), ‘Williams’ (AAA genome) and ‘Kamaramasenge’ (AAB genome); 40–50 Gy of gamma irradiation for the triploid ‘Cachaco’ (ABB genome) (Jhurree-Dussoruth et al. 2014; Jain 2010).
-
20.
Estimate the time needed to conduct the entire experiment with various tissue types. In case of time constrains and the high number of samples, split the plant material into batches for separate days, treating one tissue type per day. If this is being prepared a day before, place in vitro cuttings in a Petri plate with enough water. Induced mutations are random events, implying that even adherence to published irradiation conditions might not result in the same mutation events. A way of mitigating this uncertainty is to rely on statistical probability and to work with large population sizes. The optimal population size depends on the spectrum and density of induced mutations, and to a lesser extent on the application of the mutant population (forward or reverse genetics). A population size of 1000 M1V1 is easily accommodated in medium size laboratories and for polyploid bananas this may be considered suitable in reverse-genetics approaches to recover mutations in most target genes (Jankowicz-Cieslak et al. 2012). For forward-genetic screens a larger population size may be required because the chance of uncovering useful traits in polyploids in the M1 generation is reduced due to heterozygous state of induced mutation. A guide is to target the production of a forward-genetic population of a minimum of 5000–10,000 individuals.
-
21.
The first population after mutagenic treatment is referred to as M1V1 whereby V1 signifies the first vegetative generation after mutagenic treatment. Increasing numbers following V represent successive vegetative generations and increasing numbers after M indicate meiotic propagation. This allows tracking of generations in both facultative and obligate vegetatively propagated species.
-
22.
Ideally a line would represent a pedigree of clonally related material that begins with the first non-chimeric individual such that all subsequent material produced from this progenitor inherits the same mutations. Because the exact state where mutant in vitro plantlets are no longer chimeric is not easily determined, defining a line this way is not always possible. We therefore suggest defining a line simply as the pedigree of all material resulting from a single progenitor mutated plantlet. Materials in a line therefore may inherit the same mutations as observed in Jankowicz-Cieslak et al. (2012) but may also inherit different mutations if chimerism exists in meristematic cells at the time the progenitor is sub-cultured to produce daughter plantlets.
-
23.
We recommend three sub-culturing cycles to ensure the dissolution of chimeric materials. If too few cycles are performed, the resulting plants may still be chimeric and mutations may not be stably passed from one to another vegetative generation. It is recommended that preliminary evaluation starts at M1V3, since this generation should have solid mutants whose uniformity must be assessed. Non-uniform mutant clones must undergo a further round of propagation to reach uniformity. In this generation the evaluation of desired traits, such as TR4 screening could be undertaken or delayed until more advanced generations. From M1V4 to M1V9 uniform clones may be propagated and planted in experimental trials to test their performance for desired traits such as biotic/abiotic stress. As early as in M1V4 generation, replicated trials of selected mutants may be conducted using parental or local varieties as checks. The M1V5 and M1V6 generations can be used in multi-locational trials and tested for performance in a range of environments and agronomic traits. Final assessment can be made in M1V9 to M1V10 generations depending on plant species, the desired mutant clone or clones will be released as a new improved mutant variety (Spencer-Lopes et al. 2018).
-
24.
It is possible to slow down the growth of plants at a temperature of 15–20 ° C and a reduced day cycle of 8–12 h light. Test lower growing temperature and light conditions if they are suitable before applying them to a new variety.
-
25.
Shipment of plantlets: rooted plants in small, plastic culture tubes are very suitable for shipment. The medium containing gelling agent for single shoot development, is usually used for shipment.
-
26.
Banana requires specific growth conditions during germination and growth. For hardening use a day cycle of 12 h at 24 °C and avoid direct sunlight. High ambient humidity is very important for the initial period of cultivation. Maintain relative humidity at more than 90% during the first week of weaning and thereafter within the range of 50% (minimum daytime humidity) to 90% (maximum night-time humidity) at all times, to reduce the rate of leaf drying (Vuylsteke and Talengera 1998). If no humidity facility is available, cover pots with plastic bags. Keep plantlets covered for 2-4 weeks for acclimatization.
-
27.
Plantlets are kept in partial shade and can be acclimatized in the nursery for 2–3 months. During this period, off-types can be detected.
-
28.
Initial morphometric analysis and can be evaluation for new phenotypes can be completed in the greenhouse at the whole plant level. Figure 1.3 illustrates the strategy for in vitro mutagenesis (from mutagenized shoot meristems), handling of the mutated population and mutation recovery in a vegetatively propagated plant. The isolated putative mutants in the M1V3 generation can be evaluated for stability and multiplied to test their agronomic performance. Tissue may be collected from M1V3 individuals, DNA extracted and screened (genotypically) for any induced mutations. The inheritance of isolated mutations, both genetic and phenotypic is evaluated and confirmed in the M1V6 and subsequent generations.
References
Gosal SS, Wani SH, Kang MS (2010) Biotechnology and crop improvement. J Crop Improv 24:153–217. ISSN: 1542-7528 print/1542-7535 online. https://doi.org/10.1080/15427520903584555
IAEA (2014) Radiation protection and safety of radiation sources: international basic safety standards, IAEA Safety Standards Series No. GSR Part 3, 2014. IAEA, Vienna, Austria. ISBN 978–92–0–135310–8
Jain SM (2010) In vitro mutagenesis in banana (Musa spp.) improvement. Acta Hortic 879:605–614. https://doi.org/10.17660/ActaHortic.2010.879.67
Jankowicz-Cieslak J. and Till J.B., 2016. Chemical mutagenesis of seed and vegetatively propagated plants using EMS. Current protocols in plant biology 1(4):617–635 (Wiley). DOI: https://doi.org/10.1002/cppb.20040
Jankowicz-Cieslak J, Huynh OA, Brozynska M, Nakitandwe J, Till BJ (2012) Induction, rapid fixation and retention of mutations in vegetatively propagated banana. Plant Biotechnol J 10:1056–1066
Jhurree-Dussoruth B, Jankowicz-Cieslak J, Till BJ, Kallydin H, Burthia D (2014) Mutation induction and discovery in a Mauritian Dessert-type banana. Plant Gene Discovery & “Omics” Technologies, 16–19 February 2014, Vienna, Austria
Spencer-Lopes MM, Forster BP, Jankuloski L (eds) (2018) FAO/IAEA manual on mutation breeding – third edition. Food and Agriculture Organization of the United Nations, Rome, pp 251–266. ISBN 978-92-5-130526-3
Vuylsteke DR, Talengera D (1998) Post-flask management of micropropagated bananas and plantains. IITA, Ibadan, p 15
Acknowledgments
Authors wish to thank Mr. Danilo Moreno for assistance in performing radio-sensitivity experiments. Funding for this work was provided by the Food and Agriculture Organization of the United Nations and the International Atomic Energy Agency through their Joint FAO/IAEA Centre of Nuclear Techniques in Food and Agriculture. This work is part of IAEA Coordinated Research Project D22005.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
The opinions expressed in this chapter are those of the author(s) and do not necessarily reflect the views of the [NameOfOrganization], its Board of Directors, or the countries they represent
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 3.0 IGO license (http://creativecommons.org/licenses/by/3.0/igo/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the [NameOfOrganization], provide a link to the Creative Commons license and indicate if changes were made.
Any dispute related to the use of the works of the [NameOfOrganization] that cannot be settled amicably shall be submitted to arbitration pursuant to the UNCITRAL rules. The use of the [NameOfOrganization]'s name for any purpose other than for attribution, and the use of the [NameOfOrganization]'s logo, shall be subject to a separate written license agreement between the [NameOfOrganization] and the user and is not authorized as part of this CC-IGO license. Note that the link provided above includes additional terms and conditions of the license.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Jankowicz-Cieslak, J., Goessnitzer, F., Till, B.J., Ingelbrecht, I.L. (2022). Induced Mutagenesis and In Vitro Mutant Population Development in Musa spp.. In: Jankowicz-Cieslak, J., Ingelbrecht, I.L. (eds) Efficient Screening Techniques to Identify Mutants with TR4 Resistance in Banana. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-64915-2_1
Download citation
DOI: https://doi.org/10.1007/978-3-662-64915-2_1
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-64914-5
Online ISBN: 978-3-662-64915-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)