
Abstract
Ranaviruses (genus Ranavirus, family Iridoviridae) are large double-stranded DNA (dsDNA) viruses that infect economically and ecologically important cold-blooded vertebrates worldwide. Taxonomically, ranaviruses belong to a monophyletic group of viruses referred to as the Nucleocytoplasmic Large DNA Viruses (NCLDV). The NCLDV cluster contains viruses with the largest known viral genomes and infects a diverse array of eukaryotic hosts. The family Iridoviridae is currently divided into five genera: Iridovirus, Chloriridovirus, Megalocytivirus, Lymphocystivirus, and Ranavirus. Ranavirus taxonomy is based on restriction endonuclease fragment length polymorphism (RFLP) profiles of genomic DNA, virus protein profiles, DNA sequence analysis, and host specificity as well as whole genome dot plot analysis and phylogenetic analysis of individual and concatenated gene sequences. In this chapter, we discuss the current taxonomy of the genus Ranavirus as well as the phylogenetic relationship among currently identified ranaviruses. In addition, we discuss the future of ranavirus taxonomy in light of current phylogenetic analyses.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Nucleocytoplasmic Large DNA Viruses (NCLDV) are a monophyletic cluster of viruses that infect eukaryotes, ranging from single-celled organisms to humans, worldwide. The NCLDV group encompasses six virus families: Poxviridae, Asfarviridae, Iridoviridae, Ascoviridae, Mimiviridae, and Phycodnaviridae (Yutin and Koonin 2012; Yutin et al. 2009; Fig. 1). In addition, Marseillevirus isolates can be classified as members of the NCLDV, and there may be more viral isolates and families that will join the NCLDV cluster as our understanding of this important and complex group of dsDNA viruses expands. Recently, a proposal has been made to the International Committee on Taxonomy of Viruses (ICTV), the organization that oversees viral taxonomy, to classify NCLDV into a new order, designated Megavirales (Colson et al. 2012, 2013). Classification of NCLDV into a defined hierarchy will provide needed taxonomic structure for large dsDNA viruses. While this proposed taxonomic change will most likely be accepted in the near future, until then, our discussion will refer to this group as the NCLDV cluster of viruses.

Phylogenetic representation of the NCLDV group members. The graphical representation tree was developed from a phylogeny based on 263 amino acids from a conserved region of the DNA polymerase B gene originally published by Yutin et al. (2009). Tree is not to scale
Members within the NCLDV group have some of the largest known viral genomes. For example, members of the family Mimiviridae have genomes that are ~1.2 million base pairs (bp) in size and encode more than 1,000 viral genes (Raoult et al. 2004). They replicate within the cytoplasm of infected cells, although some members (e.g., family Iridoviridae) also include a nuclear stage during their replication cycle. As a result, NCLDV members encode many of the genes necessary for replication within the cytoplasm but still rely completely on the host translational machinery. Comparative analysis of NCLDV genomes reveals a core set of 50 viral genes that are conserved among the NCLDV (Yutin and Koonin 2012), supporting the hypothesis that this cluster of viruses originated from a common ancestor. Although the best-characterized family within the NCLDV is the Poxviridae, which includes a major human pathogen (smallpox virus), our understanding of the molecular biology, ecology, and infection dynamics of other families within the NCLDV, particularly members of the family Iridoviridae, has increased significantly in recent decades.
The family Iridoviridae is composed of five genera: the Iridovirus and Chloriridovirus genera whose members infect invertebrate hosts and the Megalocytivirus, Lymphocystivirus, and Ranavirus genera that infect cold-blooded vertebrates (Jancovich et al. 2012). Iridoviruses have linear dsDNA genomes that are circularly permutated and terminally redundant (Goorha and Murti 1982). Genome size is highly variable within the family and ranges from 140 to 303 kbp. However, because genomes are terminally redundant, unit-length genome sizes (i.e., the sum of the size of only the unique genes) are smaller and range from 105 to 212 kbp (Jancovich et al. 2012). Viruses within the family Iridoviridae share 26 core genes (Eaton et al. 2007). This cluster of core genes includes viral structural proteins as well as proteins involved in the regulation of gene expression, virus replication, and virulence (Jancovich et al. 2015; Grayfer et al. 2015). Sequence analysis of the 26 core genes has been used to generate high-resolution phylogenies (Fig. 2) for members of the family Iridoviridae as well as members of the genus Ranavirus (Jancovich et al. 2012).

Cladogram depicting the evolutionary relationships among the 11 fully sequenced ranaviruses, based on aligned deduced amino acid (AA) sequences of the concatenated 26 conserved iridovirus genes as defined by Eaton et al. (2007). The dataset contained 13,287 aligned AA positions. Maximum likelihood analysis was conducted in MEGA6 (Tamura et al. 2013). Numbers above each node represent the bootstrap values (1,000 replicates). See Table 1 for taxa abbreviations
2 Ranavirus Taxonomy
Members of the genus Ranavirus are a promiscuous group of viruses capable of infecting a wide variety of cold-blooded vertebrate hosts including fish, amphibians, and reptiles (Marschang 2011; Miller et al. 2011; Whittington et al. 2010). In addition, it has been hypothesized that ranaviruses have recently in their evolutionary history jumped from fish to amphibians and reptiles (Jancovich et al. 2010; Mavian et al. 2012a). This wide host range has been the focus of much ranavirus research, as investigators seek to understand how ranaviruses are able to infect such a wide variety of hosts (Brenes et al. 2014), when in evolutionary history did jumps from fish to other cold-blooded vertebrates occur (Chen et al. 2013; Jancovich et al. 2010; Mavian et al. 2012a), and what genetic elements contribute to ranavirus host range and pathogenesis (Jancovich et al. 2015).
There are currently six species recognized by the ICTV within the genus Ranavirus (Jancovich et al. 2012). These species include Frog virus 3 (FV3), the type species of the genus, and the best-characterized member of the family Iridoviridae; Ambystoma tigrinum virus (ATV); Bohle iridovirus (BIV); Epizootic hematopoietic necrosis virus (EHNV); European catfish virus (ECV); and Santee-Cooper ranavirus (SCRV; Jancovich et al. 2012). Moreover, there are other genetically distant ranaviruses that have not yet been recognized as species by the ICTV Iridoviridae Study Group. These include Singapore grouper iridovirus (SGIV; Song et al. 2004), grouper iridovirus (GIV), Rana esculenta virus (REV; Holopainen et al. 2009), common midwife toad virus (CMTV; Mavian et al. 2012a), Andrias davidianus ranavirus (ADRV; also known as Chinese giant salamander iridovirus; Chen et al. 2013), cod iridovirus (CoIV; Ariel et al. 2010), short-finned eel ranavirus (SERV; Holopainen et al. 2009), pike-perch iridovirus (PPIV; Holopainen et al. 2009), and Ranavirus maxima (Rmax; Ariel et al. 2010). Multiple criteria are used to delineate members within the genus Ranavirus including restriction endonuclease fragment length polymorphism (RFLP) profiles of genomic DNA, virus protein profiles, DNA sequence analysis, and host specificity (Jancovich et al. 2012). In addition to these criteria, dot plot analysis using complete genomic sequence information as well as phylogenetic analysis of individual and concatenated gene sequences have provided insight into the taxonomy of the ranaviruses (Eaton et al. 2007; Jancovich et al. 2010; Mavian et al. 2012a; Tan et al. 2004; Wang et al. 2014). Dot plot analyses offer a general overview of ranavirus genomic organization and a visual way to identify insertions, deletions, and inversions within viral genomes. Dot plot studies clearly indicate that although ranaviruses share the majority of their genes, gene order is not conserved and may serve as a way to distinguish evolutionarily related isolates or species. For example, gene order is conserved among FV3, tiger frog virus (TFV), and soft-shelled turtle virus, and distinct from that seen with ATV and EHNV (Jancovich et al. 2015).
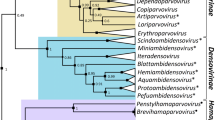
Phylogenetic analysis using the 26 core genes from completely sequenced ranaviruses has identified four distinct lineages (Fig. 2; Table 1): (1) the TFV/FV3/BIV-like viruses; (2) the CMTV/ADRV-like viruses; (3) the ATV/EHNV-like viruses; (4) the SGIV/GIV-like viruses. As suggested by analysis of the MCP, SCRV will likely constitute a fifth lineage (Fig. 3; Table 2). Furthermore, as additional ranavirus genomes are sequenced (e.g., especially those present within diverse fish species), it is likely that additional lineages will be added. Ranavirus lineages do not have a clearly defined host range. Lineages include those targeting only fish (e.g., the GIV-like and SCRV-like ranaviruses), only amphibians (e.g., CMTV/ADRV-like ranaviruses), both amphibians and fish (e.g., the ATV/EHNV-like viruses), and amphibians, fish, and reptiles (e.g., TFV/FV3/BIV-like viruses; Fig. 3). Therefore, phylogenetic analyses will enable investigators to identify and classify newly discovered ranaviruses.

Phylogram depicting the evolutionary relationships among 22 ranaviruses in the family Iridoviridae, based on the aligned full-length nucleotide (nt) sequences of the major capsid gene. The dataset contained 1,392 aligned nt positions. Maximum likelihood analysis was conducted in MEGA6 (Tamura et al. 2013). Numbers above each node represent the bootstrap values (1,000 replicates). See Tables 1 and 2 for taxa abbreviations. Branch lengths are based on the number of inferred substitutions, as indicated by the scale bar
Investigators categorize novel ranavirus isolates into viral lineages by sequencing one or more viral genes. For example, phylogenetic analysis and taxonomic classification of newly isolated ranaviruses have focused on a single, highly conserved gene (e.g., the MCP gene; Allender et al. 2013; Duffus and Andrews 2013; Geng et al. 2011; George et al. 2014; Kolby et al. 2014; Marsh et al. 2002; Waltzek et al. 2014), or on a concatenated set composed of multiple viral genes (Holopainen et al. 2009; Iwanowicz et al. 2013). While analysis of the MCP gene is convenient, the highly conserved nature of this protein may mask differences between virus isolates. Collectively, either approach provides a useful starting point to characterize and classify ranavirus isolates. However, having complete genomic sequence information available from a variety of ranavirus isolates will help in developing more rapid, sensitive, and universal approaches for the detection and classification of new ranaviruses. For example, identifying primers that flank hypervariable regions within the genome may allow viral isolates to be more readily distinguished.
There are currently 11 completely sequenced ranaviruses (Table 1). In addition, complete genomic sequence information is available for multiple strains of the same virus (e.g., FV3; Morrison et al. 2014) and closely related viruses (He et al. 2002; Huang et al. 2009; Lei et al. 2012). Comparative dot plot analysis of completely sequenced ranavirus genomes will be discussed in detail in another chapter of this book (Jancovich et al. 2015). That said, there are currently four unique genomic organizations identified among ranavirus genomes (Chen et al. 2013; Eaton et al. 2007; Jancovich et al. 2003, 2010; Mavian et al. 2012a, b; Song et al. 2004; Tan et al. 2004; Tsai et al. 2005). The SCRV group may represent a fifth type (J.K. Jancovich and T.B. Waltzek, unpublished data), and additional ranavirus genomic organizations may yet to be discovered. Interestingly, whole genome dot plot analyses show that ranaviruses with a similar genomic organization cluster together upon phylogenetic analysis using the 26 core genes. Therefore, there appears to be a direct correlation between ranavirus genomic organization and the 26 gene phylogenies.
It is unclear why there is such diversity in overall genomic architecture among ranaviruses. To that end, no other member of the NCLDV has such a diverse genomic organization. For example, all poxviruses possess genomes that display a conserved central core and variable, inverted terminal repeat regions. The core contains replicative genes common to all poxviruses, whereas the terminal repeat regions encode genes that influence host specificity and pathogenesis (Gubser et al. 2004; Upton et al. 2003). In contrast, although most genes are conserved, gene order differs among the four aforementioned ranavirus lineages. Perhaps the diverse genomic organization is a reflection of their inherently high recombination frequency (Chinchar and Granoff 1986) that leads to marked rearrangement of the viral genome. Therefore, if recombination of the viral genome increases over time, then ranaviruses showing greater sequence divergence may also show lower sequence collinearity. In view of this, future work should focus on understanding this genomic variability and diversity among ranaviruses and its relationship to viral ecology, host range, and pathogenesis.
3 The Future of Ranavirus Taxonomy: Where Do We Go from Here?
To envision the future of ranavirus taxonomy, one must first understand how the ICTV defines the different levels of virus taxonomy. The ICTV defines a species as “a monophyletic group of viruses whose properties can be distinguished from those of other species by multiple criteria” (Adams et al. 2013). The criteria for defining a viral species is determined by individual ICTV study groups and may include “natural and experimental host range, cell and tissue tropism, pathogenicity, vector specificity, antigenicity, and the degree of relatedness of their genomes or genes” (Adams et al. 2013). However, the critical component is that a viral species must be defined by “multiple” criteria, not a single distinguishing criterion. In addition, the ICTV recognizes a genus as “a group of species that share certain common criteria” (Adams et al. 2013).
As discussed above, ranavirus taxonomy has been based on RFLP profiles of genomic DNA, virus protein profiles, DNA sequence analysis, and host specificity (Jancovich et al. 2012). Unfortunately, these criteria do not allow us to quantify and differentiate between intraspecific and interspecific diversity in order to delineate one species from another. However, our understanding of ranavirus diversity has significantly increased in recent years through sequence analysis of individual ranavirus genes and sequencing of complete viral genomes. As a result, the Iridoviridae Study Group will need to reassess the criteria necessary to assign species and genera within the family. For example, the grouper iridoviruses, GIV and SGIV, appear to be the most distantly related viruses among the current isolates of the genus Ranavirus (Figs. 2 and 3). Whole genome dot plot analysis shows collinearity between the genomes of GIV and SGIV. However, grouper iridoviruses possess few regions of collinearity with other ranaviruses (Jancovich et al. 2015). In addition, GIV/SGIV lack the DNA methyltransferase gene seen among other ranaviruses, and as a result, do not have a methylated genome (Song et al. 2004; Tsai et al. 2005). Therefore, GIV/SGIV may need to be considered as a new genus, or at the very least, recognized as a distinct species in the genus Ranavirus.
Similarly, the SCRV-like ranaviruses, a group that includes doctor fish virus (DFV), largemouth bass virus (LMBV), and guppy virus 6 (GV6), are another collection of related ranaviruses that may also need to be considered as a new genus in the family (Fig. 3). Having the genomic sequence of LMBV would allow for a more complete comparison and may help delineate the taxonomic position of the SCRV group of ranaviruses. We are currently in the process of completing the genomic sequence of LMBV, DFV, and GV6. Once completed, we should be able to perform a more comprehensive analysis of this group of viruses and determine if they should be considered a unique genus in the family Iridoviridae.
Other partially characterized fish ranaviruses include the cod and turbot ranaviruses (Ariel et al. 2010), short-finned eel ranavirus (Holopainen et al. 2009) and pike-perch iridovirus (Tapiovaara et al. 1998). Although preliminary sequencing of the aforementioned viruses has been undertaken (Ariel et al. 2010; Holopainen et al. 2009), full genomic sequencing for these viruses will be needed to determine if they belong in the genus or require the formation of new genera. Therefore, the future of ranavirus taxonomy may reflect the need to “lump” currently recognized species (e.g., ATV/EHNV, TFV/FV3/BIV and CMTV/ADRV) into a single composite species while adding new species (e.g., SGIV/GIV and LMBV/DFV/GV6) or “split” species into distinct genera. To that end, the Iridoviridae Study Group will need to assess the consequences of these possible changes before taxonomic alterations can be finalized.
4 Final Thoughts
The taxonomy of ranaviruses is continually evolving, especially as new isolates are discovered worldwide. Taxonomic classification of newly discovered ranavirus isolates has been based on single and multiple viral genes as well as host, protein, serological, and morphological characteristics; however, single-gene taxonomic analysis is unlikely to be as robust as whole genome analysis or phylogenetic comparisons using the 26 core ranavirus genes. As more complete genomic sequences become available, our understanding of the diversity and complexity of ranavirus taxonomy will be delineated.
References
Adams MJ, Lefkowitz EJ, King AMQ, Carstens EB (2013) Recently agreed changes to the International Code of Virus Classification and Nomenclature. Arch Virol 158:2633–2639
Allender MC, Bunick D, Mitchell MA (2013) Development and validation of TaqMan quantitative PCR for detection of frog virus 3-like virus in eastern box turtles (Terrapene carolina carolina). J Virol Methods 188:121–125
Ariel E, Holopainen R, Olesen NJ, Tapiovaara H (2010) Comparative study of ranavirus isolates from cod (Gadus morhua) and turbot (Psetta maxima) with reference to other ranaviruses. Arch Virol 155:1261–1271
Brenes R, Gray MJ, Waltzek TB, Wilkes RP, Miller DL (2014) Transmission of ranavirus between ectothermic vertebrate hosts. PLoS One 9: e92476
Chen ZY, Gui JF, Gao XC, Pei C, Hong YJ, Zhang QY (2013) Genome architecture changes and major gene variations of Andrias davidianus ranavirus (ADRV). Vet Res 44:101–114
Chinchar VG, Granoff A (1986) Temperature-sensitive mutants of frog virus 3: biochemical and genetic characterization. J Virol 58:192–202
Colson P, de Lamballerie X, Fournous G, Raoult D (2012) Reclassification of giant viruses composing a fourth domain of life in the new order Megavirales. Intervirology 55:321–332
Colson P, De Lamballerie X, Yutin N, Asgari S, Bigot Y, Bideshi DK, Cheng XW, Federici BA, Van Etten JL, Koonin EV, La Scola B, Raoult D (2013) “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch Virol 158:2517–2521
Duffus AL, Andrews AM (2013) Phylogenetic analysis of a frog virus 3-like ranavirus found at a site with recurrent mortality and morbidity events in southeastern Ontario, Canada: partial major capsid protein sequence alone is not sufficient for fine-scale differentiation. J Wildl Dis 49:464–467
Eaton HE, Metcalf J, Penny E, Tcherepanov V, Upton C, Brunetti CR (2007) Comparative genomic analysis of the family Iridoviridae: re-annotating and defining the core set of iridovirus genes. Virol J 4:11–28
Geng Y, Wang KY, Zhou ZY, Li CW, Wang J, He M, Yin ZQ, Lai WM (2011) First report of a ranavirus associated with morbidity and mortality in farmed Chinese giant salamanders (Andrias davidianus). J Comp Pathol 145:95–102
George MR, John KR, Mansoor MM, Saravanakumar R, Sundar P, Pradeep V (2014) Isolation and characterization of a ranavirus from koi, Cyprinus carpio L., experiencing mass mortalities in India. J Fish Dis. doi:10.1111/jfd.12246
Goorha R, Murti KG (1982) The genome of frog virus-3, an animal DNA virus, is circularly permuted and terminally redundant. Proc Natl Acad Sci U S A 79:248–252
Grayfer L, Edholm E-S, De Jesús Andino F, Chinchar VG, Robert J (2015) Ranavirus host immunity and immune evasion. In: Gray MJ, Chinchar VG (eds) Ranaviruses: lethal pathogens of ectothermic vertebrates. Springer, New York
Gubser C, Hue S, Kellam P, Smith GL (2004) Poxvirus genomes: a phylogenetic analysis. J Gen Virol 85:105–117
He JG, Lu L, Deng M, He HH, Weng SP, Wang XH, Zhou SY, Long QX, Wang XZ, Chan SM (2002) Sequence analysis of the complete genome of an iridovirus isolated from the tiger frog. Virology 292:185–197
Holopainen R, Ohlemeyer S, Schutze H, Bergmann SM, Tapiovaara H (2009) Ranavirus phylogeny and differentiation based on major capsid protein, DNA polymerase and neurofilament triplet H1-like protein genes. Dis Aquat Organ 85:81–91
Huang YH, Huang XH, Liu H, Gong J, Ouyang ZL, Cui HC, Cao JH, Zhao YT, Wang XJ, Jiang YL, Qin QW (2009) Complete sequence determination of a novel reptile iridovirus isolated from soft-shelled turtle and evolutionary analysis of Iridoviridae. BMC Genomics 10:224–238
Iwanowicz L, Densmore C, Hahn C, McAllister P, Odenkirk J (2013) Identification of largemouth bass virus in the introduced Northern Snakehead inhabiting the Chesapeake Bay watershed. J Aquat Anim Health 25:191–196
Jancovich JK, Mao J, Chinchar VG, Wyatt C, Case ST, Kumar S, Valente G, Subramanian S, Davidson EW, Collins JP, Jacobs BL (2003) Genomic sequence of a ranavirus (family Iridoviridae) associated with salamander mortalities in North America. Virology 316:90–103
Jancovich JK, Bremont M, Touchman JW, Jacobs BL (2010) Evidence for multiple recent host species shifts among the ranaviruses (family Iridoviridae). J Virol 84:2636–2647
Jancovich JK, Chinchar VG, Hyatt A, Myazaki T, Williams T, Zhnag QY (2012) Family Iridoviridae. In: King AMQ (ed) Ninth report of the International Committee on Taxonomy of Viruses. Elsevier, San Diego
Jancovich JK, Qin Q, Zhang Q-Y, Chinchar VG (2015) Ranavirus replication: molecular, cellular, and immunological events. In: Gray MJ, Chinchar VG (eds) Ranaviruses: lethal pathogens of ectothermic vertebrates. Springer, New York
Kolby JE, Smith KM, Berger L, Karesh WB, Preston A, Pessier AP, Skerratt LF (2014) First evidence of amphibian chytrid fungus (Batrachochytrium dendrobatidis) and ranavirus in Hong Kong amphibian trade. PLoS One 9:e90750
Lei XY, Ou T, Zhu RL, Zhang QY (2012) Sequencing and analysis of the complete genome of Rana grylio virus (RGV). Arch Virol 157:1559–1564
Marschang RE (2011) Viruses infecting reptiles. Viruses 3:2087–2126
Marsh IB, Whittington RJ, O’Rourke B, Hyatt AD, Chisholm O (2002) Rapid differentiation of Australian, European and American ranaviruses based on variation in major capsid protein gene sequence. Mol Cell Probes 16:137–151
Mavian C, Lopez-Bueno A, Balseiro A, Casais R, Alcami A, Alejo A (2012a) The genome sequence of the emerging common midwife toad virus identifies an evolutionary intermediate within ranaviruses. J Virol 86:3617–3625
Mavian C, Lopez-Bueno A, Fernandez Somalo MP, Alcami A, Alejo A (2012b) Complete genome sequence of the European sheatfish virus. J Virol 86:6365–6366
Miller D, Gray M, Storfer A (2011) Ecopathology of ranaviruses infecting amphibians. Viruses 3:2351–2373
Morrison EA, Garner S, Echaubard P, Lesbarreres D, Kyle CJ, Brunetti CR (2014) Complete genome analysis of a frog virus 3 (FV3) isolate and sequence comparison with isolates of differing levels of virulence. Virol J 11:46–59
Raoult D, Audic S, Robert C, Abergel C, Renesto P, Ogata H, La Scola B, Suzan M, Claverie JM (2004) The 1.2-megabase genome sequence of Mimivirus. Science 306:1344–1350
Song WJ, Qin QW, Qiu J, Huang CH, Wang F, Hew CL (2004) Functional genomics analysis of Singapore grouper iridovirus: complete sequence determination and proteomic analysis. J Virol 78:12576–12590
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tan WGH, Barkman TJ, Chinchar VG, Essani K (2004) Comparative genomic analyses of frog virus 3, type species of the genus Ranavirus (family Iridoviridae). Virology 323:70–84
Tapiovaara H, Olesen NJ, Linden J, Rimaila-Parnanen E, von Bonsdorff CH (1998) Isolation of an iridovirus from pike-perch Stizostedion lucioperca. Dis Aquat Organ 32:185–193
Tsai CT, Ting JW, Wu MH, Wu MF, Guo IC, Chang CY (2005) Complete genome sequence of the grouper iridovirus and comparison of genomic organization with those of other iridoviruses. J Virol 79:2010–2023
Upton C, Slack S, Hunter AL, Ehlers A, Roper RL (2003) Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J Virol 77:7590–7600
Waltzek TB, Miller DL, Gray MJ, Drecktrah B, Briggler JT, MacConnell B, Hudson K, Hopper L, Friary J, Yun SC, Malm KV, Weber ES, Hedrick RP (2014) New disease records for hatchery-reared sturgeon. I. Expansion of the host range of frog virus 3 into hatchery-reared pallid sturgeon Scaphirhynchus albus. Dis Aquat Organ 111:219–227
Wang N, Zhang M, Zhang L, Jing H, Jiang Y, Wu S, Lin X (2014) Complete genome sequence of a ranavirus isolated from Chinese giant salamander (Andrias davidianus). Genome Announc 2:e01032–e01013
Whittington RJ, Becker JA, Dennis MM (2010) Iridovirus infections in finfish—critical review with emphasis on ranaviruses. J Fish Dis 33:95–122
Yutin N, Koonin EV (2012) Hidden evolutionary complexity of nucleo-cytoplasmic large DNA viruses of eukaryotes. Virol J 9:161–179
Yutin N, Wolf YI, Raoult D, Koonin EV (2009) Eukaryotic large nucleo-cytoplasmic DNA viruses: clusters of orthologous genes and reconstruction of viral genome evolution. Virol J 6:223–236
Acknowledgments
We would like to thank V. Gregory Chinchar and Trevor Williams for their critical review of this manuscript. This work was partially funded by the National Institutes of Health (Award No. 1R15AI101889-01) and California State University San Marcos (J.K.J.); and the University of Florida (N.S. and T.B.W.).
Open Access publication was made possible through grants provided by the University of Tennessee (Institute of Agriculture, Office of Research and Engagement, and Department of Forestry, Wildlife and Fisheries), Washington State University Libraries, Gordon State College (Office of Academic Affairs), the Association of Reptilian and Amphibian Veterinarians, and the Amphibian and Reptile Conservancy.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this chapter
Cite this chapter
Jancovich, J.K., Steckler, N.K., Waltzek, T.B. (2015). Ranavirus Taxonomy and Phylogeny. In: Gray, M., Chinchar, V. (eds) Ranaviruses. Springer, Cham. https://doi.org/10.1007/978-3-319-13755-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-13755-1_3
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-13754-4
Online ISBN: 978-3-319-13755-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)