
Abstract
Helium nanodroplets are peculiar systems, as condensed superfluid entities on the nanoscale, and as vessels for studies of molecules and molecular aggregates and their quantum properties at very low temperature. For both aspects, the dynamics upon the interaction with light is fundamental for understanding the properties of the systems. In this chapter we focus on time-resolved experiments in order to study ultrafast dynamics in neat as well as doped helium nanodroplets. Recent experimental approaches are reviewed, ranging from time-correlated photon detection to femtosecond pump-probe photoelectron and photoion spectroscopy, coherent multidimensional spectroscopy as well as applications of strong laser fields and novel, extreme ultraviolet light sources. The experiments examined in more detail investigate the dynamics of atomic and molecular dopants, including coherent wave packet dynamics and long-lived vibrational coherences of molecules attached to and immersed inside helium droplets. Furthermore, the dynamics of highly-excited helium droplets including interatomic Coulombic decay and nanoplasma states are discussed. Finally, an outlook concludes on the perspectives of time-resolved experiments with helium droplets, including recent options provided by new radiation sources of femto- or even attosecond laser pulses up to the soft X-ray range.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

10.1 Introduction
Modern time-resolved experimental techniques using ultrafast lasers offer a fascinating approach to unraveling the intriguing dynamics of helium nanodroplet systems. Although it is tempting to separate the dynamical properties from the static picture of a molecular system, such an effort in many respects is not viable at all, because statics and dynamics are directly linked from fundamental principles of interactions. Moreover, key static aspects like e.g. the structure of a complex, may even only be understood by probing its dynamics. For instance, characteristics of interaction potentials determining structural properties may not be accessible by spectroscopy but may be determined from relaxation schemes and the motion of vibrational wavepackets therein. Therefore, experimental methods employing short-pulse lasers play a key role in gaining insight both into the structure and dynamics on the atomic and molecular level.
A fundamental connection of measurements using high-resolution continuous-wave lasers in the frequency domain and measurements with pulsed lasers in the time domain can be understood from the line width of a transition which is associated to the lifetime by Fourier transformation. In this way, even quantitative aspects of decay mechanisms are already encoded in the homogeneous and inhomogeneous broadenings of measured spectra. The other way around, from time-domain “Fourier-transform” spectroscopy, detailed spectroscopic information in the frequency domain can be gained with very high resolution even when using spectrally very broad, femtosecond laser pulses. However, many important dynamical aspects can only be characterized when measuring in the time domain; in particular, if energy decay paths of a system can furcate into many degrees of freedom or corresponding states, or if a series of secondary processes can be triggered by the laser excitation processes. Typical examples include structural dynamics like e.g. desorption or fragmentation processes on the one hand, and, on the other hand, electron dynamics from non-adiabatic couplings or the interaction in a many-body system. Finally, by applying coherent multidimensional spectroscopy methods, simultaneous high-frequency and high-time resolution down to the Fourier limit is achieved. First recent results in this direction will be discussed at the end of the chapter.
With respect to helium droplets or helium in general, the superfluid properties are strongly related to dynamical processes. E.g., frictionless flow and vorticity, which are key peculiarities of superfluid behavior, are inherently interwoven with motion and dynamical aspects of the material. The key experiments probing bulk superfluid properties like the Andronikashvili experiment of rotating disks [1], or the observation of superfluid film flow, the fountain effect or heat transport (zero sound) [2], directly examine motion of the system or a motional behavior of a probe interacting with the superfluid.
When probing the dynamics on the nanoscale down to atomic/molecular dimensions, due to the shorter lengthscales on the one hand, and the lower involved masses on the other hand, the corresponding time scales shorten down to the picosecond (ps) or femtosecond (fs) time range. Electronic processes have typical time scales in the femtosecond range; vibrations and rotations of individual molecules extend their dynamics into the picosecond range; motions involving larger structures or/and weak interactions may reach nanosecond time duration. As a consequence, in order to access dynamics in nanodroplets, it is almost always inevitable to have femtosecond time resolution of the laser pulses triggering and probing the dynamics.
The combination of ultrafast methods with helium droplets is exceptionally interesting. On the one hand, the role of helium droplets as an ideal spectroscopic matrix allows for the study of nuclear motion without strong perturbation of a strongly-interacting environment. On the other hand, doped droplets serve as unique model system for the relaxation dynamics of heterogeneous nanosystems. In this direction, rare-gas clusters also gained much attention in combination with new radiation sources providing extreme high-field or/and short-wavelength laser pulses. Here, molecular and cluster beam samples in vacuum are required to keep the complexity of the condensed systems on a level that is still tractable by theoretical modelling. Finally, nanoscopic superfluidity still bears fundamental open questions, in particular, when it comes to the relevance to short-time dynamics.
A variety of time-resolved techniques have been developed over the years and applied to specific experiments involving helium nanodroplets. In overview articles, time-resolved experiments on pure and doped helium nanodroplets have already been in the focus of reviewed work [3,4,5], however, not including the recent prominent developments in ultrafast laser techniques.
Before discussing in detail specific topics on the dynamics in helium droplets, in the following chapter, time-resolved experimental techniques will be introduced and the applications to helium nanodroplets will be summarized.
10.2 Time-Resolved Techniques Applied to Helium Nanodroplets
10.2.1 Time-Resolved Photon Detection
In order to perform a time-resolved measurement, a start and stop event has to be registered with defined timing. Because of the statistical nature of spontaneous events, it is practical to start timing with a laser-induced preparation of the system employing an ultrashort pulse which provides accurate timing. A straight-forward approach is time-resolved detection of signals like e.g. the arrival of emitted photons by standard electronics. A prominent variant is the so-called time-correlated single photon counting (TCSPC), i.e. detecting single photons from fast multi-channel-plate (MCP)-amplified photon signals combined with fast digitizing of arrival times. In this approach, the timing resolution is typically limited to a few tens of picoseconds. In helium droplets experiments, TCSPC served as the initial approach to studying the dynamics of photo-induced processes of dopants.
In terms of systems that feature interesting dynamics, alkali atoms play a peculiar role because they do not submerge into helium nanodroplets but are located at the surface. The reason originates in the, compared to other atoms and molecules, large volume occupied by the valence electron, and the repulsive interaction of condensed helium to additional electrons, leading to bubble-like structures around alkali atoms. From simple arguments of surface tension and volume energy contributions, a binding motive at the surface without evolving a full bubble is energetically more stable when compared to the interior state. In other words, the alkali containing bubbles float, forming dimple-like textures on the surface of droplets. At the beginning of time-resolved studies, alkali-doped helium nanodroplets were in the focus because frequency-domain studies had manifested the surface location of dopants, desorption of dopants, the formation of exciplex molecules [6,7,8,9,10], fragmentation of dopant molecules, as well as spin flips [11, 12]. All of these aspects raised interesting questions concerning their dynamics.
The first TCSPC studies were performed on Na-doped helium nanodroplets [13] excited on the prominent \(3p \leftarrow 3s\) transition (D\(_1\) and D\(_2\) lines). Depending on the orientation of the excited p-orbital perpendicular or parallel to the surface of the helium droplet, strong repulsive forces and consequently desorption of the excited atom, or attractive forces leading to NaHe\(^*\) exciplex formation, respectively, set in upon laser interaction (Fig. 10.1). Exciplexes are complexes of a metal atom with one or a few He atoms which are stable only in an electronically excited state.

(a) Upon electronic excitation of an alkali-doped helium nanodroplet, desorption of the alkali atom (upper branch) or the formation of an exciplex molecule (lower branch) is induced, depending on the alignment of the p-orbital of the excited state (\(\Sigma \) or \(\Pi \) configuration, denoting the projection of the orbital angular momentum with respect to the internuclear axis). (b) Schematic potential diagram of diatomic states
The time-resolved fluorescence measured when exciting the repulsive \(\Sigma \)-configuration of the NaHe\(_N\) absorption band had an appearance time of 50–70 ps, significantly longer when compared to the instrument resolution of 20 ps. The latter was determined as the onset of fluorescence of gas-phase sodium atoms excited at the same transition. Shifting the laser in wavelength for the formation of NaHe exciplexes, and only collecting their red-shifted fluorescence, revealed a biexponential rise with 50–70 ps and 700 ps, which was assigned in comparison with theory to the two excited fine-structure states \(^2\Pi _{1/2}\) and \(^2\Pi _{1/3}\), respectively. These studies were extended both on the theory side, including the helium droplet interaction, and experimentally comparing different alkali metals (K, Rb) [14]. Interestingly, when going down the periodic table, the formation times of exciplexes along the \(J=1/2\) asymptote (\(n~^2P_{1/2} \leftarrow n~ ^2S_{1/2}\)) were measured to scale with the spin-orbit interaction strength, i.e. increasing into the nanosecond range. Opposed to that, upon excitation along the \(J=3/2\) path (\(n~^2P_{3/2} \leftarrow n~ ^2S_{1/2}\)), the formation times decreased, which was attributed to an enhanced long-range dispersion interaction for the heavier alkalis.
Experiments on Al atoms residing inside helium droplets provided first results on electronic relaxation dynamics [15]. A fast nonradiative quenching of the excited \(3 ^2D\) state into the \(4 ^2S\) state, releasing about 7000 cm\(^{-1}\) in energy, was measured to take place within 50 ps, which unfortunately matched the time-resolution in that measurement. Here, TCSPC was only able to provide an upper bound for the relaxation time.
Studies on photo-induced nonadiabatic dynamics of alkali trimers were also commenced with the technique of TCSPC. The peculiar properties of helium droplets to isolate van der Waals-bound high-spin quartet states [11, 16, 17] enabled to observe spin dynamics, i.e. forming covalently-bound alkali molecules upon intersystem crossing [18]. E.g., in the case of sodium, an intersystem-crossing time of 1.4 ns was determined, which significantly decreased for higher excitation energies approaching the access point to the doublet manifold. At the same time, vibronically-resolved data gave insight into the vibrational cooling of the trimers, which appeared to be on the same time scale as the spin-flip dynamics.
The influence of the helium droplet environment on spin-flip dynamics and predissociation was extended in detailed studies on the excitation of alkali dimers in triplet states (\(1~^3\Pi _g \leftarrow 1~^3\Sigma _u^+\)) [19]. The appearance of both molecular and atomic fragment emission was measured having a rise time \(<80\) ps, independent of the addressed vibrational excitation of the upper state. Predissociation and intersystem crossing appear to be in competition and on the same time scale. The intersystem crossing time in this case was deduced to be of the order of 10 ps which is surprisingly fast, considering the weak interaction to the helium surface which induces the process.
10.2.2 Pump-Probe Fluorescence Detection
To overcome the limitations of time resolution given by electronics, the femtosecond pump-probe technique was introduced many years ago in order to study details of molecular dynamics. For his pioneering work, Ahmed H. Zewail was awarded the Nobel Prize in 1999. In pump-probe studies, two or more ultrashort laser pulses are employed. In the simplest variant the process to be studied is triggered by an ultrashort laser pulse and the dynamics is probed by triggering a detection process, again with an ultrashort pulse delayed in time. The time resolution is only given by the properties of the laser pulses and the precision of setting a delay between the two pulses; for both, attosecond timing can be reached, covering the range needed for molecular processes and even electronic dynamics. An obvious extension of the TCSPC approach would be a pump-probe laser-induced fluorescence scheme. When using high-intensity femtosecond pulses, high photoionization rates can be achieved even in multi-photon processes. Alternatively to fluorescence detection, photoion or photoelectron detection can be advantageous because of the high detection efficiency of charged particles and mass and/or kinetic energy selectivity of detected particles can be obtained. For this reason, most of the results discussed in the following include ionization of the sample in the probe step.
10.2.3 Time-Resolved Spectroscopy by Photoion Detection
The first femtosecond pump-probe studies of doped He nanodroplets were carried out by the groups of Stienkemeier and Schulz at the Max-Born-Institut in Berlin in the late 1990s. Using the output of a mode-locked Ti:Sa laser in combination with mass-resolved ion detection using a quadrupole mass spectrometer, the yields of photoions where measured as a function of the delay between pairs of near-infrared (NIR) laser pulses. Owing to their large resonant absorption cross sections and extremely low ionization energies, alkali atoms, molecules, and clusters are well suited for this photoionization scheme and have been studied in detail in a series of such experiments [4, 10, 20,21,22,23,24,25,26]. Most importantly, due to their weak binding to the surface of He droplets, alkali atoms and molecules ionized by a resonant multiphoton process tend to detach from the droplets. This facilitates their detection as bare ions or as complexes with one or a few attached He atoms.
In photoionization experiments, the dynamics are determined by the interaction of both the neutral and ionic species with the He droplet which can qualitatively differ. Ionized dopants experience a much stronger attractive interaction towards the helium density than neutrals in their ground or excited states. This is in contrast to the above discussed fluorescence experiments, which solely focus on neutral species. For the case of an alkali-atom dopant, a schematic representation of the pump-probe photoionization scheme is shown in Fig. 10.2. Resonant excitation by the pump pulse induces the desorption of the atom from the droplet surface. Upon photoionization by the probe pulse, the dopant-droplet interaction suddenly changes from repulsive to attractive (Fig. 10.2b). Subsequently, the photoion either falls back into the droplet where it is solvated by forming a dense He shell around it, or it continues to move away from the droplet to be detected as a bare ion. In a series of femtosecond pump-probe experiments supported by TDDFT simulations, Mudrich and coworkers have obtained detailed insights into the competing dynamics between desorption and re-absorption of surface-attached alkali dopants [27,28,29,30] (Sect. 10.3.1). A similar behavior is also observed for other species immersed inside helium droplets by Koch and coworkers [31,32,33] (Sect. 10.3.2). From these experiments as well as theoretical predictions, it seems that the interplay between the de-solvation of excited neutrals and the solvation of the ionized species is a general trend in pump-probe photoionization experiments of alkalis and other dopants.

Femtosecond pump-probe photoionization scheme for the example of alkali-atom dopants. (a) Resonant dopant excitation by the pump pulse leads to a dopant-droplet repulsion, initiating the desorption of the dopant from the droplet surface. Photoionization of the dopant by the probe pulse causes the ion either to fall back into the droplet where it is solvated by forming high-density He solvation layers around it (upper branch), or to detach as a free ion (lower branch). (b) Generic pseudodiatomic alkali-He\(_N\) potential curves illustrating the energetics of the involved states
In addition, photoinduced processes on the intra and inter-dopant level overlay and interplay with the dopant-droplet interaction dynamics, to which pump-probe photoionization experiments provide access as well. Examples are the femtosecond and picosecond dynamics of exciplex formation for potassium (K) and rubidium (Rb) atoms attached to He droplets [10, 21]. Also, highly regular sub-fs oscillations were observed in the pump-probe traces due to electronic wavepacket interference. This phenomenon and a derived new spectroscopic scheme will be discussed in Sect. 10.6. Subsequent measurements on alkali dimers (Na\(_2\), K\(_2\), Rb\(_2\)) and trimers (K\(_3\), Rb\(_3\) and K-Rb heterotrimers) revealed essentially unperturbed vibrational wavepacket dynamics of the free molecules after their detachment from the droplets [22, 23, 25, 34], see Sect. 10.4.1. In a few instances, indications for the interaction of the He droplet with the vibrating molecules, causing dephasing and relaxation, were found and discussed in the framework of a quantum mechanical oscillator coupled to a superfluid bath [35, 36]. Most importantly, long-lasting vibrational coherences were measured, facilitated by the ultracold He droplet environment that prepares the molecules in their vibrational ground state prior to excitation. In this way, it was possible to measure highly resolved vibrational spectra of alkali dimers, trimers, and alkali-He exciplexes [22, 24, 25, 34, 37]. In these early experiments, a high laser pulse repetition rate (80 MHz) was used, which, in principle, introduces some ambiguity due to the possibility to excite and probe each droplet multiple times as it moves through the interaction region. As such, signals from species attached to the He droplet surface as well as from atoms and molecules already desorbed off the droplets into the gas phase may both have contributed in these experiments. Therefore, later experiments were carried out using amplified pulses at a repetition rate in the range 5-200 kHz where this ambiguity can be excluded. These experiments have mainly focused on the desorption dynamics of excited alkali atoms, molecules [38], and alkali-He exciplexes [27,28,29,30].
Another line of fs pump-probe experiments was pursued by the group of Tiggesbäumker and Meiwes-Broer in Rostock, based on their vast experience in strong-field ionization of metal clusters. When being exposed to intense NIR pulses, metal clusters (free or embedded in He nanodroplets) are multiply ionized and charge states of the exploding ions as high as \(Z=10\) for silver (Ag), \(Z=12\) for lead (Pb), and \(Z=13\) for cadmium (Cd) clusters were observed [39]. In addition to highly charged atomic and fragment clusters ions, mass spectra of strong-field ionized metal-doped He nanodroplets display regular progressions due to complexes of metal ions with attached He atoms [39,40,41]. For magnesium (Mg) ions, up to 150 attached He atoms were detected [40]. These are indicative for the formation of so-called ‘snowballs’, stable ion-He complexes first observed in bulk liquid He [42]. The term derives from the fact that for some species, the density of the local He shell around the cation adopts a regular structure and surpasses that of solid He. The pump-probe dynamics of snowball complexes of Ag were found to be opposite to that of the bare metal ions, indicating that He droplets can feature a cage effect causing the re-aggregation of fragments [40].
Initially, He nanodroplets were mostly regarded as an alternative method for forming metal clusters and the focus had been on the charging dynamics and Coulomb explosion of the dopant metal atoms, see Sect. 10.5.3 [43,44,45,46]. Later, the important role of the He shell in the ionization dynamics was recognized [40, 45, 47, 48] and the focus shifted more towards the nanoplasma dynamics of the He nanodroplets themselves [49,50,51,52]. In particular, resonant heating and charging of the nanoplasma manifests itself by enhanced yields of singly and even doubly charged He ions at a pump-probe delay around 0.5 ps, see Sect. 10.5.3 [51,52,53].
Particularly peculiar photoionization dynamics was observed by the Rostock group for Mg-doped He nanodroplets [54, 55]. Based on linear absorption spectra and on fs pump-probe resonant ionization traces of multiply doped He droplets, it was concluded that Mg atoms aggregate in He nanodroplets in an unusual way to form a foam-like structure where the metal atoms arrange themselves in a regular 10 Å-spaced network separated by He atoms. This structure collapses upon electronic excitation to form metallic clusters. Thus, the transient mass spectra reveal a sharp drop of the yield of Mg\(^+\) and small Mg\(_n^+\) cluster ions within 350 fs due to the decreased ionisation cross-section of Mg as the electronic properties evolve from the atomic to a bulk-like state. Subsequent slow recovery of the Mg ion signals within \(\approx 50\) ps was associated with the escape dynamics out of the He droplets. The formation of Mg foam in He droplets was essentially confirmed by theoretical model calculations [56].
The group of Gessner and Neumark in Berkeley performed seminal studies on the ultrafast dynamics of pure He nanodroplets using resonant excitation by XUV pulses. Using electron and ion imaging detection, intricate relaxation dynamics of highly excited He droplets were observed, including the emission of Rydberg atoms, small He clusters, and very low-energy electrons on various time scales. These studies have recently been reviewed [5] and will be discussed in more detail in Sect. 10.5.1.
More recently, the group of Koch in Graz succeeded in measuring excited-state dynamics of indium (In) atoms and dimers embedded inside He nanodroplets, see Sect. 10.3.2 [31,32,33]. Similarly to surface-bound alkali atoms, the delay-dependent ion yield revealed the ejection dynamics of the excited atom out of the He droplet. Indium dimers featured long-lasting vibrational coherences similarly to alkali dimers, despite their initial state of solvation inside the He droplets.
10.2.4 Time-Resolved Photoelectron Spectroscopy
Photoelectrons (PE) are an important observable for tracking ultrafast processes in He\(_N\) with time-resolved pump–probe photoionization. In contrast, photo-ions are in many situations hard to detect and/or add additional dynamics due to their strong attractive interaction with the droplet, as discussed in the previous section. Time-resolved photoelectron spectroscopy (TRPES) is a well established pump–probe photoionization technique for measuring the temporal evolution of the kinetic energy and the yield of the generated PEs [57,58,59]. The probe pulse couples (or projects) the excited state wavefunction onto the ionic state, a process that is governed by electronic selection rules and the Franck–Condon overlap of the involved vibrational states. The evolution of the excited state energy and its ionization probability provides insight into the dynamics of electrons and nuclei, which are often coupled non-adiabatically. The interpretation of transient signals therefore relies on quantum-chemical simulations. Photophysical and photochemical processes that can be observed include, among others, intra- and intermolecular electron and proton transfer, non-adiabatic energy relaxation, quantum beats and wave packets of electronic, vibrational and rotational degrees of freedom, or photodissociation and -association.
The applicability of TRPES for the observation of ultrafast molecular processes inside He\(_N\) stands or falls with the influence of the He environment on the PE observable. While this influence is moderate at picosecond timescales after photoexciation, in agreement with early frequency-domain PE studies [60,61,62], it can be significantly stronger within the first picosecond after photoexcitation, especially for atomic and small molecular chromophores. However, numerous femtosecond time-resolved experiments have shown that the coupled electronic and nuclear dynamics of chromophores in He\(_N\) can be observed with TRPES, as demonstrated for bare droplets [5, 63,64,65,66] (Sect. 10.5.1), as well as with surface-located [29, 30] (Sect. 10.3.1) and fully solvated dopants [31,32,33] (Sect. 10.3.2 & 10.4.2). Even if the photoexcitation process drives the chromophore–droplet system strongly out of equilibrium [31,32,33, 66], accurate insight can be obtained.

The He influence on photoelectron spectra in femtosecond pump—probe photoionization for different time delays [31]. The spectra are obtained with In atoms located inside He\(_N\) (pump pulse for In-He\(_N\): 376 nm, 3,30 eV; pump pulse for bare In: 410 nm, 3.02 eV; probe pulse: 405 nm, 3.06 eV; In ionization energy: 5.79 eV, pump–probe cross correlation time: 150 fs). (a) Evolution of a PE peak due to dynamics of the solvation shell. (b) Comparison of PE peaks for solvated atoms with equilibrated solvation shell (1000 fs pump-probe time delay, red trace) and bare atoms (red trace)
In the following we discuss the increased He influence on PE spectra within the first picosecond after photoexcitation for the In–He\(_N\) system [31], which is shown in Fig. 10.3. The corresponding potential energy curves can be seen in Fig. 10.8b. Within the first picosecond the PE peak energy decreases by 290 meV (from 610 meV to 320 meV) due to significant rearrangement of the He solvation shell around the In atom in response to photoexcitation. These dynamics are accompanied by the transfer of electronic energy of the dopant to kinetic energy of the surrounding He atoms, as discussed in Sect. 10.3.2. Additionally, larger linewidths and increased peak areas are observed for short delays (Fig. 10.3a). During the pump–probe cross correlation time (150 fs for this experiment) the simultaneous presence of pump and probe photons leads to saturation and peak distortion. Afterwards, up to \(\approx 500\) fs, the line width is increased because of two reasons: (i) the combination of transient peak shift (\(\approx 1\) meV/fs) and 150 fs cross correlation and, (ii) the Franck–Condon overlap of the excited and ionic states, which are distorted inside the He\(_N\) (see Fig. 10.8).
The He-related influence on the PE spectrum within the first picosecond will be superimposed on the TRPES signal of intrinsic nuclear and electronic dynamics of embedded molecules. The magnitude of this influence corresponds to the distortion of the excitation band, as observed by frequency domain spectroscopy (see also Sect.10.3.2). Accordingly, sharp electronic transitions (zero-phonon lines) frequently observed for larger molecules [67, 68] indicate that this initial influence might be less severe for such systems.
After \(\approx 1\) ps, when the He solvation shell has equilibrated, the PE signal (Fig. 10.3b, red curve) peaks at slightly higher energies (30 meV increase) compared to the bare atom peak (red), representing the reduced ionization potential inside the droplet [61]. The increased width of the in-droplet peak compared to the bare-atom peak (62 meV versus 35 meV) is again related to the Franck–Condon overlap of the distorted excited and ionic states inside the droplet (see Fig. 10.8). The PE peak also exhibits a wing extending to lower energies, even below the bare-atom band, which is a signature of energy relaxation of the photoelectrons due to binary collisions with individual He atoms on the way out of the droplet, as previously observed in PE spectroscopy experiments with nanosecond laser pulses [61].
On the droplet surface, the PE peak-shift is slower by a factor of 2–3 [29, 30] due to the lower He density (Sect. 10.3.1). In pump-probe photoionization of nanoplasmas inside He\(_N\), the photoelectron kinetic energy was recently used as observable for the temporal evolution of the plasma (Sect. 10.5.3). After plasma ignition with a strong-field pulse, the kinetic energy of electrons released by the probe pulse corresponds to the electron temperature in the plasma [52]. Strong-field probing revealed the appearance of photoelectron spectra characteristic for above-threshold ionization, indicating electron recombination into high-lying Rydberg states [53].
10.2.5 Time-Resolved Correlation Spectroscopy
The detection of photoelectrons or -ions can be extended by establishing correlation between the ionization products, such as ion–electron or ion–ion correlations. The assignment of electron spectra to ion fragments, for example, allows for the identification of different pathways in strong-field ionization of molecules [69]. In femtosecond pump-probe photofragmentation experiments bond breaking may occur in the electronically excited state (after pump pulse excitation) or in the ionic state (after probe pulse ionization); two ionization channels that can be disentangled through electron–ion correlation but remain indistinguishable by sole detection of electrons or ions [70,71,72]. Correlation can be established either by coincidence detection [73, 74], where pairs of charged particles are detected for single ionization events, or by covariance-mapping, where correlations are revealed based on statistical fluctuations [75]. While coincidence detection requires disadvantageously low count rates (typ. \(<0.3\) ionization events per laser shot) in order to avoid so-called false coincidences [76], covariance-mapping allows for much larger signal rates. Recently, the analysis of photoelectron-photoion coincidence measurements with Bayesian probability theory was demonstrated to enable high count rates and provides additional advantages, such as an increased signal-to-noise ratio, exclusion of false coincidences, proper pump-only signal subtraction, and confidence intervals of the spectrum [77, 78].
The hurdle for correlated detection in combination with He\(_N\) is the trapping mechanism of ions inside the droplets due to strong attractive forces (Sect.10.2.3). Ion trapping can be overcome if the ions are provided with sufficient kinetic energy to escape the droplet, as it is the case in Coulomb explosion after double ionization [79, 80], or for vibrational IR excitation of molecular ions [81]. In addition, dopant ions are ejected from the droplets to some extent when being indirectly created through Penning or charge-transfer ionization via excited or ionized He, respectively [82,83,84]. Ion–ion and ion–electron coincidence detection enabled the identification of interatomic autoionization processes inside He\(_N\) such as interatomic Coulombic decay (ICD) in pure He\(_N\) [79] and double ionization of alkali dimers through ICD or through electron-transfer mediated decay (ETMD) [80, 85, 86] (see Sect. 10.5.2)
A very recent example of time-resolved electron–ion covariance spectroscopy of the In\(_2\)–He\(_N\) system is shown in Fig. 10.4. Pump–probe photoionization of In\(_2\) inside He\(_N\) leads to an unexpected strong In–He\(_n^+\) (\(n=0,...,\approx 30\)) ion signal within the first tens of picoseconds [33], whereas ion yields are typically zero within \(\approx 50\) ps after pump excitation due to trapping (see Fig. 10.13). Comparison of electron–ion covariance measurements at short (0.8 ps) and long (200 ps) time delays sheds light on the process (Fig. 10.4): At long delays (red trace) the prominent PE peak at \(\approx 0.65\) eV is correlated to In\(_2^+\), identifying photoionization of excited In\(_2^*\) after ejection from the droplet. The In–He\(_n^+\) PE peak in question at short delays (blue trace) appears at slightly higher energies (\(\approx 0.80\) eV), whereas the PE peak of In* would be expected at lower energies (\(\approx 0.32\) eV, see Fig. 10.3), and should also be sharper. This indicates that the In–He\(_n^+\) signal originates actually from unfragmented In\(_2^*\) molecules, and that fragmentation occurs after ionization in the In\(_2^+\) ionic state. A strong increase of the In–He\(_n^+\) yield with probe power supports this assumption and suggests that ion ejection is caused by probe-pulse induced In\(_2^+\) excitation to a repulsive In\(_2^{+*}\) state. Note that a sole TRPES measurement would not be able to screen out the photoelectrons associated to In–He\(_n^+\) since they have the same energy as those leading to In\(_2^+\).

Covariance detection of electrons and ions after pump-probe photoionization of In\(_2\)–He\(_N\). The PE spectrum at 0.8 ps (blue) is correlated to the detection of InHe\(_n^+\) (\(n=0,...,\approx 30\)), while the 200 ps PE spectrum (red) is correlated to In\(_2^+\). Both spectra are normalized. The signal peaking at 1.5 eV in both spectra is likely due to a erroneous correlation of electrons from pump-only ionization and the respective ions
Having discussed the major experimental tools suitable for time-resolved spectroscopy of pure and doped He droplets, we will highlight some recent application examples in the next section. Before we come to that, we conclude this section by giving an overview of the theoretical work on the dynamics of pure and doped He nanodroplets, as many of the time-resolved experiments have greatly benefited from model calculations.
10.2.6 Time-Dependent Density-Functional Theory Simulations
The structure of pure and doped He nanodroplets has been studied theoretically by various methods, the most accurate being Quantum Monte Carlo (QMC) [87]. However, the computational cost quickly exceeds currently available computer resources when it comes to simulating experimentally relevant nanodroplet systems. Furthermore, QMC cannot describe the dynamic evolution of superfluid He in real time. These limitations can be overcome by time-dependent density functional theory (TDDFT) which can be applied to much larger systems than QMC and allows for a time-dependent formulation. A promising recent development is a hybrid path-integral molecular dynamics/bosonic path-integral Monte Carlo method [88]. This method provides the theoretical foundation of simulating fluxional molecules and reactive complexes in He environments seamlessly from one He atom up to bulk He at the accuracy level of coupled cluster electronic structure calculations.
TDDFT is the only method to date that can successfully reproduce results from a wide range of time-resolved experiments in superfluid He on the atomic scale. The great benefit of these simulations is that detailed insights into the structural dynamics of the entire system is obtained, including density modulations of the He such as surface or bulk density waves and solvation shells, which are not directly accessible experimentally. Likewise, the He dynamics induced by ions such as the formation of snowballs and the nucleation of vortices, which have so far eluded experimental detection, are still amenable to TDDFT simulations. Thus, during the last decade, TDDFT has emerged as a powerful tool to describe the structure and dynamics of doped He droplets, thereby valuably complementing the experimental advances. This work has been summarized in two review articles [7, 89]. The method has mostly been developed and promoted by M. Barranco and his group in Barcelona, and the code is freely available [90].
Inspired by experiments, a variety of metal atoms and ions have been studied in view of the structure and dynamics of their complexes with He\(_N\) [7, 89]. Upon electronic excitation of either surface-bound alkali atoms [91,92,93] or initially submerged atoms (silver, Ag) [94], in most cases the excited atoms were ejected from the droplets within a few ps or a few tens of ps, respectively. Depending on the excited state, either bare atoms were ejected or exciplexes were formed, which in turn either desorbed from the droplets or remained attached to them [29, 94]. Ag atoms in the lowest excited state were ejected from the droplet with a speed consistent with the Landau velocity \(v_\mathrm {L}=58\) m/s, which was measured experimentally for excited Ag and other molecular dopants [95]. It was concluded that excited dopants interacting repulsively with the He droplets are expelled towards the droplet surface while repeatedly exciting pairs of rotons such that their speed stays below \(v_\mathrm {L}\).
The microscopic dynamics of metal ions located near the He droplet surface have so far only become accessible through simulations as ions tend to remain tightly bound in the He droplet interior and therefore elude detection. For the Ba\(^+\)He\(_N\) system, it was found that due to the relatively strong attractive ion-He interaction, the velocity of the Ba\(^+\) cation during the solvation process temporarily exceeds \(v_\mathrm {L}\), leading to the nucleation of a quantized ring vortex [96]. When formed at the He droplet surface, the Ba\(^+\) ion moves towards the center of the droplet. After about 8 ps, the Ba\(^+\) is fully surrounded by He and a few ps later a dense solvation layer of He forms with transiently appearing spots where He localizes; but eventually the He shell remains smooth. Thereafter, the solvated ion keeps oscillating inside the droplet without friction at a velocity \(<v_\mathrm {L}\). Due to their large masses and stronger attractive interactions with the He, Rb\(^+\) and Cs\(^+\) ions initially located at the droplet surface form snowballs at the droplet surface within 10 ps [97]. At longer times the snowballs become solvated by the He droplet which rearranges itself around the stationary ion. Large density fluctuations induced by the cation solvation process lead to the nucleation of quantized vortices. In the case of Cs\(^+\), the initial phase of snowball formation prevents the ion from penetrating into the He droplet. The snowball therefore forms at the surface of the droplet in \(\approx 30\) ps. Due to the effective shielding of the Cs\(^+\) charge by the surrounding He atoms, it is only weakly bound to the droplet. For relatively small He droplets consisting of 1000 atoms, the Cs\(^+\) snowball even detached after 90 ps from the droplet due to He density fluctuations.

TDDFT simulations of the dynamics of a Rb atom on a He nanodroplet consisting of 1000 He atoms. At \(t=0\), the Rb atom is excited into the lowest excited state (5\(p\) \(\Pi _{1/2}\)) and at \(t=20\) ps it is ionized. Based on results reported in [28]
To compare with recent fs pump-probe experiments, TDDFT calculations were performed that simulated the sequence of pump-probe excitation and ionization at a variable delay. In this way it was possible to reproduce the combined process of desorption of the excited atom and the subsequent fallback and solvation of the ion [28, 98]. Figure 10.5 shows snapshots of the simulated evolution of a Rb atom excited into its lowest excited state 5\(p\) \(\Pi _{1/2}\) at \(t=0\) (green dot turning blue). At \(t=20\) ps it is ionized (red dot). At \(t=2\) ps, the excited Rb atom departs from the droplet leaving behind He density waves traveling through the droplet. At \(t=45\) ps, the Rb\(^+\) is at its largest elongation away from the droplet, before falling back into the droplet to form a snowball (\(t=130\) ps). The time constants obtained from the simulation are in good agreement with the experimental results [28]. Similar simulations were performed to complement recent XUV pump-probe studies of the photodynamics of pure He nanodroplets [66, 99]. Here, one or two excited He atoms (He\(^*\)) in a He droplet take the role of the dopants. Surprisingly, the response of the He droplet strongly resembles that of excited metal atoms in the sense that a bubble forms around the He\(^*\) within \(\lesssim 0.5\) ps, followed by the ejection of He\(^*\) from the droplet. In the case two He\(^*\) are located near each other, the two bubbles merge, which causes the He\(^*\) to decay by ICD, see Sect. 10.5.2.
More recently, the structure and dynamics of rotating He nanodroplets has been a focus of TDDFT simulations [100,101,102]. In particular, the formation of quantized vortices in \(^4\)He nanodroplets and their ability to capture dopant atoms was investigated in detail [103, 104]. Furthermore, collisions of atoms with He droplets as they occur in the experiments during the pick-up process where addressed [105]. Even the merging of two He nanodroplets was simulated, with particular focus on vorticity and quantum turbulence [106].
The Gonzalez group recently developed a hybrid method using DFT to describe the He droplet and a quantum wave packet treatment of the dopant [107] that allows to investigate the He influence on intramolecular processes. With this approach, they obtained predictions of the femtosecond time-resolved dynamics of dimer molecules inside He droplets, including photodissociation of Cl\(_2\) [107,108,109,110], molecule formation of Ne\(_2\) [111, 112], vibrational energy relaxation of I\(_2\) [113], and rotational energy relaxation of H\(_2\) [114].
10.3 Dynamics of Atomic Dopants
The weak influence of the He environment on dopants often results in a negligible perturbation of the ground-state structure of dopants [115], as well as in minor influence on their vibrational and rotational degrees of freedom [67]. Photoexcitation of electronic transitions, in contrast, can lead to a considerable rearrangement of the He solvation shell triggered by a change of the repulsive interaction between the chromophore dopant and the He atoms. The solvent-related response to photoexcitation can best be investigated with atomic dopants in order to avoid complications related to internal degrees of freedom. Since fully solvated dopants experience a stronger but symmetric He interaction, compared to the weaker and asymmetric interaction of surface-located dopants, these situations will be discussed separately.
10.3.1 Surface-Located Atoms
While most atoms and molecules are submerged in the interior of He nanodroplets, alkali atoms and small alkali clusters reside in weakly bound dimple states at the droplet surface [6, 7]. Upon electronic excitation, all alkali atoms promptly detach from the He droplet surface due to enhanced Pauli repulsion acting between the diffuse excited valence electron and the He. The only exceptions are Rb and Cs in their lowest excited states where small photon excess energies are insufficient to induce direct desorption [116] and indirect desorption through M\(^*\)-He exciplex formation is prevented by a barrier along the M\(^*\)-He potential [9, 117]. In contrast, alkali ions tend to form strongly bound snowball complexes in the bulk of the He droplets as a result of attractive polarization forces [118, 119].
The kinematics of the desorption of atoms induced by optical excitation was first studied experimentally by nanosecond (ns) electron and ion imaging spectroscopy and theoretically by TDDFT [91, 92, 120,121,122]. Likewise, the dynamics of solvation of alkali ions formed by photoionization was treated by TDDFT and experimentally using ns ion imaging and mass spectrometry [121, 123, 124]. The observed linear dependence of the mean kinetic energy of the desorbed excited atoms on the laser frequency points at an impulsive desorption process [92, 120, 122]. This process is well described by one-dimensional pseudo-diatomic potential curves which quantify the effective interaction between the dopant and the He droplet as a whole [6, 117, 125]. Even the angular distributions of ion images agreed very well with the description of the alkali-droplet complex in terms of a pseudo-diatomic molecule. In some cases, in particular for highly excited states, electronic relaxation occurred in the course of desorption due to curve crossings induced by the interaction with the He droplet [92, 121, 126]. The energy partitioning between the He and the desorbing atom depends on the alkali species and on the quantum state, and appears to be related to the size and shape of the electron orbital [92, 122]. Alkali-He exciplexes were formed either directly by laser-excitation into bound molecular states [120, 122], or by a tunneling process [14, 29, 126]. The desorption of the exciplexes occurred either promptly as for excited atoms, or by a thermal process driven by vibrational or spin relaxation [29, 120, 126].
The early fs pump-probe experiments with He droplets doped with alkali metals mostly focused on the formation of alkali-He exciplexes [10, 21, 24, 127] and on electronic and vibrational coherences of alkali atoms and molecules, respectively [22,23,24,25, 34,35,36]. As dual fs pulses at high repetition rate (80 MHz) were used, the exact location of the dopants, attached to the droplets or in the vacuum, has remained somewhat uncertain; resonant absorption from multiple laser pulses may have induced the desorption prior to the pump-probe process.
Time-resolved measurements of the desorption dynamics of excited alkali atoms and exciplexes have so far only been reported for Rb and RbHe exciplexes. Using amplified fs pulses of the Ti:Sa laser and harmonics thereof, yields, kinetic energies, and angular distributions of electrons and ions for various excited states of the RbHe\(_N\) complex have been traced [27,28,29,30]. This is achieved by the method of velocity-map imaging (VMI), where the velocity distribution of electrons or ions is mapped onto a two-dimensional spatial distribution in the plane of a position-sensitive detector [128]. Given cylindrical symmetry with respect to an axis perpendicular to the spectrometer axis (usually the laser polarization), the measured two-dimensional distribution can be converted into the three-dimensional velocity distribution by inverse Abel transformation. The radial part reflects the kinetic energy spectrum and the angular part contains information about the symmetry of the state that is photoionized.

Velocity-map ion images of [RbHe]\(^+\) and Rb\(^+\) created by fs pump-probe photoionization of Rb-doped He nanodroplets at a laser wavelength of 415 nm [excitation of the 6\(p\) \(\Pi \) state, (a)] at various delays, and at 403 nm [6\(p\) \(\Sigma \) state, (b)] for a delay of 4.8 ps. Based on results reported in [30]. The bottom panel schematically illustrates the corresponding photoinduced processes
As an example, Fig. 10.6 shows typical velocity-map ion images recorded upon excitation of the 6\(p\) \(\Pi \) state [panel (a)] and the 6\(p\) \(\Sigma \) state [panel b)] [28, 30]. In the 6\(p\) \(\Pi \) state, a large fraction of Rb atoms form RbHe exciplexes prior to desorption. As it is expected for promptly desorbing atoms, the [RbHe]\(^+\) ions feature a typical ring-like intensity distribution \(I_\mathrm {RbHe}\) with an angular dependence \(I_\mathrm {RbHe}\propto \sin ^2\theta \) with respect to the polarization of the laser pulses which is characteristic of a \(\Sigma \rightarrow \Pi \) perpendicular dissociative transition. The increase of the mean radius of this distribution as a function of delay clearly shows that desorption of RbHe exciplexes proceeds as a prompt, pseudo-diatomic dissociation process. A schematic representation is shown at the bottom of Fig. 10.6. Excitation of the RbHe\(_N\) complex into the 6\(p\) \(\Sigma \) state results in a \(I_\mathrm {Rb}\propto \cos ^2\theta \)-angular intensity distribution according to a \(\Sigma \rightarrow \Sigma \) parallel transition causing prompt dissociation, see panel (b).

(a) Illustration of the pump-probe scheme for probing the desorption dynamics of Rb atoms excited to 6p-correlated states, based on the RbHe\(_N\) pseudo-diatomic potential curves. Based on results reported in [27]. (b) Detected Rb\(^+\) ion yield at a laser wavelength of 403 nm (6\(p\) \(\Sigma \) excitation); (c) Rb\(^+\) ion kinetic energies; (d) electron energy. Based on results reported in [30]
Figure 10.7a depicts the pseudo-diatomic potential curves involved in the two processes. Owing to the repulsive character of the excited states, the Rb atoms promptly desorb as free atoms (6\(p\) \(\Sigma \) state) or as a mixture of Rb atoms and RbHe exciplexes (6\(p\) \(\Pi \) state). In contrast, the ionic potential curve is attractive, causing Rb\(^+\) or [RbHe]\(^+\) to fall back into the droplet when created near the droplet surface, i.e. at short pump-probe delay. The condition for the ion to fall back into the droplet or to move away is given by the balance of the kinetic energy gained by repulsion in the neutral excited state on the one hand, and the potential energy barrier in the ionic state on the other. Indeed, the yield of detected Rb\(^+\) at a laser wavelength of 403 nm (6\(p\) \(\Sigma \) excitation) nearly vanishes at short delay and steeply rises around 0.5 ps, see Fig. 10.7b. The yield of large [RbHe\(_{n}\)]\(^+\), \(n\approx 5000\), complexes (not shown) features the opposite behavior [27]. This confirms the concept that ions fall back into the droplet when created near the surface. The yield of photoelectrons (not shown) displays no significant pump-probe dependence, indicating that electrons are emitted from the dopants irrespective of the dopants’ position with respect to the droplet surface. A transient maximum of the [RbHe]\(^+\) yield around 1 ps was interpreted in terms of associative photoionization of [RbHe]\(^+\), i.e. the direct optical excitation of a bound cationic state [27].
The Rb\(^+\) ion kinetic energy monotonously rises within \(\approx 1\) ps [Fig. 10.7c] due to the acceleration of the excited Rb atom away from the droplet surface. Concurrently, the photoelectron energy drops by about 1200 cm\(^{-1}\) [panel (d)] due to the increase of the potential-energy difference between the pseudo-diatomic excited state and the ionized state as the Rb atom moves away from the droplet.
Similar delay-dependent electron and ion signals were observed in a two-color pump-probe experiment where Rb atoms were excited to the 5p-correlated states. The main difference was a slower dynamics by a factor of nearly 100. Ion yields and kinetic energies continuously rose on the 100 ps time scale. This is due to less repulsive pseudo-diatomic potential curves in these states compared to the 6p-correlated states [117]. Both the dynamics in the 6p and 5p states were well reproduced by TDDFT simulations [28]. A detailed ion and electron-imaging study of RbHe exciplexes formed in the 5\(p\) \(\Pi \) state, combined with TDDFT simulations, revealed that the desorption of the RbHe exciplexes, proceeding within \(\approx 700\) ps, is induced by \(^2\Pi _{3/2}\rightarrow ^2\Pi _{1/2}\) spin relaxation. The formation time of the RbHe exciplex was found to range between 20 and 50 ps [29].
These studies were further extended to Rb\(_2\) dimers formed on He nanodroplets [38]. Similarly to alkali atoms, Rb\(_2\) excited to intermediate states were found to promptly desorb off the droplets. However, both angular and energy distributions of detected Rb\(_2^+\) ions appear to be most crucially determined by the Rb\(_2\) intramolecular symmetries rather than by the symmetries of the Rb\(_2\)He\(_N\) pseudo-diatomic complex. The pump-probe dynamics of Rb\(_2^+\) was found to be slower than that of Rb\(^+\) in the same wavelength range of the pump pulse, pointing at a weaker effective guest-host repulsion for excited molecules than for atoms.
To summarize this section, as general trends, an excited alkali atom or molecule tends to promptly desorb off the He droplet surface, in good agreement with a pseudodiatomic dissociation model. In contrast, an ion, formed at the droplet surface, sinks into the bulk of the droplet where a dense He shell form around it. Pump-probe photoionization signals manifest the competing dynamics of desorption of the excited neutral and the falling back of the ion into the droplet. Another trend is that a resonantly excited metal atom tends to form a metal-He exciplex with variable abundance depending on the symmetry of the excited state. These metal-He exciplexes tend to promptly desorb as well; exceptions are the lowest excited states where the repulsion between the excited alkali and the He droplet is weak. The combination of fs pump-probe photoionization spectroscopy with VMI of electrons and ions reveals detailed information about the dynamics and kinematics of the desorption process for specific excited states. In future experiments, it would be interesting to extend these studies to larger alkali oligomers and other types of metals which are initially located deeper within the He droplet surface (Mg, Ca), or in the droplet interior (Ag, Al, Cr, Cu,...). Likewise, direct measurements of the dynamics of ejection of excited ions [81, 124] would by highly desirable.
10.3.2 Solvated Atoms—Solvation Dynamics
In their electronic ground state, atomic dopants inside He\(_N\) are surrounded by a He solvation shell that forms through equilibration of attractive dopant–He forces and repulsive Pauli interactions between the dopant’s valence electrons and the closed-shell helium. Electronic excitation of the dopant often causes its valence electron to expand radially, inducing a strong interaction with the solvation shell. The energy related to this expansion process has to be provided as excess photon energy, represented by photoexcitation bands that are typically blue-shifted and broadened by several hundred wave numbers with respect to the bare-atom transition (see Fig. 10.8a), as observed in frequency-domain experiments for atomic dopants such as Al [15], Ag [62], or Cr [129]. Additionally, these experiments have revealed dopant ejection after photoexcitation, indicating the heliophobic character of the excited state. Electronic relaxation through nonradiative population transfer to lower states induced by curve crossings due to interaction with surrounding He atoms was observed [15, 60, 62, 129,130,131], as well as excipex formation. These frequency-domain results raise a number of questions about the nature of dopant photoexcitation inside a He\(_N\): (i) Which primary solvent-related processes are triggered by photoexcitation, (ii) what are their characteristic time scales, (iii) how is the excess energy dispersed into the system, and (iv) what is the dependence of these processes on experimental parameters such as droplet size or excitation energy?

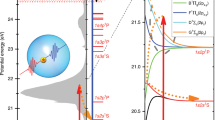
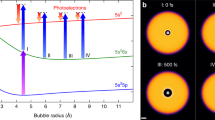
Fast solvent response to photoexcitation of In–HeN: Expansion of the solvation shell and energy dissipation. (a) Photoexcitation spectra of In–HeN and bare In in the range of the 6\(s\) \(\leftarrow \)5p transition [132]. Vertical bars indicate phoexcitation energies corresponding to the spectrograms in (c). (b) Sketch of the In–HeN potential energy surfaces as function of the bubble radius for In in its ground [5s25p (2P1/2), blue], lowest excited [5\(s\) \(^2\)6s (\(^2 S_{1/2}\)), green] and ionic ground state [5\(s\) \(^2\) (1\({S}_{0}\)), red]. Pump excitation at different photon energies (c.f., (a)) and probe ionization at 405 nm are indicated. Red arrows correspond to the PE kinetic energy measured by TRPES. (c) PE spectrograms showing the initial bubble expansion obtained with different photoexcitation energies [32], as indicated in (a) and (b). The simulated PE peak shift (\(E_{\text {He}_{\text {N}}\text {-In}{*}} - E_{\text {He}_{\text {N}}\text {-In}^{+}}\) from (e) is shown as dashed line. (d) He density distributions of In–\(\text {He}_{4000}\) at selected times after photoexcitation, as calculated with TDDFT [31]. (e) Interaction energy \(E_{\text {He}_{\text {N}}\text {-In}{*}}\) of the 5s26s excited state (green curve) and interaction energy \(E_{\text {He}_{\text {N}}\text {-In}^{+}}\) of the 5s2 ionic state (red curve) [31]. Additionally, the kinetic energy of the He atoms, \(E_{\text {kin, He}}\), is plotted as dashed line
In the following we discuss answers to these questions that were obtained with TRPES of indium (In) atoms inside He\(_N\) [31, 32]. In TRPES inside He\(_N\) the valence electron, which is electronically excited by the pump pulse, is exploited to sense the temporal evolution of the He environment by retrieving the ionization energy with the probe pulse (see Sect.10.2.4). Rearrangement of the solvation shell around the dopant through nuclear relaxation can be followed as transient PE peak shift as the potential energies of the excited and the ionic state depend on the dopant distance to neighboring He atoms. The observed processes can be distinguished by their time scale into a fast expansion of the solvation shell to form a He bubble (\(\approx 500\) fs), as well as a slower bubble oscillation and dopant ejection from the droplet (\(\approx 50\) ps), which will be discussed separately.
10.3.2.1 Fast Solvent Response: Expansion of the Solvation Shell
The electronic photoexcitation process of In atoms inside He\(_N\) is depicted in Fig. 10.8b, which shows the In–He\(_N\) pseudo-diatomic potential energy curves as function of the bubble radius: Photoexcitation of the In atom in its ground-state solvation shell (4.5 Å radius) leads initially to an increased excited state energy, which is represented by the broadened and blue-shifted excitation spectrum (Fig. 10.8a) [132]. Relaxation towards an expanded solvation shell causes energetical shifts in both the excited and the ionic state, which can be tracked by probe-pulse ionization at increasing time delays and observation of the resulting PE energy.
Corresponding PE spectra for different excitation wavelengths are shown in Fig. 10.8c [31]. Each spectrum shows a rapid initial shift of the PE peak to lower energies, that levels off in all three cases within 1 ps to the same value of 320 meV. The 1 ps PE-energy is approximately 0.03 eV above the gas phase value due to the reduced ionization potential of solvated atoms.
With increasing photoexcitation energy, the PE peak maximum at time zero shifts to higher values, from 0.65 eV for 380 nm to 0.79 eV, for 360 nm [32]. Additionally, the PE peak shift proceeds faster for higher excitation energies, as becomes evident by comparison the initial slopes for the three spectra in Fig. 10.8c. This trend is expected from the increasing steepness of the excited state potential energy curve in Fig. 10.8b, suggesting a stronger acceleration of the excited state wave packet for higher excitation energies. In combination, these results show that the excitation excess energy is fully transferred to kinetic energy of the solvation shell within 1 ps. Concerning variation of the droplet size, no influence is found on the PE spectra (\(\bar{N}=2600-40000\)), showing that bubble expansion is a purely local process that only depends on the its environmental fluid density [32].
Additional insight into the ultrafast In–He\(_N\) photoexcitation dynamics is obtained from TDDFT simulations using the BCN-TLS-HeDFT computing package [90], based on In–He pair potentials of the ground, excited and ionic state [31, 132]. He density plots at selected times after photoexcitation (Fig. 10.8d) show that the solvation shell almost doubles in radius within the first picosecond from 4.5 to 8.1 Å. Based on this time evolution of the He density the time-dependencies of the excited and ionic states can be computed by integrating the corresponding pair potentials over the corresponding droplet densities. Figure 10.8e shows that the He environment increases the excited state energy (\(E_{\text {He}_N\text {In*}}\)), while it decreases the ionic state energy (\(E_{\text {He}_N\text {In}^+}\)). The difference of both energy deviations (\(E_{\text {He}_N\text {In*}}-E_{\text {He}_N\text {In}^+}\)) corresponds to the transient PE shift and is in excellent agreement with the TRPES measurement (Fig. 10.8c, dashed line). The TDDFT simulation thus sheds light onto the dissipation process of the excess energy, as it allows to quantify the individual contributions to the ionization energy measured by TRPES. Only with the TDDFT results it becomes clear that the excess energy of the photoexcitation process is initially stored as potential energy of the excited state and subsequently converted into kinetic energy of the surrounding He atoms (\(E_\text {kin,He}\) in Fig. 10.8e) within the first picosecond, leading to He density waves propagating through the droplet (see Fig. 10.9b).

Slow solvent response to photoexcitation of In–He\(_N\): Bubble oscillation and dopant ejection [32]. (a) Transient PE energies for different droplet sizes, showing an overall decrease due to dopant ejection with a local maximum due to bubble contraction (all curves are vertically offset by the same amount). (b) He density distributions of In–He\(_{4000}\) at selected times after photoexcitation, as calculated with TDDFT. (c) Transient ion yield for different droplet sizes representing dopant ejection (all curves are vertically offset by the same amount). (d) Simulated PE transients for trajectories originating at different distances to the droplet center (droplet size \(N=4000\), 36 Å radius)
10.3.2.2 Slower Solvent Response: Bubble Oscillation and Dopant Ejection
After adaption of the He environment to the electronically excited dopant within the first picosecond, the heliophobic character of the excited state leads to dopant ejection on a \(10-100\) ps timescale. Additionally, the impulsive stimulation of the He solvation layer initiates a collective oscillation of the He bubble. Although the two processes overlap in time, they can be sensed and distinguished with TRPES, underlining its sensitivity.
Figure 10.9a shows the transient shift of the PE peak position up to 200 ps, exhibiting a gradual \(\approx 20\) meV decrease to the bare-atom PE energy. This PE peak shift represents dopant ejection from the droplet and is influenced by the distributions of both droplet sizes and starting locations within the observed ensemble. Superimposed on the PE energy reduction, a temporary increase is observed at \(\approx 30\) ps that is caused by the increase of He density around the dopant in consequence of the first contraction of the bubble oscillation. Importantly, neither the PE peak shift nor the temporal increase exhibit a dependence on the droplet size (Fig. 10.9a), which indicates that the In atoms are not equally distributed within the droplet but rather are confined within a small spherical shell beneath the surface. Also, the excitation energy has no influence on ejection signal and only weakly increases the bubble oscillation period (not shown) [32], which is in line with the interpretation that the excitation excess energy is fully transferred to kinetic energy of the solvation shell within 1 ps. The momentum of the dopant is thus not changed upon photoexcitation inside the droplet due to the symmetry of the bubble expansion, in contrast to surface-located dopants [133].
Complementary information on the ejection dynamics is obtained from photo-ions, which can only be detected if ionization by the probe pulse takes place outside the droplet at sufficient distance to its surface [133] (see Sect.10.2.3). The transient ion yield (Fig. 10.9c) remains essentially zero up to \(\approx 50\) ps as this observable is insensitive to dynamics inside the droplet (bubble expansion and oscillation). The signal onset at 50 ps and its rise up to 200 ps represents In ejection and support the TRPES results (Fig. 10.9a); in particular, the ion transients are also nearly independent of the droplet size.
Further insight into the dynamics on the 10-100 ps time scale is obtained from TDDFT simulations, which predict correct time-scales for both the bubble oscillation and ejection from the droplet, as can be seen from the corresponding He density distributions in Fig. 10.9b). Computed PE transients (Fig. 10.9d) strongly depend on the location within the droplet where the photoexcitation takes place: center-located atoms experience a periodic PE increase of \(\approx 30\) meV, representing the bubble contraction, while off-center locations show a limited number of bubble oscillations followed by gradual \(\approx 20\) meV energy decrease due to ejection. In particular, the single bubble contraction predicted for the trajectory originating at 20 Å distance from the droplet center (16 Å beneath the droplet surface) supports the assumption that the In atoms are contained within a small region beneath the droplet surface.
These results on the In–He\(_N\) system are in agreement with a recent study on pure He\(_N\), triggered by XUV pulses from a free electron laser [66], see Sect. 10.5.1. There, the TRPES results show signatures for creation of a He bubble around a localized excitation, He\(^*\), on a very similar timescale (\(\approx 500\) fs) and much faster ejection of the He\(^*\) (2.5 ps), which might be related to near-surface excitation and stronger acceleration of He atoms compared to the heavier In.
In summary, the In–He\(_N\) experiments provide the first real-time study of the solvation dynamics triggered by photoexcitation of an impurity embedded inside a helium droplet, in contrast to the surface-dopants discussed above (Sect. 10.3.1). The atomic impurities used do not show any internal dynamics on the relevant time scales and are thus ideal probes for the response of the superfluid helium solvent. As a remarkable result, similar solvation and ejection dynamics are found for impurities embedded inside the droplets, impurities attached to the surface or even for single, directly excited helium atoms in the helium droplet. For molecular dopants, the described solvent-related dynamics—solvation shell expansion, bubble oscillation and dopant ejection—will be superimposed on intramolecular dynamics because electronic excitation is the primary process in photochemical reactions. A mechanistic description of these processes will thus be key to the conception and interpretation of ultrafast photochemical studies inside He\(_N\). For larger molecules one can expect less pronounced solvation shell dynamics since excited molecular orbitals may experience less contact to the He surrounding, as indicated by sharp electronic transitions (zero-phonon lines) that are frequently observed for these dopants [67, 68]. A systematic characterization of these processes for different classes of molecules in future experiments will be essential.
10.3.3 Dynamics of Superfluid Droplets Compared to Normalfluid \(^3\)He Droplets
A fascinating property of helium is its superfluid nature and related dynamics, posing fundamental questions on the existence of such phenomena in confined, nanoscale droplets. Non-dissipative flow has been observed measuring ro-vibrational spectra, confirming superfluidity in nanodroplets [134]. Even in molecular complexes containing only a few helium atoms non-classical inertia has been verified [135]. The experimental results triggered significant interest from theory, leading to a deeper understanding of nanoscopic superfluidity [136,137,138]. Ultrafast time-resolved studies came into play measuring the vibrational relaxation of attached molecules, indicating evidence for a Landau-critical velocity on the molecular level connected to vibrational motion [35]. Indeed, the Landau velocity was then confirmed from measuring the velocity of ejected dopants [95].
For all such studies, one ideally compares the Bose-Einstein-condensed superfluid \(^4\)He droplets with \(^3\)He droplets representing a Fermi fluid. The latter can readily be formed in droplet beam sources (see [3] and references therein). Comparing studies were seminal confirming superfluidity in helium droplets with infrared spectroscopy [134], furthermore, in electronic spectra connected to the structure of zero-phonon lines and phonon wings [139, 140], as well in recent X-ray diffraction imaging experiments [141] and corresponding theory [7, 89, 102]. Femtosecond time-resolve experiments comparing \(^3\)He droplets have been studied with alkalies as probe species. In laser-induced fluorescence (LIF) spectra of Na atoms, the effect on changing the helium isotope becomes apparent as a significant shift of the spectra [142]. However, in comparison with theory this is well understood from the decreased density of \(^3\)He and respective binding energies to the helium surface dimple. Differences are even more pronounced in excitation spectra of alkaline earth dopants, providing a sensitive probe of their location [143]. Nevertheless, superfluid dynamics did not show up in LIF data since observing the electronic excitation probes the environment “frozen” in the ground state configuration.
In time-resolved measurements the formation of RbHe exciplexes has been probed with femtosecond time resolution [21] revealing a faster formation of \(^4\)HeRb (8.5 ps) compared to \(^3\)HeRb (11.6 ps). This was a surprising result because intuitively the lighter \(^3\)He is expected to evolve a faster dynamic. Also from the interaction potentials and a theoretical model, based on the helium tunneling into the bound exciplex configuration, a 40-fold acceleration of forming a \(^3\)HeRb was calculated [21]. Apparently, the tunneling model does not give the right picture. It was speculated that a difference in the vibrational relaxation when entering the bound molecular potential could be responsible for the different formation times.

Generation and observation of vibrational wave packets in a pump-probe experiment
10.4 Vibrational Dynamics of Molecular Dopants
Vibrational wave packets (WP) were among the first dynamical processes investigated by time-domain spectroscopy. A vibrational WP in a molecule can be seen as quantum mechanical analogue to classical vibration that, due to its coherent nature, provides insight into both the intrinsic structure of the molecule and its interaction with the environment. In a pump-probe experiment, the pump pulse can launch a WP in the excited electronic state (or the ground state) by simultaneously populating several vibrational levels with a well-defined initial phase. The pulse thus creates a coherent superposition of nuclear eigenstates (Fig. 10.10) [144] that evolves in time and can be tracked by photoionization with the probe pulse, projecting the WP onto the ion continuum. Dependence of the ionization probability and ionization energy on the reaction coordinate (e.g., internuclear distance) yields a periodic modulation of the PE yield and energy, as well as the photoion yield, all at characteristic frequencies that correspond to the energetic distances between excited vibrational states. Dispersion in an anharmonic potential energy curve leads to dephasing and rephasing of the WP at characteristic revival times.
If the molecule is embedded in a dissipative environment, collisions with solvent molecules may cause vibrational relaxations and decoherence (deterioration of the phase relation within the observed ensemble of molecules), resulting in an irreversible loss of modulation contrast. Information about decoherence, energy relaxation or even deformation of the potential energy curve by the solvation shell, can be retrieved from a spectrogram, as obtained from Fourier transformation of the oscillating signal within a sliding time window. Vibrational WPs of small molecules (mostly dimers) in He\(_N\) have been used to probe the He influence on nuclear structure and dynamics, both at the droplet surface and in its interior, which will be discussed separately in the following.
10.4.1 Vibrational Wavepackets in Alkali Dimers and Trimers
Alkali diatomic molecules were among the first molecules to be studied by time-resolved laser spectroscopy due to their strong electronic transitions in the NIR and visible (VIS) ranges of the spectrum. Low ionization potentials make alkali dimers accessible to photoionization spectroscopy using comparatively low photon energies and laser intensities. Besides, potential-energy curves can be calculated with high precision, thus facilitating the interpretation of spectroscopic data.
A number of interesting phenomena have been studied using these simple molecules, e.g., wavepacket propagation in spin-orbit-coupled states [145, 146], fractional revivals of vibrational wavepackets [147, 148], the competition of different ionization pathways [149, 150], and isotope-selective ionization [151]. Detailed insights into the vibrational dynamics have been obtained by applying new experimental techniques such as photoelectron spectroscopy [152] and optimal control schemes using shaped laser pulses [153].
Alkali dimers have newly attracted interest due to the recent advances in the formation of ultracold molecules out of ultracold atomic ensembles by means of Feshbach resonances [154] and photoassociation [155]. These studies require the knowledge of molecular spectra with great precision. However, conventional molecular spectroscopy usually probes molecules in their singlet ground state; triplet states, which play important roles in the physics of ultracold molecule physics, are more difficult to access experimentally.
Alkali dimers are efficiently formed by aggregation of atoms picked up by He nanodroplets. Due to the lower binding energy of alkali dimers in their lowest metastable triplet state (\(\approx 300\) cm\(^{-1}\)) compared to the singlet ground state (\(\approx \) 3000 cm\(^{-1}\)), preferentially triplet dimers remain attached to the surface of the droplets [12, 156]. While triplet dimers are oriented parallel to the He surface, singlet states tend to adopt a more erect configuration [157,158,159]. For Li\(_2\) in its triplet state, drastically enhanced vibrational quenching rates were predicted for the triplet state as compared to the singlet ground state [158, 160].
Vibrational WP dynamics in triplet states of an alkali dimer attached to a He nanodroplet was observed for the first time using Na\(_2\) [23]. No influence of the He droplet on the WP dynamics was observed, likely due to the desorption of the dimer off the droplet prior to the actual pump-probe process. For K\(_2\) in singlet states, indications for the influence of the He droplet on the vibrational dynamics [22] were found. Transient modulations of both amplitudes and frequencies of vibrational frequency components were observed, from which the time constant for the desorption dynamics was estimated to range between 3 and 8 ps.

(a) Pump-probe trace of vibrational WPs in high-spin Rb\(_2\) molecules formed on the surface of He droplets recorded at an excitation wavelength of \(\lambda = 1060\) nm. (b), (c) Fourier spectra inferred from (a) and other traces. Based on results reported in [25]
For Rb\(_2\) in the triplet states \(a^3\Sigma _u^+\) and \((1)^3\Sigma _g^+\), long-lived vibrational coherences were observed up to pump-probe delays \(\gtrsim 1.5\) ns, see e.g. Fig. 10.11a [25]. Likely, the fast desorption of the excited Rb\(_2\) and its low internal temperature facilitates the detection of WP interferences with high contrast, including full and fractional revivals. Fourier analysis of the time traces provides high-resolution vibrational spectra (see Fig. 10.11b, resolution about 900 MHz), which are in excellent agreement with ab initio calculations and of interest for ultracold molecules experiments [161]. Even individual beat frequencies for the two isotopologs, \(^{85}\)Rb\(_2\) and \(^{87}\)Rb\(_2\) were resolved [Fig. 10.11c]. This shows that high-resolution spectroscopic data can be extracted from fs pump-probe experiments on doped He nanodroplets.
By comparing the measured data with theoretical results based on dissipative quantum dynamics calculations, it was found that the most important effect of the He environment is vibrational relaxation causing dephasing and energy dissipation [35, 36]. Alternatively, rotational wavepacket dynamics was considered as a cause of the observed decay of the oscillation amplitude [162]. However, unphysically high rotational temperatures would have to be assumed. Besides, no rotational revival structure was observed, which would show up at delay times around 0.6 ns [36]. The strong dependence of the measured dephasing time on the laser wavelength cannot be rationalized by rotational dynamics, either. However, contributions of rotational dynamics to the fast decay observed at short delays \(\lesssim 0.3\) ps cannot be excluded.
In the K\(_2\) case, the best agreement between theory and experiment was achieved when damping of the WP motion was neglected for slowly moving WPs [35]. Likewise, the WP dynamics of Rb\(_2\) was best described by low damping rates for the WP motion in the lower vibrational states \(\nu \lesssim 15\) of the \((1)^3\Sigma _g^+\) state, whereas higher vibrations were more strongly damped [36]. It is tempting to relate these findings to the critical Landau velocity \(v_\mathrm {L}\) for frictionless motion in superfluid He [95]. However, more systematic measurements, in particular for molecules immersed in the bulk of the droplets, are needed to unambiguously assess the role of superfluidity in the damping of molecular vibrations in or on He nanodroplets.
One great advantage of the He nanodroplet isolation technique is that heterogeneous, polyatomic complexes can be formed rather easily and with some degree of control [67]. By multiply doping He droplets with different species, small heterogeneous clusters [34, 163,164,165] up to core-shell nanoparticles can be aggregated [166, 167]. In this way, the fs spectroscopy studies of alkali dimers were extended to alkali trimers, specifically Rb\(_3\), Rb\(_2\)K, RbK\(_2\), and K\(_3\) in quartet states [34]. Similarly to the alkali dimers, long-lived vibrational coherences were observed in certain regions of the spectral range accessible by the Ti:Sa laser. Thus, vibrational spectra with a spectral resolution of the order of 0.2 cm\(^{-1}\) were measured. A typical sliding window Fourier spectrum, or spectrogram, recorded on the mass of Rb\(_3\), is shown in Fig. 10.12a. The power spectrum of the full pump-probe scan is shown in panel (b). Clearly, distinct frequencies are visible which persist over delay times ranging between \(\approx 10\) and \(\approx 100\) ps.
In contrast to the straight-forward assignment of pump–probe power spectra recorded for alkali dimers, the interpretation of the trimer spectra turns out to be much more involved. This is due to the concurrent effect of Jahn-Teller and spin-orbit-couplings in these rather heavy species. Nevertheless, the measured frequencies were assigned to beats of vibrational normal modes in the \((1)^4A_2'\) and \((2)^4E'\) states by comparing to ab initio calculations. The most prominent lines are the asymmetric stretch and bending modes \(Q_{x/y}\) of the lowest quartet state excited by impulsive Raman scattering, followed by the \(Q_{x/y}\)-modes of the excited electronic states. The symmetric stretch mode \(Q_s\) is only visible in the spectrum of Rb\(_3\), whereas it is significantly broadened due to fast dephasing within a few ps. Intramolecular vibrational relaxation, intersystem-crossing or vibrational relaxation by coupling to the He droplet may be the cause of this fast damping.

(a) Sliding window Fourier spectrum of Rb\(_3\) measured by fs pump-probe photoionization spectroscopy at a laser wavelength of 850 nm. The corresponding electronic states as well as vibrational modes are indicated. (b) Fourier spectrum including the whole delay range. Based on results reported in [34]
In summary, very long-lived vibrational coherences over hundreds of picoseconds up to nanoseconds were observed for alkali-molecules attached to the surface of helium nanodroplets. This is in stark contrast to molecules in solution or thin films were decoherence is generally much stronger. Nevertheless, signs of dephasing and decoherence induced by the molecule-droplet interactions were found. Due to the general tendency of excited alkali molecules to desorb from the droplet surface, it is hard to pinpoint decoherence effects based on vibrational interference spectra, though. The high spectral resolution achievable by coherent wavepacket interference spectroscopy combined with the synthesis advantages of helium nanodroplets provide a valuable tool for high-resolution spectroscopy of rare molecular species, as has been shown for high-spin alkali molecules and heterogeneous alkali complexes which are not accessible by other methods.
10.4.2 Vibrational Wave Packets in Solvated Dimers
While vibrational dynamics of alkali molecules on the droplet surface were found to be only weakly influenced by the He environment, as discussed in the previous chapter, the perturbation of fully solvated molecules can be assumed to be stronger, which might lead to complete suppression of intramolecular dynamics [168]. Recently it could be demonstrated, however, that the He influence on vibrational dynamics of solvated dimer molecules can also be low. Figure 10.13 shows the time-resolved pump-probe photoionization spectra of In\(_2\)–He\(_N\), which contains characteristic signatures of both solvent-related and intramolecular dynamics. By comparison with the In atom dynamics (Sect.10.3.2), the initial PE peak shift from 0.75 to 0.60 eV within the first picosecond in Fig. 10.13a can be identified as bubble expansion in response to valence electron expansion due to photoexcitation. Also, the ion transients (Fig. 10.13b) show that the electronically excited In\(_2^*\) is ejected from the droplet. The slower ion rise compared to the atom, is related to a reduced interaction of the molecular valence electron with the He environment and the larger mass of the molecule.

In\(_2\) vibrational wave packet dynamics inside He\(_N\) [33]. (a) PE spectrum showing the initial oscillations after photoexcitation. (b) Comparison of the transient In\(^+\) and In\(_2^+\) ion yields. (c) Spectrogram showing the oscillation frequency obtained from sliding-window Fourier transformation of the initial WP signal (left) and the full revival (right). (d) Integrated PE yield (blue) and oscillation amplitue from sliding-window Fourier transformation (red)
Intramolecular vibrational WP dynamics are represented as modulation of the PE signal with a periodicity of 0.42 ps (2.42 THz oscillation frequency), as can be seen in the PE spectrum (Fig. 10.13a). The integrated PE yield (Fig. 10.13d, blue curve) shows that the initial WP modulation decays within 10 ps and reappears as full revival between 280 and 300 ps, although with reduced amplitude. The time-dependency of the oscillating signal is shaped by a combination of WP dispersion in the anharmonic potential and decoherence due to He interaction and can be analyzed by sliding-window Fourier transformation. The corresponding spectrograms are shown in Fig. 10.13c and the (normalized) time-dependent amplitude in Fig. 10.13d (red trace).
Since the full revival at 290 ps is observed at times where the excited dimers have left the droplets (see Fig. 10.13b), direct comparison of WP oscillation of solvated In\(_2\) inside He\(_N\) and bare In\(_2\) in gas phase is possible and reveals the decoherence imposed by the He environment. In the He environment, both dispersion and decoherence are active so that the WP oscillation decay proceeds twice as fast [50% decrease within (\(4\pm 1\)) ps], as compared to the bare In\(_2\) [(\(7\pm 1\)) ps] where solely dispersion is active. This comparison allows to estimate a lower limit of the decoherence half-life of about 10 ps. The oscillation amplitude of the revival, however, suggests a longer half-life since the delayed and slow ion yield increase (Fig. 10.13b) indicates average In\(_2\)–He interaction of several tens of picoseconds, while the oscillation amplitude of the revival has decayed to only 20% of the initial value. The fact that the same oscillation frequencies are observed for solvated and bare In\(_2\) indicates that the distortion of the excited potential energy curve is not significant for this excitation energy, and that no phase-conserving vibrational relaxation takes place. Further experiments are required in order to test for potential energy distortions at higher energies and to identify the contributions of elastic depahsing and vibrational energy relaxation [113].
Comparison of these long decoherence times (tens of picoseconds) to the much faster decoherence in conventional solvents, typically hundreds of femtoseconds to few picoseconds in special cases [169], underlines the superiority of superfluid He as a solvent. This low interference with nuclear motion can be rationalized by considering the In\(_2\) molecule and its surrounding He bubble as coupled oscillators that interact only weakly due to their different oscillation periods (In\(_2\): 0.42 ps, bubble: \(\approx 30\) ps, see Sect.10.3.2). Also, the In\(_2\) oscillation energy of 80 cm\(^{-1}\) significantly exceeds the elementary excitations of He\(_N\) (phonons and ripplons) [67]. These results indicate that molecular systems that exhibit a slow decay of nuclear coherence in gas-phase can now be investigated in a thermal bath environment.
In summary, the In\(_2\)–He\(_N\) experiments provide an impressive example for the preservation of vibrational coherence in a fully solvated molecule despite the disturbance of the helium bath. The vibrational coherence is preserved even while the molecule propagates through the medium and gets ejected. This enabled a direct comparison of the coherent WP motion for the solvated and free gas-phase molecule. In future experiments it will be interesting to examine the influence of additional mediator atoms or molecules on the WP properties of the In\(_2\)-He\(_N\) system. It will be straight forward to monitor alterations of the decoherence time and probably oscillation frequency as function of amount and interaction strengths of mediator particles.
10.5 Dynamics of Highly Excited Helium Droplets
He nanodroplets are particularly attractive as model systems for studying the photodynamics of finite-size condensed-phase systems, both experimentally and theoretically [5, 66, 89]. (i) He atoms have a simple electronic structure which simplifies electron spectra and model calculations; (ii) interatomic binding is extremely weak which allows one to neglect chemical binding; He droplets can essentially be treated as assemblies of unperturbed atoms; (iii) the structure of He nanodroplets is homogeneous and nearly size-independent due to their superfluid nature; this avoids the congestion of spectra by multiple phases and structures as it is the case for solid clusters [170, 171]. Furthermore, exploring transient phenomena associated with superfluidity is a particularly fascinating aspect of time-resolved He nanodroplet spectroscopy [5, 31, 66, 95, 172].
There are essentially two experimental approaches to studying the dynamics of excited and ionized He nanodroplets: Electron bombardment and resonant absorption or ionization by extreme ultraviolet radiation (XUV). Electron bombardment in combination with mass spectrometry is a well-established technique; positive and negative ions of both He and dopants can be created relatively easily [173,174,175,176]. However, the energy resolution is rather limited in such experiments and so far no time-resolved measurements have been reported.
The first XUV experiments were performed by the group of Toennies et al. in Berlin using synchrotron radiation (BESSY I) [177]. This mass spectrometric study established the key aspects of photoionization dynamics of He droplets: Ionisation occurs not only by direct electron emission at photon energies exceeding the ionization energy of He atoms (\(E_i=24.6\) eV) but also by autoionization at photon energies in the range \(23~\mathrm {eV}\lesssim h\nu \le E_i\). The dominant ionisation products in this regime are He\(_2^+\) ions and small He\(_n^+\) clusters as well as large cationic clusters with \(n\gtrsim 10^3\). In doped droplets, the dopants are ionized indirectly by a Penning ionization-like process through He\(^*\) ‘excitons’ whereas no evidence for direct photoionization of dopants was found.
At \(h\nu \le E_i\), He\(^+\) ions are formed in the droplets which first undergo resonant charge hopping (19-35 fs per hop) over a distance of 3.1 Å before localizing by forming He\(_2^+\) [178, 179]. Alternatively, when a dopant is present in the droplet, the He\(^+\) can localize by charge transfer ionization of the dopant [180, 181]. The excess energy is carried away by emission of a photon (radiative charge transfer, RCT [182]). In many cases, this reaction leads to the ejection of the dopant ion or a complex of the dopant ion with He atoms. A more intricate variant of charge transfer ionization is electron-transfer mediated decay (ETMD) [183], where the excess energy is transferred back to the electron-donating dopant or to a third particle nearby. RCT and ETMD have been studied in doped He nanodroplets for various alkali and alkaline earth dopants [82, 84, 86, 184].
These studies were refined in a series of synchrotron experiments carried out by D. Neumark and coworkers in Berkeley. By applying photoion and electron imaging detection, further insights into the relaxation of photoexcited or ionized He droplets were obtained [185,186,187]. In particular, extremely low-energy electrons were observed in the regime of He droplet autoionization, whereas for \(h\nu > E_i\), electrons had as much as 0.5 eV higher kinetic energy than those from atomic He at the same photon energy. By implementing photoelectron–photoion coincidence (PEPICO) imaging detection at the synchrotron facility ELETTRA in Trieste, these studies were further extended in the direction of interatomic Coulombic decay (ICD) processes [79, 80, 82,83,84, 86, 184, 188,189,190], see Sect. 10.5.2 as well as photoelectron spectroscopy [189,190,191]. In these studies, the role of the He droplets was to serve as an inert substrate that prepares a molecular complex in its vibronic groundstate; the reaction was then initiated by the interaction with an energetic He\(^*\) or He\(^+\) created in the droplet.
Complementary synchrotron experiments were carried out by the group of Möller et al. in Hamburg using fluorescence detection of excited pure He nanodroplets. Sharp atomic and molecular lines in the emission spectra indicated the localization of droplet excitation on an excited He atom (He\(^*\)) or He\(_2^*\) excimer, and the formation of void bubbles around them [192,193,194]. Relaxation into lower-lying singlet and even triplet states was observed; the latter were induced by electron-ion recombination in the droplet autoionization regime [192]. Upon excitation of high-lying electronically excited states, unusual Rydberg states located at the surface of He nanodroplets were evidenced by time-correlated fluorescence spectroscopy [195].
10.5.1 Time-Resolved XUV Spectroscopy of Pure He Nanodroplets
Time-resolved spectroscopy of He nanodroplets first became possible thanks to the development of XUV light sources based on high-harmonics generation of intense NIR laser pulses. In a series of XUV-pump and NIR-probe experiments, the Berkeley group succeeded in tracing the relaxation dynamics of resonantly excited pure He nanodroplets [5]. By imaging the emitted electrons [63,64,65] and ions [65, 196, 197] following excitation of the 1s4p-correlated state of the droplet (\(h\nu = 23.6\pm 0.2\) eV), intra-band and inter-band relaxation into lower lying droplet states was inferred, leading to the expulsion of free He\(^*\) Rydberg atoms. Specifically, He atoms in orbitally aligned 1s4p-states and in unaligned 1s3d states were found to be the dominant fragments. The ejection timescales of atoms in 1s4p and 1s3d-states were \(\lesssim 120\) fs and \(\approx 220\) fs, respectively. The component of very low-energy electrons associated with droplet autoionization was observed with a rise time of 2–3 ps.
With the advent of intense XUV and X-ray radiation sources provided by free-electron lasers (FELs), it has become possible to excite, ionize, and even directly image He nanodroplets by intense XUV or X-ray pulses [101, 198,199,200]. In the following we discuss recent experiments that systematically studied the relaxation of He nanodroplets resonantly excited into the lowest absorption band using tunable XUV-FEL pulses [66]. Owing to the use of UV probe pulses (3\(^{rd}\) harmonic of the Ti:Sa laser), all final states of the relaxation could be detected. The He droplets in the 1s2p-correlated state was found to undergo ultrafast interband relaxation to the 1s2s state. Subsequently, the excitation localizes by forming a bubble around a localized excited atom, He\(^*\), in either of the two metastable 1s2s \(^1\) \(S\) or \(^3\) \(S\) states. Eventually, the He\(^*\) is transported to the droplet surface where it is ejected or where it resides in a dimple prior to forming a He\(^*_2\) excimer [173]. The interpretation of the measured high-resolution TRPES is supported by TDDFT simulations carried out by the group of M. Barranco. They essentially confirm a three-step relaxation process: Ultrafast electron localization, electronic relaxation into metastable states, and the formation of a bubble, which eventually bursts at the droplet surface, thereby ejecting a single excited He atom.

(a) XUV pump—UV probe scheme used for TRPES of 1s4p-excited pure He nanodroplets. (b) Snapshot of the 2D He density profile obtained from time-dependent density-function simulations. Adapted from [66], published under a CC BY 4.0 license. (c) Transient photoelectron spectra. All features at delay times \(>0.3~\)ps are due to two-photon ionization by the 3.1 eV-probe pulse or autoionization (AI). Based on results reported in [201]
Figure 10.14a and b schematically depict the pump-probe scheme used to excite pure He nanodroplets into the 1s4p \(^1P\) state at \(h\nu =23.8\) eV. The tunable XUV pulse is generated by the seeded FEL FERMI, Trieste [202]. The relaxation dynamics is probed by TRPES using a time-delayed near-UV pulse with a photon energy \(h\nu ' = 3.1~\)eV. The grey shaded area in (a) depicts the absorption spectrum of medium-sized He nanodroplets measured by fluorescence emission [203]. The pump and probe photons are represented as red and blue vertical arrows, respectively. The dotted curved arrows indicate the relaxation into lower excited states of the droplet or free atoms.
Typical photoelectron spectra recorded for pump-probe delays up to 150 ps are shown in Fig. 10.14c [201]. The bright red dot at zero delay and electron energy \(\approx 2.3\) eV is due to 1+1 resonant two-photon ionization of the droplet 1s4p excitation, whereas all features at delays \(>0.3\) ps are due to 1+2 three-photon ionization as the excited-state population relaxes into 1s2s, p states within \(\lesssim 1~\)ps. Thereafter, the population accumulates in the metastable 1s2\(s\) \(\,^{1,\,3}\) \(S\) atomic states. The 1s2\(s\) \(\,^{3}\) \(S\) metastable triplet state is most likely formed by electron-ion recombination following droplet autoionization [192, 195]. The low-energy component in the electron spectrum is due to He-droplet autoionization [185]. The subsequent relaxation of the 1s2s states proceeds as in the aforementioned case of direct optical excitation [66]. Thus, despite the extremely weak binding of the He atoms in the droplets and the superfluid nature thereof, energy dissipation is very efficient; up to 4 eV of electron energy is dissipated within 1 ps by intraband and interband relaxations, i. e. the coupling of electronic and nanofluid nuclear degrees of freedom [66]. Note that the electron spectra contain replicas of the discussed features at multiples of the probe photon energy (not shown). These peaks are due to above-threshold ionization (ATI) induced by the absorption of multiple probe photons by helium nanodroplets containing several electronically excited states. When comparing to ATI electron spectra of excited helium atoms, ATI is found to be drastically enhanced in excited helium nanodroplets. The enhancement of ATI in multiply excited helium nanodroplets is attributed to laser-assisted electron scattering and a collective coupling between the excited helium atoms within the droplets [204].
In future experiments and model calculations, the initial step of the localization of the droplet excitation on one individual atomic center should be studied in more detail. At present, neither the range of delocalization, nor the time scale of the collapse of the initial delocalized state is known. Only for small He clusters has the vibronic relaxation dynamics been addressed theoretically [205]. Shorter pulses, as provided by XUV attosecond sources, will be instrumental for resolving this type of excitonic dynamics in He nanodroplets and in other nanoclusters [206].
10.5.2 Interatomic Coulombic Decay Processes in Doped Helium Nanodroplets
A number of indirect ionization processes have been evidenced in recent synchrotron studies, all of which are related to interatomic Coulombic decay (ICD) [207,208,209]. This term, first introduced by Cederbaum et al. in 1997 [210], subsumes various autoionization channels occurring in weakly bonded matter, in which not only the initially excited state, but also neighbouring atoms or molecules take part. ICD has mostly been studied using rare-gas dimers and clusters as model systems, but more relevant condensed phase systems such as liquid water comes more and more to the fore, in particular in view of the possible relevance of ICD for radiation damage in biological matter [211,212,213].
In He nanodroplets, these processes rely on the interatomic transfer of charge or energy within the droplet [79, 179, 214] or between the excited or ionized He droplet and a dopant particle [82,83,84, 187,188,189]. Some of these processes lead to efficient double ionization of the He droplet or the dopant following absorption of a single photon [80, 86, 184]. Recent experiments at the XUV FEL FERMI, Trieste, further extended these works to collective autoionization processes of multiply excited He nanodroplets irradiated by intense, resonant XUV pulses [215,216,217,218]. Pure and doped He nanodroplets have also attracted the interest of theoreticians as testbeds for ICD investigations [219,220,221].
When only a few excitations are present in one He droplet, autoionization proceeds according to the ICD mechanism He\(^*\) + He\(^*\rightarrow \) He + He\(^+\) + \(\mathrm {e}^-_\mathrm {ICD}\), first proposed by Kuleff et al. for the neon dimer [222]. The ICD electrons are created with a characteristic energy around 16 eV, as this process predominantly involves pairs of 1s2s-relaxed He\(^*\) located at short interatomic separation. TDDFT simulations indicate that the ICD rate is enhanced by the merger of the bubbles around the two adjacent He\(^*\), thereby pushing them even closer together [99].
Indeed, electron spectra recorded for multiply excited He nanodroplets display a clear peak at 16.6 eV, see Fig. 10.15a. Note that for increasing XUV intensity, this peak broadens and shifts towards lower energy. In addition, a low-energy component gains in intensity, which is indicative for thermal electron emission. Both features mark the transition of the multiply-excited He nanodroplet into a nanoplasma, which in this case is induced by collective autoionization [215, 218]. The collective Coulomb potential of the evolving nanoplasma tends to down shift the energy of emitted electrons [53].
To directly measure the ICD rate, the same pump-probe scheme as described before was applied for slightly higher pulse energies [Fig. 10.14a]. By photoionizing the He\(^*\) with the probe pulse, the population of He\(^*\) pairs was depleted, thereby interrupting the ICD process. Indeed, the yield of ICD electrons becomes minimal around a delay of 200 fs, while the yield of electrons emitted by photoionizing He\(^*\) reaches a maximum, see Fig. 10.15b. From the subsequent rise of the ICD signal the time scale of ICD in this system was deduced by comparing with a simple Monte-Carlo simulations [solid lines in Fig. 10.15b]. ICD was found to be surprisingly fast (400-900 fs) and only weakly depended on the initial number of excitations per He droplets, which were controlled by the XUV intensity and the He droplet size [99]. This counterintuitive result is rationalized by the relatively strong interatomic attraction acting between two He\(^*\) and the merger of bubbles around two interacting He\(^*\). The strong distance dependencies of these two effects makes the ICD rate extremely sensitive to the initial separation of the He\(^*\) pairs; those pairs with small initial separations \(\lesssim 10\) Å decay very effectively, whereas all other He\(^*\) are likely to be ejected out of the droplets before getting sufficiently close to one another to decay by ICD.
In summary, He nanodroplets are attractive targets for studying ICD processes as they bridge the gap between van der Waals molecules and condensed phase systems. Both homogeneous and heterogeneous ICD processes have been evidenced in pure and doped He droplets, respectively. Using intense ultrashort XUV-FEL pulses, the dynamics of a resonant ICD process was measured for the first time in a condensed-phase system using He nanodroplets. The unexpectedly short ICD time was rationalized by the peculiar quantum fluid dynamics of He droplets. In future experiments it appears promising to apply the technique of pump-probe depletion of excited-state populations for elucidating the dynamics of other types of ICD processes, including those that involve dopant atoms and molecules.

(a) Electron spectra recorded for He nanodroplets irradated by resonant XUV pulses at variable intensity. The peak around 16 eV indicates ICD of pairs of He\(^*\). Peak broadening at increasing intensity is due to the formation of a collective Coulomb potential as the droplet evolves into a nanoplasma. Based on results reported in [218]. (b) Yields of electrons created by photoionization (PI) of He\(^*\) and of ICD electrons as a function of XUV-pump and UV-probe delay. The minimum in the ICD trace is due to the transient depletion of He\(^*\), thereby suppressing ICD. Based on results reported in [99]
10.5.3 Dynamics of Helium Nanoplasmas
Helium nanodroplets are mostly used for isolating molecules at low temperature in a transparent and extremely inert environment. The particularly favourable properties of He droplets originate in the extremely high excitation and ionisation energy in combination with the extremely low droplet temperature and the resulting superfluid state. These properties make He droplets the ‘ideal spectroscopic matrix’ [3, 67]. In contrast, doped He nanodroplets can turn into a highly reactive medium, a so-called nanoplasma, when illuminated by intense (\(\gtrsim 10^{15}\) Wcm\(^{-2}\)) NIR laser pulses. Alternatively, multiple excitation by intense resonant XUV pulses also leads to the formation of a nanoplasma due to collective autoionization—a combination of ICD and inelastic scattering processes [215,216,217,218].
At first glance, He droplets appear less suited for studying nanoplasmas due to the high threshold intensity needed for singly ionizing He at 800 nm wavelength (\(1.5\times 10^{15}\) Wcm\(^{-2}\) [223]), and due to the small number of two electrons each He atom can at most contribute to building up a nanoplasma. However, He droplets doped with heavier species have recently revealed a diverse strong-field ionization dynamics resulting from the extremely large differences in ionization energies of the dopants and the He host medium. The controlled location of dopants inside or at the surface of the droplets adds another control parameter for nanoplasma ignition [224].
Initiated by tunnel-ionization of the dopant atoms acting as seeds, a He nanodroplet evolves into a nanoplasma, a highly ionized collective state, which can greatly enhance ionisation and fragmentation of the embedded dopants [47, 49, 50, 52, 225]. Alternatively, an ionizing XUV pump pulse preceding the driving NIR pulse can be used to ignite the nanoplasma, as recently demonstrated for pure argon clusters [226]. The intense NIR probe pulse acts as a strong time-varying external driving field which forces the seed electrons into oscillatory motion within the cluster. The resulting electron-impact ionization in the field of the created ionic cores enhances the build-up of a confined plasma-like state. During this laser-driven ionization process, a large fraction of electrons released from their parent atoms remain trapped in the space charge potential of the cluster (inner ionisation). The critical phase of light-matter interaction sets in as the plasma expands and the dipolar eigenfrequency of the plasma (‘plasmon resonance’) meets the frequency of the driving laser field [227, 228]. Under such resonance conditions, the nanoplasma becomes highly light absorbing. Consequently, the nanoplasma heats up dramatically and emits electrons and ions (outer ionisation). Very high ion charge states, electron energies up to multi-keV and ion kinetic energies up to MeV, and even XUV and X-ray photons have been detected [227, 228].
The first strong-field ionization experiments were carried out by the Rostock group using metal cluster-doped He nanodroplets. The focus was mainly on the charging of the embedded metal cluster rather than on the dynamics of the He nanoplasma. Single intense laser pulses of variable duration as well as dual pulses were used, and soon a significant influence of the He environment on the ionization dynamics of the metal core was realized [45, 229]. The condition for resonant charging was reached earlier in time than for the bare metal cluster, due to more efficient initial non-resonant charging of the metal core in the presence of the He environment that supplied additional electrons generated by electron-impact ionization of He shell. This lead to a faster expansion and thus to an earlier resonant matching of the plasmon and the photon energies. As a further consequence of the metal–He interaction in large droplets, caging of fragments was observed, which induced the reaggregation of the metal clusters [45].

(a) Yields of He ions as a function of delay between two intense NIR pulses of \(\approx 10\) fs duration. The mean droplet size was 15, 000 He atoms, and the mean number of Xe dopants is 15; based on results reported in [51]. (b) He ion yields for He droplets doped with about 30 Ar atoms as a function of the delay between a soft X-ray (250 eV) pump pulse and an intense NIR probe pulse
The active role of the He shell in the strong-field ionization process of doped He nanodroplets was confirmed by classical molecular dynamics (MD) simulations [47,48,49, 51, 224, 225]. A direct manifestation of the strong dopant-He coupling in the nanoplasma state is the observed efficient charging of Xe dopants up to Xe\(^{21+}\), by far exceeding the charge states reached for free Xe atoms or Xe clusters of the size of the dopant cluster at the used NIR intensities \(\approx 10^{15}\) Wcm\(^{-2}\) [52, 225]. The delay-dependent nanoplasma absorption and the electron energies were predicted to feature two maxima due to distinct resonance conditions of the dopant and the He nanoplasma components which are met at different times in the course of the expansion [47, 48].
In NIR pump-probe experiments, a plasmon resonance feature was clearly visible in the transient He\(^+\) and He\(^{2+}\) ion yields [51], see Fig. 10.16a, and in TR-PES [52]. The maximum shifted from a delay time of 100 fs for small He nanodroplets, He\(_N\), \(N\approx 6000\), to about 500 fs for \(N\approx 15,000\), in good agreement with MD simulations [51]. The efficiency of the dual pulse scheme for igniting and driving a nanoplasma in doped He nanodroplets was confirmed by applying an optimal control scheme to enhance the strong-field induced emission of highly charged atomic ions from embedded silver clusters [46].
More detailed insight into the dynamics of a NIR-ignited He nanoplasma was recently obtained by following in time the energy of Auger electrons emitted by a correlated electronic decay process akin to ICD inside the nanoplasma [53]. Similar correlated decay processes have recently been observed for nanoplasmas induced in heavier rare gas clusters [230, 231]. The delay-dependent shifting of Auger electron energies and above-threshold ionization (ATI) peaks by more than 15 eV reflects the evolution of the collective Coulomb potential created by the He nanoplasma through electron emission on the time scale of tens of ps. Single-shot electron velocity-map images of He nanoplasmas display large variety of signal types, most crucially depending on the cluster size [232]. The common feature is a two-component distribution for each single-cluster event: a bright inner part with nearly circular shape corresponding to electron energies up to a few eV, surrounded by an extended background of more energetic electrons.
In a recent experiment carried out using the FEL FLASH at DESY in Hamburg, a nanoplasma was ignited by irradiating doped He nanodroplets with soft x-ray (\(h\nu =250\) eV) pump pulses and time-delayed NIR probe pulses [233]. Fig. 10.16b shows the measured He\(^+\) and He\(^{2+}\) ion yields. In contrast to the experiments that employ NIR dual pulses, here the X-ray pump pulse selectively inner-shell ionized the dopant cluster owing to the much larger absorption cross section of the dopants (Ar, Kr, Xe) compared to He. Classical MD simulations indicated that the pronounced maximum of the He ion yields at a delay of about 200 fs was partly due to the plasmon resonance, and partly to electron migration from the He shell to the highly charged dopant-cluster core leading to a transient increase of the total number of quasi-free electrons present in the cluster volume due to electron-He collisions. The MD simulations, which reproduced the experimental pump-probe curves, also showed that the expansion of an X-ray-ionized Ar cluster embedded in a He nanodroplet is strongly damped compared to a free Ar cluster of the same size. Thus, He droplets act as efficient tampers that slow down the explosion of embedded nanostructures, a property that could be exploited for improving coherent diffraction images [234].
In another recent FEL-based experiment, the dynamics of strong-field induced nanoplasmas in He droplets were probed using single-shot, single-particle fs time-resolved X-ray coherent diffractive imaging (CDI) at the Linac Coherent Light Source (LCLS) [235]. NIR-induced nanoplasma formation and subsequent droplet evolution were probed by delayed X-rays pulses (\(\approx 100\) fs, \(h\nu '=600\) eV). Delay-dependent CDI patterns revealed distinct dynamics evolving on multiple timescales.
To summarize this section, a He nanodroplet can be turned from a weakly-interacting cryo-matrix into a highly charged, highly reactive nanoplasma by irradiation by intense NIR or XUV pulses. The strong-field ionization dynamics turns out to be extremely non-linear and highly sensitive to the presence of dopants owing to the large difference in ionization energies between dopants and the He host droplet. The characteristic pump-probe dynamics is a time-delayed absorption resonance associated with the evolution of a collective plasmon resonance. Still open questions pertain to the mechanisms and dynamics of the early phase of nanoplasma ignition and the late stage of recombination of electrons and ions during the expansion of the nanoplasma. In the latter phase, highly excited atoms and ions are populated which can in turn interact and decay by ICD-like processes. Furthermore, the enhanced emission of highly directional energetic electrons by plasmonic enhancement effects [236, 237], as recently observed with heavier rare-gas clusters, might be efficient for He nanodroplets as well. The property of He nanodroplets to act as a tamper that protects embedded molecules and nanostructures against ultrafast charging and fragmentation makes them interesting for single-shot X-ray coherent-diffraction imaging, a new technique that bears enormous potential for bio-molecular imaging and nanoscience [238, 239].
10.6 Coherent Multidimensional Spectroscopy in Helium Nanodroplets
Regarding the ultrafast spectroscopy concepts applied to helium nanodroplets, we have so far discussed time-resolved photoion and photoelectron spectroscopy. In these experiments, the attainable time and frequency resolution is directly given by the duration and spectral width of the pump and probe pulses. Furthermore, a well-defined phase relation between pump and probe pulses is not required which simplifies the demands on the optical setup.
This is in contrast to ultrafast coherent control and quantum interference spectroscopy methods [240, 241]. Here, phase-locked pulse sequences are applied and the quantum interference between different excitation pathways is probed or controlled. While coherent control is a topic of high interest in many fields [242], the focus of this contribution lies on spectroscopic applications. Quantum interference spectroscopy bears the advantages of a high time-frequency resolution as well as the capability to selectively probe specific signal contributions by appropriate design of the pulse sequences. Examples are the detection of multiple-quantum coherences which provide a highly sensitive probe for inter-particle interactions [37, 243] or photon-echos giving insight into ensemble inhomogeneities [244]. Established methods involve WP interferometry and coherent multidimensional spectroscopy (CMDS).
10.6.1 Spectroscopic Concepts of Wave Packet Interferometry and Coherent Multidimensional Spectroscopy
The concept of WP interferometry has been applied in many different experiments to probe and control the dynamics of various quantum systems, as discussed in two review articles [241, 245]. The terms WP interferometry and quantum interference spectroscopy are often used equivalently. Depending on the investigated system, the experiment may be more intuitively described by the interference of WPs or quantum pathways excited in the system. In the following we will apply the quantum pathway picture. Figure 10.17 shows the basic concept. Pump and probe pulses each excite a specific pathway in the system (Fig. 10.17a) leading to the same final state population. Since both pathways propagate along different states during the pump-probe delay \(\tau \), they accumulate different phases, giving rise to an alternating constructive and destructive interference pattern in the signal with a periodicity of \(2\pi / (\omega _e-\omega _g)\) (Fig. 10.17b). At the same time the overall decay of the signal amplitude reflects dephasing and decoherence effects. Hence, the fringe pattern contains the information of an absorption spectrum, which is obtained by a Fourier transform of the signal (Fig. 10.17c). With this approach, the spectral resolution is given by the length of the time-domain signal and is thus decoupled from the spectral width of the laser spectrum. As such, state-resolved information can be gained even for broadband pulse spectra covering many resonances in the system.

Wave packet interferometry scheme. (a) Pump-probe pulses excite two different quantum pathways leading to the same final state. The interference between the pathways depends on the different phase factors accumulated during the pump-probe delay \(\tau \). (b) Schematic time-domain signal for the case of six excited states, as in (a). Oscillations reflect the constructive/destructive pathway interference. (c) Fourier transform of the signal, yielding the absorption spectrum of the system (blue) along with the laser spectrum (gray)
There is a conceptual similarity to the detection of coherent vibrational WP oscillations discussed in several examples in Sect. 10.4. In these experiments, the propagation of vibrational wave packets along an electronic potential energy surface is probed. A Fourier analysis of the WP beating provides in analogy spectral information beyond the frequency resolution given by the femtosecond pulses. This scheme is primarily sensitive to the system’s vibrational and rotational degrees of freedom, whereas WP interferometry also maps the electronic properties of the system including vibrational-electronic couplings.
In terms of dynamics, WP interferometry provides limited information. The method can only monitor changes of WPs within the Frank-Condon window between the ground and excited state. Processes such as the decay of WPs into new states or the transient change of the potential energy surface due to chemical reactions or perturbations by the environment may be hidden. In contrast, non-interferometric pump-probe experiments as discussed further above can offer a much enhanced observation window and dynamics can be monitored over a large parameter space. The situation is different if additional probe laser pulses are added to the WP interferometry scheme. These pulses may then probe the state of the system outside of the ground-excited state Frank-Condon window and thus extend the observation window for the system dynamics.
CMDS is an example for a particular powerful nonlinear extension of WP interferometry. This method greatly improves the information content deductible from ultrafast spectroscopy experiments [246,247,248]. CMDS combines the resolution advantage of WP interferometry with the extended sensitivity to dynamics known from classical pump-probe spectroscopy. The result is a nonlinear spectroscopy scheme which features several spectroscopic advantages not simultaneously present in any other technique. These include the direct spectroscopic access to couplings and relaxation pathways in the probed sample, the high time-frequency resolution as well as the capability to reveal system-bath interactions and inhomogeneities in real-time.

The principle of 2D spectroscopy. (a) Pulse sequence exciting the sample. Pulses 1, 2 and 3, 4 form phase-locked pulse pairs to perform two correlated WP interferometry experiments (WPI 1,2), whereas the time evolution of the system is probed in between the pulse pairs. (b) Model energy-level system along with a selection of possible nonlinear signals induced by the four-pulse sequence. SE: stimulated emission, GSB: ground-state bleach, ESA: excited-state absorption. (c) 2D frequency spectrum obtained from a 2D Fourier transform of the signal. Peaks A and B on the diagonal represent the \( \left| {g} \right\rangle \leftrightarrow \left| {a} \right\rangle , \left| {b} \right\rangle \) resonances. Their 2D lineshapes reflect the inhomogeneous and homogeneous linewidth along the diagonal and antidiagonal, respectively. Peak C denotes an excited state absorption from \( \left| {a} \right\rangle \) to the higher-lying state \( \left| {c} \right\rangle \). AB and BA denote cross peaks which reflect couplings between states \( \left| {a} \right\rangle \) and \( \left| {b} \right\rangle \). (d) Projection of the 2D spectrum onto the x-axis, resulting in a one-dimensional spectrum as it would be measured with conventional absorption spectroscopy
There are many variants of CMDS in terms of spectral range, detection scheme and number of excitation pulses [249]. A detailed description of all aspects is beyond the scope of this book. Here, we will restrict our discussion on population-detected two-dimensional (2D) spectroscopy in the VIS spectral domain, which probes electronic transitions. This is the only variant so far applied to helium nanodroplet samples [250] and can be readily explained in the framework of WP interference.
In the 2D version of CMDS, the method basically correlates two WP interferometry measurements, each performed by a phase-locked pulse pair (Fig. 10.18a). Performing a 2D Fourier transform of the data with respect to the time delays \(\tau \) (between pulse 1 and 2) and t (between 3, 4), yields a 2D frequency-correlation spectrum (Fig. 10.18c), hence the name multidimensional spectroscopy. These spectra show the frequency-resolved absorption (x-axis) directly correlated to the frequency-resolved/detection (y-axis) of the sample. In addition, the time delay T in between the two WP interferometry experiments probes the time evolution of the system. Due to the underlying interferometric measurement scheme a high time-frequency resolution is achieved which automatically adapts to the time scales and spectral linewidths of the system [251].
The interaction of the quantum system with the four-pulse sequence gives rise to a multitude of nonlinaer signals (examples shown in Fig. 10.18b). To categorize the signals, it is convenient to adapt the common terminology from transient absorption spectroscopy: stimulated emission (SE) and excited state absorption (ESA) signals both probe the excited state, whereas ground state bleach (GSB) signals probe the ground state properties. ESA pathways involve transitions to higher-lying states and contribute with negative amplitude to the spectra, while SE and GSB contribute both with positive amplitudeFootnote 1.
The 2D spectra can be interpreted as follows: (i) spectral peaks on the diagonal reflect the linear absorption spectrum, however with the additional information of 2D line shapes, directly dissecting the inhomogeneous (along diagonal) and homogeneous (along anti-diagonal) broadening in the system. Hence, time-resolved information about the system-bath interactions can be directly gained from the line shape analysis [244, 254]. (ii) Off-diagonal peaks (termed cross peaks), indicate couplings between different states and energy relaxation (peaks AB and BA in Fig.10.18c). This greatly simplifies the identification of relaxation pathways and allows to follow the relaxation dynamics in real-time [255,256,257]. (iii) Transitions to higher-lying states (ESA) contribute with negative amplitude and are thus readily identified (peak C in Fig.10.18c). All this information is difficult to deduce from one-dimensional spectroscopy, where 2D lineshape information is not available and cross peaks overlap spectrally with diagonal features. This is schematically expressed by a projection of the 2D spectrum onto a single axis (Fig. 10.18d).
CMDS has been so far mainly applied in the condensed phase where the method has achieved considerable success, as highlighted in several review articles [246,247,248, 254, 258, 259]. Certainly, the advantages of CMDS are also very beneficial in the gas phase, in particular in more complex gas phase samples, such as species embedded in helium nanodroplets offering the study of intra/inter-molecular dynamics and peculiar system-bath interactions. However, an extension of CMDS to the gas phase has been so far vastly impeded by the difficult signal-to-noise challenge due to the low sample densities predominant in gas phase experiments. Over the last years, Bruder and Stienkemeier et al. have developed a specialized experimental approach [250] to solve this issue, which is outlined below.
10.6.2 Resolving the Experimental Challenges
The implementation of CMDS experiments faces two major signal-to-noise challenges. On the one hand, CMDS is a nonlinear spectroscopy scheme. Thus, a high dynamic range is required to uncover the weak nonlinear signals from dominant linear signals and general background noise. To give some numbers, in CMDS experiments of doped helium nanodroplet species, background signals are typically one to three orders of magnitude larger than the CMDS signal. On the other hand, the underlying interferometric measurement scheme adds an additional noise source stemming from phase jitter between the optical pulses. Sub-cycle phase stability is demanded, which implies a reduction of optical pathlength fluctuations to \(< \lambda /50\) [260], a value that is very hard to achieve with conventional interferometers. This requirement also applies to coherent control and WP interferometry experiments, however, in CMDS it scales with higher order due to the nonlinear WP interferometry scheme.
Several techniques were developed which solve these issues in the condensed phase, as summarized in Ref. [249]. Out of these methods, the phase modulation technique developed by Marcus and coworkers [261, 262] is most suitable for the application in helium droplet beam experiments for several reasons. It provides efficient phase stabilization to reduce phase jitter, extraordinary sensitivity by incorporating lock-in detection and it can be combined with efficient photoionization detection schemes [37, 243]. It is also compatible with high power, high repetition rate (\(> 100\,\) kHz) laser systems which improve statistics while avoiding saturation of optical transitions and detectors.

Phase modulation scheme to improve phase stability and sensitivity in 2D spectroscopy experiments. A sequence of four laser pulses excites the sample. Four acousto-optical modulators (AOMs) shift the carrier envelope phase \(\phi _i\) of the individual laser pulses in each laser cycle by a well-defined value, which results in a continuous modulation of the quantum interference signals. A lock-in amplifier is used for demodulation. An optical interference signal is coupled-out from the optical setup for referencing the lock-in demodulation process
The phase modulation technique is shown in Fig. 10.19. Precise phase beatings are imprinted in the optical pulse sequence which transfers to a characteristic amplitude modulation of the interference signals in the time domain. Background signals are not modulated or appear at other modulation frequencies, enabling selective, highly efficient lock-in amplification of the interference signals. Moreover, heterodyne detection with an optical interference signal is implemented which leads to a cancellation of the optical phase jitter and rotating frame detection. The latter results in a downshift of quantum interference frequencies by several orders of magnitude.

Performance comparison between phase-modulated and conventional WP interferometry. Time domain WP interferometry signals obtained with the phase modulation technique (a) and without (b) showing the first 4 ps of the signal. (c), (d) Respective Fourier transforms of the full data set (50 ps length). In (c), the scales on the top/bottom show the rotating frame and the up-shifted frequency axis, respectively. Dashed vertical lines indicate the atomic resonances: \(5S_{1/2}\rightarrow 5P_{3/2}\) and \(5P_{3/2}\rightarrow 5D_{3/2,5/2}\).
The advantages of the phase modulation technique are demonstrated in Fig. 10.20, showing a comparison between conventional and phase-modulated WP interferometry of a rubidium-doped helium nanodroplet sample [37]. With the spectral bandwidth of the femtosecond laser, the \(5S_{1/2}\rightarrow 5P_{3/2}\) (D\(_2\) line) as well as the \(5P_{3/2}\rightarrow 5D_{5/2,3/2}\) atomic transitions in Rb are resonantly excited and detected by 1+2 REMPI combined with mass-resolved ion detection. The quantum interference signals in the time domain exhibit rapid oscillations corresponding to the constructive and destructive interference of excitation pathways induced as a function of the pump-probe delay. As a striking feature, the oscillation period in the phase-modulated WP interferometry measurements is more than a factor of 100 larger compared to the conventional technique (Fig. 10.20a,b), which is due to the rotating frame detection. Hence, much sparser sampling of the signal is possible while deducing the same amount of information.
A Fourier transform yields the absorption spectrum revealing a drastic difference in the signal-to-noise performance of both experiments. In the conventional WP interferometry, the atomic resonances can only be qualitatively identified due to the strong phase jitter on the signal. In contrast, the phase modulation approach delivers a highly resolved spectrum with excellent signal-to-noise ratio. Obviously, the frequency spectrum precisely resembles the signature of free gas-phase Rb atoms without any sign of droplet-induced perturbations, implying that the current experiment is predominantly sensitive to already desorbed atoms. As mentioned above, this is explained by the high laser repetition rate (80 MHz), supporting the desorption of the atoms and subsequent probing in the gas phase in the same experiment. Quantum interference experiments with lower repetition rate are presented further below in Sect. 10.6.4. In summary, the phase modulation experiment in Fig. 10.20 featuring a great signal-to-noise improvement for highly dilute helium droplet samples marks an important milestone and opened-up the door for CMDS experiments of helium nanodroplet samples.
10.6.3 High Resolution Wave Packet Interferometry
The previous example indicates the prospective of using the phase modulation technique for high resolution spectroscopy. In WP interferometry, the frequency-resolution limit \(\Delta \nu \) of the experimental apparatus is directly connected with the scanned pump-probe delay range \(\Delta \tau _\mathrm {range}\) by \(\Delta \nu = 1 / \Delta \tau _\mathrm {range}\). With mechanical delay stages scanning ranges of \(<2\) ns are realistic [36] which corresponds to a resolution limit of 500 MHz. Frequency-comb-based approaches can in principle extend the scanning range to reach a frequency resolution of \(\approx 100\,\)MHz [263]. This option however rises considerably the demands on the laser source and is not further discussed here. In general, much higher spectral resolution is achieved with continuous-wave lasers. However, the advantage of WP interferometry is a flexible time-frequency resolution to study spectral and temporal aspects of the target system. Moreover, the intense femtosecond pulses improve the signal strength in multiphoton probing schemes. This applies to photoionization schemes but also to some exotic molecules such as alkali-helium (AkHe) exciplexes.

As already discussed above (Sect. 10.2.1, 10.3.1), AkHe molecules exhibit an anti-bonding ground state whereas some excited electronic states support bound configurations. As an example, the Rb atomic levels along with the RbHe pair potentials are given in Fig. 10.21. Upon electronic excitation of the AkHe\(_\mathrm {N}\) system, exciplexes may form and desorb from the droplet. Desorbed metastable AkHe\(_n\) complexes up to \(n\le 4\) have been observed with mass spectrometry [10]. The formation, probing and detection thus requires a multiphoton experiment which is highly favorable with femtosecond laser sources, making WP interferometry the ideal spectroscopic tool to study the level structure of these systems.
Despite many experimental and theoretical studies devoted to these peculiar molecules [8,9,10, 13, 14, 21, 24, 126, 127, 264, 265] some questions about the formation mechanism and associated formation times remain unsolved[14, 21, 127]. Moreover, until recently high resolution spectral data has not been available, partly due to the difficult accessibility by standard absorption/emission spectroscopy [8, 9] and the limited resolution given by other techniques [24, 126, 127]. The first highly resolved vibronic spectrum of an AkHe molecule has been obtained with the phase modulation technique [37].
In the RbHe molecule, WPs between the electronic states correlated to the 5P and 5D atomic asymptotes of rubidium were induced and probed via subsequent photoionization (Fig. 10.21). The resulting Fourier transform spectrum is shown in Fig. 10.22, revealing a clean, highly resolved vibronic spectrum with a resolution of 0.3 cm\(^{-1}\). In contrast to the vibrational WP studies (Sect. 10.4) probing purely vibrational modes, the experiment here detects vibronic resonances between different electronic states. The strong spin-oribit coupling in the RbHe system results in a complex manifold of many closely spaced electronic states which explains the highly structured spectrum in Fig. 10.22. Reasonable good agreement with a theoretical model is found [37] which is remarkable considering the high degree of spectral details and the experimental resolution being clearly beyond the precision of current models. The spectroscopic potential of the novel WP interferometry technique is shown by a comparison with previous experiments based on picosecond pulse shaping and conventional WP interferometry. While good agreement is found between the different experimental techniques, the new method provides a factor of \(\ge 10\) higher resolution. This example hence underlines the advantage of femtosecond spectroscopy techniques in the spectral study of metastable molecules and provides new benchmark spectroscopic data for the development of ab-initio methods.

High resolution Rb\(^*\)He spectrum recorded with phase-modulated WP interferometry (black) [37] along with femtosecond-pump, picosecond-probe photoionization measurements (red) [127]. Beat frequencies deduced from conventional WP interferometry (blue) [24] are shown on the top. Vertical dashed lines indicate the atomic \(5P_{3/2}\rightarrow 5D_{3/2,5/2}\) transitions. The asterix marks an artificial peak coming from low frequency noise.
10.6.4 Ultrafast Droplet-Induced Coherence Decay in Alkali Dopants
The peculiarities of the alkali-helium droplet interaction have been already discussed. The repulsive character of most excited AkHe\(_N\) states induces a line-broadening in the order of 10–100 wavenumbers [125]. Accordingly, ground-excited state coherences are expected to decay on the order of 0.1–1 ps. Quantum interference spectroscopy should provide a real-time analysis of this process. In fact, time resolved quantum interference studies were among the first femtosecond experiments of doped droplet species [20]. In these early attempts, insufficient phase stability prohibited a Fourier analysis of the transient interference signals. Instead, indications about the droplet interaction were directly deduced from the coherence decay times which were in the order of few hundred femtoseconds for low excited states in KHe\(_N\).
With the novel phase-modulated WP interferometry technique a high resolution study of the decoherence process becomes possible. While the experiment in Fig. 10.20 had probed already desorbed Rb atoms and thus renders insensitive to droplet-induced dynamics, Fig. 10.23 shows data from the same target system using a modified experimental setup, now clearly revealing the ultrafast decoherence induced by the guest-host interaction [266]. The experimental modifications comprise of a lower laser repetition rate (80 MHz \(\rightarrow \) 200 kHz) and a separate, delayed ionization laser (\(\lambda =520\,\)nm, delay \(\sim \) ns) to ionize the species after full desorption. Moreover, very broad bandwidth femtosecond pulses (FWHM = 1600 cm\(^{-1}\)) are used to simultaneously cover the absorption lines of Rb atoms and Rb\(_2\), Rb\(_3\) molecules adsorbed to the droplet surface.

Droplet-induced decoherence in Rb atoms and Rb\(_2\), Rb\(_3\) molecules tracked by phase-modulated WP interferometry. (a) Pump-probe transient in the time domain, effusive atomic background is subtracted. The pronounced spike at 0 fs stems from the optical pump-probe cross-correlation mapped to the continuum by three-photon ionization. (b) Fourier transform. Labels indicate the excited resonances in the Rb molecules. Dashed vertical lines mark the atomic D\(_{1,2}\) transitions
The time domain signal reveals an extremely fast coherence decay within \(\approx 150\,\)fs, followed by a much weaker but persistent interference signal extending beyond delays of 1.5 ps. A Fourier transform uncovers a rich absorption spectrum showing the absorption bands of the Rb trimer: \(1\,^4A_2^{'} \rightarrow 2\,^4E^{'}\) at 11600 cm\(^{-1}\), the dimer: \(a\,^3\Sigma _u^+ \rightarrow 1\,^3\Pi _g\) at 13500 cm\(^{-1}\) and monomer: \(5s\,^2\Sigma _{1/2} \rightarrow 5p\,^2\Pi _{1/2, 3/2}\) at 12600 cm\(^{-1}\) and 12830 cm\(^{-1}\), \(5s\,^2\Sigma _{1/2} \rightarrow 5p\,^2\Sigma _{1/2}\) at 12850 cm\(^{-1}\). These features are in good agreement with previous steady-state absorption spectroscopy [9, 267, 268]. In particular, the monomer response now resembles the blue-shifted strongly broadened absorption profile characteristic for the pseudo-diatomic model [125]. The broad blue shoulder of the Rb\(_3\) resonance (11800–12600 cm\(^{-1}\)) was also observed in femtosecond absorption mass-spectrometry, where a clear correlation to the Rb\(_3^+\) ion yield was determined [25].
This example shows the sensitivity of WP interferometry to the guest-host interaction in doped droplet beam experiments and the capability to probe complex spectra extending over a broad spectral range and many resonances from different species. The experiment serves as precursor study for 2D spectroscopy on these samples. An extension to 2D spectroscopy facilitates the direct correlation of absorption and emission of each spectral feature/dopant species and allows to follow their dynamical evolution in real time as discussed below.

CMDS results for Rb\(_2\) and Rb\(_3\) molecules formed on the surface of helium nanodroplets. 2D spectra detected via photoelectrons (a), (b) and photoions (c) for different evolution times \(T=0\, , 200\, , 700\) fs as labeled. X-axis: absorption, y-axis: detection frequency. Labels indicate the molecular transitions and spin-orbit splittings. Two distinct ESA features (ESA\(_{1,2}\)) and a cross peak (CP) are marked, as well. (d) Ab-initio Rb\(_2\) potential energy curves and concluded photodynamics. Labels of probe-transitions correspond to the ones in (a-c). The helium perturbation on the \(0_g^+\) state is schematically drawn as dashed curve. (e) Time evolution of the ESA amplitudes reveal coherent vibrational WP oscillations with a phase shift of \(\pi \) between the traces.
10.6.5 Coherent Multidimensional Spectroscopy of Doped Helium Nanodroplets
The unique properties of 2D spectroscopy render it a powerful tool for the study of ultrafast dynamics and guest-host interactions in doped helium droplet samples. The latter effect is particularly pronounced for alkalis, which hence provide an ideal test system for 2D spectroscopy experiments. Figure 10.24 shows 2D spectroscopy data for rubidium-doped helium droplets. In these experiments, a four-pulse sequence induces the 2D signal (cf. Figs. 10.18, 10.19) which is detected via photoionization. The ionization step is performed either by an additional interaction with the fourth laser pulse or by a delayed fifth pulse and is combined either with electron (Fig. 10.24a,b) or ion detection (c). The different ionization and detection schemes explain the different appearance of peak amplitudes in the spectra. The 2D maps show clear, pronounced peaks well separated from the noise floor, which is remarkable considering the challenging signal-to-noise conditions in these experiments. These measurements constitute the first 2D spectroscopy study of isolated cold molecules [269].
The 2D spectra directly disclose the correlations between the absorption and emission of the Rb\(_2\) and Rb\(_3\) molecules, revealing various cross peaks and ESA signals, which were not observed in previous experiments. For the Rb\(_2\) molecule, exemplary the excitation and probing scheme is shown in Fig. 10.24d. The Rb\(_2\) data exhibits two strong ESA features (labeled ESA\(_1\), ESA\(_2\)) which extend into the complex Rb\(_2\) Rydberg manifold, featuring a high density of electronic states (not shown in Fig. 10.24d). Despite the complex level structure, some clear conclusions can be drawn with the help of 2D spectroscopy. The position of the ESA peaks along the detection axis show the spectral position of the Frank-Condon windows to the Rydberg states. At the same time, the coherent vibrational WP oscillations reflected in the ESA peaks (Fig. 10.24e) pin down the location of the Frank-Condon windows. From the clear \(\pi \)-phase shift between both traces the existence of two Frank-Condon windows located at the inner and outer turning point of the excited state potential, respectively, becomes apparent (see sketch Fig. 10.24d).
The 2D data also permits a refined interpretation of a Stokes shift observed in the Rb\(_2\) emission (cross peak red-shifted by 600 cm\(^{-1}\), labeled CP in Fig. 10.24c). This feature was previously interpreted as the emission from vibrationally relaxed free gas-phase Rb\(_2\) molecules [268]. In contrast, the high time-frequency resolution in the 2D spectroscopy experiment uncovers an ultrafast intra-molecular relaxation within \(< 100\) fs (not shown) into the outer potential well of the \(1\,^3\Pi _g\) state, catalyzed by the helium perturbation (sketched in Fig. 10.24d) [269].

Real-time observation of the helium surface repulsion. (a) Sketch of the Rb\(_3\)–He\(_N\) interaction potentials. Steps 1–3 show the repulsion of the helium density following the impulsive molecular excitation. The excited state relaxation process is traced by the SE signal. The GSB probes the ground state where no system-bath dynamics occur. (b) Time-evolution of the spectra showing the cut-out of the Rb\(_3\) \(1\,^4A_2^{'} \rightarrow 1\,^4A_{1,2}^{''}\) excitation. A clear dynamic Stokes shift (spectral splitting of SE and GSB signals) is visible, which converges to a constant red-shift of 150 cm\(^{-1}\) within \(\approx 2.5\,\)ps.
While the Rb\(_2\) molecule offered rich intra-molecular dynamics on femtosecond time scales, the Rb\(_3\) molecule serves as a sensitive probe for the dynamics of the quantum fluid droplet. Many of the above discussed ultrafast dynamics studies investigated guest-host interactions in doped droplets with the goal to deduce properties about the quantum fluid itself. To avoid less available femtosecond XUV light sources, often impurities are embedded as probes that are optically accessible. In time-resolved photoionization studies of impurities, the droplet response is inferred from the transient energy shift between the neutral excited and ionic state of the guest-host interaction potential (Fig. 10.8). 2D spectroscopy offers an alternative approach which probes the matrix-shift along the ground and excited states of the purely neutral interaction potential (Fig. 10.25a).
For the large inertia of mass of the Rb\(_3\) molecule, any short-time dynamics along the interaction coordinate may be solely attributed to the response of the helium density. Hence the molecule provides an ideal probe for the dynamics of the quantum fluid at the droplet surface. Upon impulsive excitation of the molecule with the femtosecond laser pulses, the helium density will repel and the system will relax along the interaction coordinate (Fig. 10.25a). The process can be followed in real time in the 2D spectroscopy data (Fig. 10.25b). Here, a pronounced dynamic Stokes shift is observable, which stems from the SE signal probing the system’s relaxation along the excited state of the interaction potential. The energy shift reaches an asymptotic value of \((150\pm 19)\,\mathrm {cm}^{-1}\) within \(\approx 2.5\) ps which marks the time-scale for the ultrafast rearrangement of the helium density to reach equilibrium. In comparison, the desorption of Rb atoms and molecules for the lowest excitations commonly takes place on much longer time scales (Sect. 10.3.1). A similar dynamic is expected for the Rb\(_2\) molecule, which is, however, covered by the persistent ESA\(_2\) peak.
These 2D spectroscopy experiments have demonstrated the power and added value of applying CMDS to doped helium droplet species. Ak molecules attached to He droplets have been extensively studied in recent years, both with high-resolution steady-state spectroscopy and time-resolved pump-probe experiments. Yet, the high time-frequency resolution and the ability to directly correlate absorption and emission spectra in 2D spectroscopy has still brought new insight into these systems which shows great promise for future studies on other systems. In particular, the spectra of many organic molecules dissolved inside helium nanodroplets show only weak perturbation [270, 271]. Hence, high-resolution 2D spectroscopy of organic compounds are at hand which would provide invaluable complementary information to condensed phase studies. Very recently, Bruder and Stienkemeier et al. have performed the first CMDS experiments of an organic molecule fully dissolved in helium droplets [272] which marks the highest resolution so far achieved in a molecular 2D spectrum and thus underlines this potential.
10.7 Conclusions and Outlook
The various ultrafast spectroscopy methods presented in this chapter have uncovered a rich variety of structural and electronic dynamics in pure and doped helium nanodroplets with unique features that are not found in other quantum systems. On the one hand, helium nanodroplets offer the possibility for high resolution studies of species dissolved in a weakly-perturbing environment which is in stark contrast to studies in the condensed phase. Naively, high spectral resolution is associated with continuous-wave laser spectroscopy. However, as has been discussed in this chapter, femtosecond laser pulses open-up the preparation and study of coherent WPs which can provide high spectral information through Fourier analysis with comparable resolution to steady-state methods. Moreover, the direct WP analysis in the time-domain provides access to phase information and decay processes which may not be accessible in steady-state spectroscopy. These aspects have been well demonstrated in the observation of extremely long-lived vibrational WPs in alkali molecules attached to the helium droplet surface (Sect. 10.4.1), the recurrence of vibrational revivals in ejected In\(_2\) molecules (Sect. 10.4.2) or in the identification of multiple ionization pathways from phase shifts in the WP motion (Sect. 10.6.5). Intense femtosecond pulses also open-up nonlinear multiphoton experiments which provide access to strong-field effects, to the study of higher-lying states, and the properties of metastable species. Examples were the formation and high-resolution study of metastable exciplex molecules (Sect. 10.6.3), or nanoplasmas ignited inside helium nanodroplets with remarkable efficiency (Sect. 10.5.3). These studies, however, also demonstrate that the interaction with high-intensity or/and high photon energy laser pulses in most cases leads to the deposition of large amounts of energy into almost all degrees of freedom, in this way wiping out the low-temperature quantum properties of superfluid droplets.
On the other hand, time-resolved experiments have provided insight into the dynamics of the droplets themselves. Novel coherent XUV light sources have for the first time enabled the direct study of the multifaceted relaxation dynamics inside helium nanodroplets in real-time (Sect. 10.5.1). Ultrafast electronic relaxation, bubble formation and ejection of metastable He atoms from the droplets were directly measured. Furthermore, insight into structural dynamics of the helium density have been gained from time-resolved studies with impurities embedded inside the droplets or attached to their surface. The experiments have uncovered a general behavior of the quantum fluid which can be categorized into a fast change of the helium solvation shell and a somewhat slower transport dynamics. The primary, fast response of the helium density in the local environment of the impurity (solvation shell) takes place on a few hundred femtoseconds to a few picoseconds as a response to the impulsive electronic excitation of the impurity. While this process is accurately predicted by theory (Sect. 10.2.6), only very recently TRPES (Sect. 10.3.2) and 2D spectroscopy experiments (Sect. 10.6.5) have provided the first experimental access to these dynamics. The experiments permit a comparison of the time scales for the helium repulsion inside and at the surface of the droplets, revealing a slower equilibration at the surface. While a different behavior of the quantum fluid inside the droplet compared to surface states is expected, the complex aspects of the dynamics with respect to the dopant properties and density modes of the droplet will require further experiments in this direction for more general conclusions.
The secondary transport dynamics often occur on a longer time scale in the picosecond to nanosecond domain. As a typical feature of helium nanodroplets, electronic excitation of impurities almost always triggers a propagation along the impurity-droplet interaction potential leading to the ejection of the dopant from the droplet or to the trapping of the dopant inside snowball structures in the droplet core. These dynamics directly manifest themselves in pump-probe measurements of alkali atoms and molecules which first desorb from the droplet surface when being resonantly excited, and then fall back into the droplet when being ionized (Sect. 10.3.1). The impurity transport and detachment may be accompanied by helium density oscillations which are observable in TRPES measurements (Sect. 10.3.2). Time-dependent density functional simulations nicely visualize the full evolution of the dopant-droplet system, and even reproduce the experimentally observed dynamics quantitatively (Sect. 10.2.6). In addition, the dynamics of the quantum fluid superimpose and interplay with the intra-molecular and inter-molecular dynamics of the impurities which may comprise of intra-molecular vibrational energy redistribution (IVR), spin and electronic relaxation as well as dissociation and charge transfer processes (Sect. 10.4.2, 10.3.1, 10.5.2, 10.6.5) and often take place on a comparable time scale as the dynamics of the helium density.
This interplay of dopant and helium bath dynamics makes helium nanodroplets a challenging, however at the same time, a fascinating target system for spectroscopic studies. While pure and doped helium droplets form an enclosed nanosystem which is still amenable to theoretical models, these systems feature intriguing aspects of fundamental molecular dynamics, system-bath interactions and unique quantum fluid properties as it is not found in any other system. The here discussed time-resolved studies have provided a first glimpse into the ultrafast dynamics of these systems and have founded the experimental and theoretical basis for further exploration. In this view, two major routes are identified which concern the vast synthesis abilities provided by helium nanodroplets as well as the rapid technological developments in XUV light sources and XUV spectroscopy methods.
First, the ability of He droplets to generate microsolvation environments [273, 274] will provide new options to the field of femtochemistry. Flexible pickup possibilities allow to design the environment around a molecule of interest in terms of number of solvent molecules and their interaction strength (polarizability, dipole moment, hydrogen bond). The advantages provided by time-domain spectroscopy, including coherence phenomena and phase information, can now be applied to these systems. In particular, building up the solvation shell piece-by-piece will allow to track ultrafast dynamics as the environmental conditions bridge from isolation to full solvation, shedding light on the gap between accurate gas-phase studies and real-world systems in solution. Furthermore, the repertoire of time-domain techniques ranging from pump-probe to multidimensional spectroscopy, can be applied to droplet-specific systems that have previously evaded time-domain investigations, including tailor-made complexes [275, 276], fragile agglomerates [11, 277], or highly reactive species [278]. For example, diverse combinations of donor-acceptor pairs can be prepared for charge-transfer studies, where the time-domain approach will be essential to disentangle nuclear and electronic dynamics. Another field of interest would be the investigation of exciton dynamics in molecular aggregates [279], including migration, fission and annihilation mechanisms. In view of fundamental photophysical reactions and the development of efficient photo-switches, the real-time study of isomerization inside the quantum fluid [280] and inside microsolvation environments would contribute new insights.
Second, the novel developments in ultrafast XUV light sources open up fascinating perspectives in the time-resolved study of pure and doped helium nanodroplets as they offer direct access to the optical properties of superfluid helium. The static and dynamics properties of helium droplets have been studied in recent years using all types of XUV light sources [64, 66, 184, 192, 215]. Still, questions remain, in particular, about collective phenomena such as correlated electronic decay processes observed in these systems. As one example, a variety of highly efficient ICD processes in doped and pure droplets have been evidenced in recent years. To capture the full kinematics and dynamics of such processes, covariance and coincidence detection methods are instrumental. With the advent of high-repetition-rate intense femtosecond lasers and XUV radiation sources, combining coincidence detection with femtosecond time-resolved spectroscopy of helium nanodroplets is in reach.
Furthermore, CMDS and related coherent nonlinear spectroscopy methods provide selective probes for collective properties [243] and variations in the local environment of many-body quantum systems [244, 254]. As such, XUV-CMDS experiments would shed new light on the inhomogeneity and many-body nature of the helium droplet absorption spectrum. Furthermore, with a transfer of coherent nonlinear methods to the X-ray domain, localized core resonances would be accessible, thus facilitating the study of dopant complexes inside helium droplets with unprecedented atomic sensitivity. Recently, XUV-WP interferometry of helium atoms was demonstrated which shows that the phase stability issue in XUV quantum interference experiments can be solved [281]. Moreover, coherent XUV and X-ray wave mixing experiments were established detecting nonlinear mixing signals with nanometer resolution and site-specificity [282,283,284]. These developments open-up the perspective for XUV and X-ray coherent nonlinear and even multidimensional spectroscopy experiments.
Yet, the most direct probing of the structural dynamics of nanoparticles is achieved through the new technique of X-ray single-shot coherent diffraction imaging (CDI) [198]. Currently, this technique relies on radiation provided by one of the few existing XUV and X-ray FEL facilities. However, tremendous progress is being made in generating intense and femtosecond and even attosecond pulses in the XUV and X-ray ranges [285]. New radiation sources such as high-harmonic generation based on high-power table-top femtosecond lasers will make it possible to directly visualize the structural dynamics of helium nanodroplets and other nanoparticles using pump-probe and possibly more sophisticated CDI schemes [235, 286].
References
E.L. Andronikashvili, J. Phys. U.S.S.R. 10, 201 (1946)
C. Enss, S. Hunklinger, Low-Temperature Physics (Springer-Verlag, Berlin Heidelberg, 2005)
F. Stienkemeier, K. Lehmann, J. Phys. B 39, R127 (2006)
M. Mudrich, F. Stienkemeier, Int. Rev. Phys. Chem. 33, 301 (2014)
M.P. Ziemkiewicz, D.M. Neumark, O. Gessner, Int. Rev. Phys. Chem. 34, 239 (2015)
F. Stienkemeier, J. Higgins, C. Callegari, S.I. Kanorsky, W.E. Ernst, G. Scoles, Z. Phys. D. 38, 253 (1996)
M. Barranco, R. Guardiola, S. Hernández, R. Mayol, J. Navarro, M. Pi, J. Low Temp. Phys. 142, 1 (2006)
J. Reho, J. Higgins, C. Callegari, K.K. Lehmann, G. Scoles, J. Chem. Phys. 113, 9686 (2000)
F.R. Brühl, R.A. Trasca, W.E. Ernst, J. Chem. Phys. 115, 10220 (2001)
C.P. Schulz, P. Claas, F. Stienkemeier, Phys. Rev. Lett. 87, 153401 (2001)
J. Higgins, C. Callegari, J. Reho, F. Stienkemeier, W.E. Ernst, K.K. Lehmann, M. Gutowski, G. Scoles, Science 273, 629 (1996)
J. Higgins, C. Callegari, J. Reho, F. Stienkemeier, W.E. Ernst, M. Gutowski, G. Scoles, J. Phys. Chem. A 102, 4952 (1998)
J. Reho, C. Callegari, J. Higgins, W.E. Ernst, K.K. Lehmann, G. Scoles, Faraday Discussion 108, 161 (1997)
J. Reho, J. Higgins, C. Callegari, K.K. Lehmann, G. Scoles, J. Chem. Phys. 113, 9694 (2000)
J.H. Reho, U. Merker, M.R. Radcliff, K.K. Lehmann, G. Scoles, J. Phys. Chem. A 104, 3620 (2000)
J. Nagl, G. Auböck, A. Hauser, O. Allard, C. Callegari, W. Ernst, J. Chem. Phys. 128, 154320 (2008)
O. Bünermann, F. Stienkemeier, Eur. Phys. J. D 61, 645 (2011)
J. Reho, J. Higgins, M. Nooijen, K.K. Lehmann, G. Scoles, M. Gutowski, J. Chem. Phys. 115, 10265 (2001)
J. Reho, J. Higgins, K.K. Lehmann, Faraday Discussion 118, 33 (2001)
F. Stienkemeier, F. Meier, A. Hägele, H.O. Lutz, E. Schreiber, C.P. Schulz, I.V. Hertel, Phys. Rev. Lett. 83, 2320 (1999)
G. Droppelmann, O. Bünermann, C.P. Schulz, F. Stienkemeier, Phys. Rev. Lett. 93, 0233402 (2004)
P. Claas, G. Droppelmann, C.P. Schulz, M. Mudrich, F. Stienkemeier, J. Phys. B 39, S1151 (2006)
P. Claas, G. Droppelmann, C.P. Schulz, M. Mudrich, F. Stienkemeier, J. Phys. Chem. A 111, 7537 (2007)
M. Mudrich, G. Droppelmann, P. Claas, C. Schulz, F. Stienkemeier, Phys. Rev. Lett. 100, 023401 (2008)
M. Mudrich, P. Heister, T. Hippler, C. Giese, O. Dulieu, F. Stienkemeier, Phys. Rev. A 80, 042512 (2009)
M. Schlesinger, W.T. Strunz, Phys. Rev. A 77, 012111 (2008)
J. von Vangerow, O. John, F. Stienkemeier, M. Mudrich, J. Chem. Phys. 143, 034302 (2015)
J. Von Vangerow, F. Coppens, A. Leal, M. Pi, M. Barranco, N. Halberstadt, F. Stienkemeier, M. Mudrich, J. Phys. Chem. Lett. 8, 307 (2017)
F. Coppens, J. Von Vangerow, M. Barranco, N. Halberstadt, F. Stienkemeier, M. Pi, M. Mudrich, Phys. Chem. Chem. Phys. 20, 9309 (2018)
N.V. Dozmorov, A.V. Baklanov, J. von Vangerow, F. Stienkemeier, J.A.M. Fordyce, M. Mudrich, Phys. Rev. A 98, 043403 (2018)
B. Thaler, S. Ranftl, P. Heim, S. Cesnik, L. Treiber, R. Meyer, A.W. Hauser, W.E. Ernst, M. Koch, Nat. Commun. 9, 4006 (2018)
B. Thaler, P. Heim, L. Treiber, M. Koch, J. Chem. Phys. 152, 014307 (2020)
B. Thaler, M. Meyer, P. Heim, M. Koch, Phys. Rev. Lett. 124, 115301 (2020)
C. Giese, F. Stienkemeier, M. Mudrich, A.W. Hauser, W.E. Ernst, Phys. Chem. Chem. Phys. 13, 18769 (2011)
M. Schlesinger, M. Mudrich, F. Stienkemeier, W.T. Strunz, Chem. Phys. Lett. 490, 245 (2010)
B. Grüner, M. Schlesinger, P. Heister, W.T. Strunz, F. Stienkemeier, M. Mudrich, Phys. Chem. Chem. Phys. 13, 6816 (2011)
L. Bruder, M. Mudrich, F. Stienkemeier, Phys. Chem. Chem. Phys. 17, 23877 (2015)
A. Sieg, J. von Vangerow, F. Stienkemeier, O. Dulieu, M. Mudrich, J. Phys. Chem. A. 120, 7641 (2016)
J. Tiggesbäumker, F. Stienkemeier, Phys. Chem. Chem. Phys. 9, 4748 (2007)
T. Döppner, T. Diederich, S. Göde, A. Przystawik, J. Tiggesbäumker, K.-H. Meiwes-Broer, J. Chem. Phys. 126, 244513 (2007)
S. Müller, M. Mudrich, F. Stienkemeier, J. Chem. Phys. 131, 044319 (2009)
K. Atkins, Phys. Rev. 116, 1339 (1959)
T. Döppner, S. Teuber, M. Schumacher, J. Tiggesbäumker, K. Meiwes-Broer, Appl. Phys. B. 71, 357 (2000)
T. Döppner, T. Diederich, J. Tiggesbäumker, K.-H. Meiwes-Broer, Eur. Phys. J. D 16, 13 (2001)
T. Döppner, T. Diederich, A. Przystawik, N.X. Truong, T. Fennel, J. Tiggesbäumker, K.H. Meiwes-Broer, Phys. Chem. Chem. Phys. 9, 4639 (2007)
N. Truong et al., Phys. Rev. A 81, 013201 (2010)
A. Mikaberidze, U. Saalmann, J.M. Rost, Phys. Rev. A 77, 041201 (2008)
C. Peltz, T. Fennel, Eur. Phys. J. D 63, 281 (2011)
A. Mikaberidze, U. Saalmann, J.M. Rost, Phys. Rev. Lett. 102, 128102 (2009)
S. Krishnan et al., Phys. Rev. Lett. 107, 173402 (2011)
S.R. Krishnan et al., New J. Phys. 14, 075016 (2012)
M. Kelbg, A. Heidenreich, L. Kazak, M. Zabel, B. Krebs, K.-H. Meiwes-Broer, J. Tiggesbäumker, J. Phys. Chem. A 122, 8107 (2018)
M. Kelbg, M. Zabel, B. Krebs, L. Kazak, K.-H. Meiwes-Broer, J. Tiggesbäumker, Phys. Rev. Lett. 125, 093202 (2020)
A. Przystawik, S. Göde, T. Döppner, J. Tiggesbäumker, K.-H. Meiwes-Broer, Phys. Rev. A 78, 021202 (2008)
S. Göde, R. Irsig, J. Tiggesbäumker, K.-H. Meiwes-Broer, New J. Phys. 15, 015026 (2013)
A. Hernando, M. Barranco, R. Mayol, M. Pi, F. Ancilotto, Phys. Rev. B 78, 184515 (2008)
A. Stolow, A.E. Bragg, D.M. Neumark, Chem. Rev. 104, 1719 (2004)
I.V. Hertel, W. Radloff, Rep. Prog. Phys. 69, 1897 (2006)
T. Weinacht, B.J. Pearson, Time-Resolved Spectroscopy: An Experimental Perspective (CRC Press, Taylor & Francis Group, Boca Raton, FL 33487–2742, 2019)
P. Radcliffe, A. Przystawik, T. Diederich, T. Döppner, J. Tiggesbäumker, K.-H. Meiwes-Broer, Phys. Rev. Lett. 92, 173403 (2004)
E. Loginov, D. Rossi, M. Drabbels, Phys. Rev. Lett. 95, 163401 (2005)
E. Loginov, M. Drabbels, J. Phys. Chem. A. 111, 7504 (2007)
O. Kornilov, C.C. Wang, O. Bünermann, A.T. Healy, M. Leonard, C. Peng, S.R. Leone, D.M. Neumark, O. Gessner, J. Phys. Chem. 114, 1437 (2010)
O. Kornilov, O. Bünermann, D.J. Haxton, S.R. Leone, D.M. Neumark, O. Gessner, J. Phys. Chem. 115, 7891 (2011)
M.P. Ziemkiewicz, C. Bacellar, K.R. Siefermann, S.R. Leone, D.M. Neumark, O. Gessner, J. Chem. Phy. 141, (2014)
M. Mudrich et al., Nat. Commun. 11 (2020)
J.P. Toennies, A.F. Vilesov, Angew. Chem. Int. Ed. 43, 2622 (2004)
C. Callegari, W.E. Ernst, in Handbook of High Resolution Spectroscopy, edited by F. Merkt, M. Quack (John Wiley & Sons, Chichester, 2011)
A.E. Boguslavskiy, J. Mikosch, A. Gijsbertsen, M. Spanner, S. Patchkovskii, N. Gador, M.J.J. Vrakking, A. Stolow, Science 335, 1336 (2012)
P. Maierhofer, M. Bainschab, B. Thaler, P. Heim, W.E. Ernst, M. Koch, J. Phys. Chem. A 120, 6418 (2016)
M. Koch, B. Thaler, P. Heim, W.E. Ernst, J. Phys. Chem. A 121, 6398 (2017)
M. Koch, P. Heim, B. Thaler, M. Kitzler, W.E. Ernst, J. Phys. B: At., Mol. Opt. Phys. 50, 125102 (2017)
R.E. Continetti, Ann. Rev. Phy. Chem. 52, 165 (2001)
T. Arion, U. Hergenhahn, J. Elec. Spectros. Relat. Phen. 200, 222 (2015)
L.J. Frasinski, K. Codling, P.A. Hatherly, Science 246, 1029 (1989)
V. Stert, W. Radloff, C. Schulz, I. Hertel, Eur. Phys. J. D 5, 97 (1999)
M. Rumetshofer, P. Heim, B. Thaler, W.E. Ernst, M. Koch, W. von der Linden, Phys. Rev. A 97, 062503 (2018)
P. Heim, M. Rumetshofer, S. Ranftl, B. Thaler, W.E. Ernst, M. Koch, W. von der Linden, Entropy 21, 93 (2019)
M. Shcherbinin, A.C. LaForge, V. Sharma, M. Devetta, R. Richter, R. Moshammer, T. Pfeifer, M. Mudrich, Phys. Rev. A 96, 013407 (2017)
A. LaForge, M. Shcherbinin, F. Stienkemeier, R. Richter, R. Moshammer, T. Pfeifer, M. Mudrich, Nat. Phys. 15, 247 (2019)
S. Smolarek, N.B. Brauer, W.J. Buma, M. Drabbels, J. Am. Chem. Soc. 132, 14086 (2010)
D. Buchta et al., J. Phys. Chem. A 117, 4394 (2013)
M. Shcherbinin, A.C. LaForge, M. Hanif, R. Richter, M. Mudrich, J. Phys. Chem. A 122, 1855 (2018)
L. Ben Ltaief et al., J. Phys. Chem. Lett. 10, 6904 (2019)
A.C. LaForge et al., Phys. Rev. Lett. 116, 203001 (2016)
L.B. Ltaief et al., Phys. Chem. Chem. Phys. 22, 8557 (2020)
Microscopic Approaches to Quantum Liquids in Confined Geometries, Series on Advances in Quantum Many-Body Theory, edited by E. Krotscheck, J. Navarro (World Scientific Pub Co, Singapore, 2002), p. 450
F. Brieuc, C. Schran, F. Uhl, H. Forbert, D. Marx, J. Chem. Phys. 152, 210901 (2020)
F. Ancilotto, M. Barranco, F. Coppens, J. Eloranta, N. Halberstadt, A. Hernando, D. Mateo, M. Pi, Int. Rev. Phys. Chem. 36, 621 (2017)
M. Pi, F. Ancilotto, F. Coppens, N. Halberstadt, A. Hernando, A. Leal, D. Mateo, R. Mayol, M. Barranco, \(^4\)He-DFT BCN-TLS: a computer package for simulating structural properties and dynamics of doped liquid helium-4 systems. https://github.com/bcntls2016/ (2016)
A. Hernando, M. Barranco, M. Pi, E. Loginov, M. Langlet, M. Drabbels, Phys. Chem. Chem. Phys. 14, 3996 (2012)
J. von Vangerow, A. Sieg, F. Stienkemeier, M. Mudrich, A. Leal, D. Mateo, A. Hernando, M. Barranco, M. Pi, J. Phys. Chem. A 118, 6604 (2014)
M. Martinez, F. Coppens, M. Barranco, N. Halberstadt, M. Pi, Phy. Chem. Chem. Phy. 21, 3626 (2019)
D. Mateo, A. Hernando, M. Barranco, E. Loginov, M. Drabbels, M. Pi, Phys. Chem. Chem. Phys. 15, 18388 (2013)
N.B. Brauer, S. Smolarek, E. Loginov, D. Mateo, A. Hernando, M. Pi, M. Barranco, W.J. Buma, M. Drabbels, Phys. Rev. Lett. 111, 153002 (2013)
D. Mateo, A. Leal, A. Hernando, M. Barranco, M. Pi, F. Cargnoni, M. Mella, X. Zhang, M. Drabbels, J. Chem. Phys. 140, 131101 (2014)
A. Leal, D. Mateo, A. Hernando, M. Pi, M. Barranco, A. Ponti, F. Cargnoni, M. Drabbels, Phys. Rev. B 90, 224518 (2014)
F. Coppens, J. von Vangerow, A. Leal, M. Barranco, N. Halberstadt, M. Mudrich, M. Pi, F. Stienkemeier, Eur. Phys. J. D 73, 94 (2019)
A.C. LaForge et al., Phys. Rev. X 11, 021011 (2021)
F. Ancilotto, M. Barranco, M. Pi, Phy. Rev. B 97, 184515 (2018)
S.M. O’Connell et al., Phys. Rev. Lett. 124, 215301 (2020)
M. Pi, F. Ancilotto, J.M. Escartín, R. Mayol, M. Barranco, Phys. Rev. B 102, 060502 (2020)
A. Leal, D. Mateo, A. Hernando, M. Pi, M. Barranco, Phys. Chem. Chem. Phys. 16, 23206 (2014)
F. Coppens, F. Ancilotto, M. Barranco, N. Halberstadt, M. Pi, Phys. Chem. Chem. Phys. 19, 24805 (2017)
F. Coppens, A. Leal, M. Barranco, N. Halberstadt, M. Pi, J. Low Temp. Phys. 187, 439 (2017)
J.M. Escartín, F. Ancilotto, M. Barranco, M. Pi, Phys. Rev. B 99, 140505 (2019)
A. Vilà, M. González, R. Mayol, J. Chem. Theory Comput. 11, 899 (2015)
A. Vilà, A., M. González, R. Mayol, Phys. Chem. Chem. Phys. 17, 32241 (2015)
A. Vilà, M. González, R. Mayol, Phys. Chem. Chem. Phys. 18, 2409 (2016)
A. Vilà, M. González, Phys. Chem. Chem. Phys. 18, 27630 (2016)
A. Vilà, M. González, Phys. Chem. Chem. Phys. 18, 31869 (2016)
M. Blancafort-Jorquera, A. Vilà, M. González, Phys. Chem. Chem. Phys. 21, 24218 (2019)
A. Vilà, M. Paniagua, M. González, Phys. Chem. Chem. Phys. 20, 118 (2018)
M. Blancafort-Jorquera, A. Vilà, M. González, Phys. Chem. Chem. Phys. 21, 21007 (2019)
M. Koch, G. Auböck, C. Callegari, W.E. Ernst, Phys. Rev. Lett. 103, 035302 (2009)
G. Auböck, J. Nagl, C. Callegari, W.E. Ernst, Phys. Rev. Lett. 101, 035301 (2008)
C. Callegari, F. Ancilotto, J. Phys. Chem. A 115, 6789 (2011)
D.E. Galli, M. Buzzacchi, L. Reatto, J. Chem. Phys. 115, 10239 (2001)
M. Rossi, M. Verona, D. Galli, L. Reatto, Phys. Rev. B 69, 212510 (2004)
L. Fechner, B. Grüner, A. Sieg, C. Callegari, F. Ancilotto, F. Stienkemeier, M. Mudrich, Phys. Chem. Chem. Phys. 14, 3843 (2012)
E. Loginov, C. Callegari, F. Ancilotto, M. Drabbels, J. Phys. Chem. A 115, 6779 (2011)
E. Loginov, M. Drabbels, J. Phys. Chem. A 118, 2738 (2014)
M. Theisen, F. Lackner, W.E. Ernst, J. Phys. Chem. A 115, 7005 (2011)
X. Zhang, M. Drabbels, J. Chem. Phys. 137, 051102 (2012)
O. Bünermann, G. Droppelmann, A. Hernando, R. Mayol, F. Stienkemeier, J. Phys. Chem. A 111, 12684 (2007)
E. Loginov, A. Hernando, J.A. Beswick, N. Halberstadt, M. Drabbels, J. Phys. Chem. A 119, 6033 (2015)
C. Giese, T. Mullins, B. Grüner, M. Weidemüller, F. Stienkemeier, M. Mudrich, J. Chem. Phys. 137, (2012)
A.T.J.B. Eppink, D.H. Parker, Rev. Sci. Instrum. 68, 3477 (1997)
A. Kautsch, M. Hasewend, M. Koch, W.E. Ernst, Phys. Rev. A 86, 033428 (2012)
A. Kautsch, M. Koch, W.E. Ernst, J. Phys. Chem. A 117, 9621 (2013)
M. Koch, A. Kautsch, F. Lackner, W.E. Ernst, J. Phys. Chem. A 118, 8373 (2014)
B. Thaler, R. Meyer, P. Heim, S. Ranftl, J.V. Pototschnig, A.W. Hauser, M. Koch, W.E. Ernst, J. Phys. Chem. A 123, 3977 (2019)
J. von Vangerow, F. Coppens, A. Leal, M. Pi, M. Barranco, N. Halberstadt, F. Stienkemeier, M. Mudrich, J. Phy. Chem. Lett. 8, 307 (2017)
S. Grebenev, J.P. Toennies, A.F. Vilesov, Science 279, 2083 (1998)
A.R.W. McKellar, Y. Xu, W. Jäger, Phy. Rev. Lett. 97, 183401 (2006)
P. Sindzingre, M.L. Klein, D.M. Ceperley, Phys. Rev. Lett. 63, 1601 (1989)
F. Paesani, A. Viel, F.A. Gianturco, K.B. Whaley, Phys. Rev. Lett. 90, 073401 (2003)
S. Moroni, A. Sarsa, S. Fantoni, K.E. Schmidt, S. Baroni, Phys. Rev. Lett. 90, 143401 (2003)
S. Grebenev, B. Sartakov, J.P. Toennies, A.F. Vilesov, Science 289, 1532 (2000)
N. Poertner, J.P. Toennies, A.F. Vilesov, G. Benedek, V. Hizhnyakov, EPL (Europhysics Letters) 88, 26007 (2009)
D. Verma et al., Phys. Rev. B 102, 014504 (2020)
F. Stienkemeier, O. Bünermann, R. Mayol, F. Ancilotto, M. Barranco, M. Pi, Phys. Rev. B 70, 214509 (2004)
A. Hernando, R. Mayol, M. Pi, M. Barranco, F. Ancilotto, O. Bünermann, F. Stienkemeier, J. Phys. Chem. A 111, 7303 (2007)
G. Wu, P. Hockett, A. Stolow, Phys. Chem. Chem. Phys. 13, 18447 (2011)
S. Rutz, R. de Vivie-Riedle, E. Schreiber, Phys. Rev. A 54, 306 (1996)
B. Zhang, N. Gador, T. Hansson, Phys. Rev. Lett. 91, 173006 (2003)
S. Rutz, E. Schreiber, Chem. Phys. Lett. 269, 9 (1997)
M.J.J. Vrakking, D.M. Villeneuve, A. Stolow, Phys. Rev. A 54, R37 (1996)
R. de Vivie-Riedle, K. Kobe, J. Manz, W. Meyer, B. Reischl, S. Rutz, E. Schreiber, L. Wöste, J. Phys. Chem. 100, 7789 (1996)
C. Nicole, M.A. Bouchène, C. Meier, S. Magnier, E. Schreiber, B. Girard, J. Chem. Phys. 111, 7857 (1999)
A. Lindinger, J.P. Toennies, A.F. Vilesov, J. Chem. Phys. 121, 12282 (2004)
M. Wollenhaupt, V. Engel, T. Baumert, Ann. Rev. Phys. Chem. 56, 25 (2005)
T. Baumert, G. Gerber, in Femtosecond Laser Spectroscopy, edited by P. Hannaford (Springer Verlag, , 2005), p. chap. 9
C. Chin, R. Grimm, P. Julienne, E. Tiesinga, Rev. Mod. Phys. 82, 1225 (2010)
K.M. Jones, E. Tiesinga, P.D. Lett, P.S. Julienne, Rev. Mod. Phys. 78, 483 (2006)
F. Stienkemeier, W.E. Ernst, J. Higgins, G. Scoles, J. Chem. Phys. 102, 615 (1995)
F. Ancilotto, G. DeToffol, F. Toigo, Phys. Rev. B 52, 16125 (1995)
S. Bovino, E. Coccia, E. Bodo, D. Lopez-Durán, F. A. Gianturco, J. Chem. Phys. 130, (2009)
G. Guillon, A. Zanchet, M. Leino, A. Viel, R.E. Zillich, J. Phys. Chem. A 115, 6918 (2011)
S. Bovino, E. Bodo, E. Yurtsever, F.A. Gianturco, J. Chem. Phys. 128, 224312 (2008)
J.G. Danzl, E. Haller, M. Gustavsson, M.J. Mark, R. Hart, N. Bouloufa, O. Dulieu, H. Ritsch, H.-C. Nägerl, Science 321, 1062 (2008)
M. Gruebele, A.H. Zewail, J. Chem. Phys. 98, 883 (1993)
G. Droppelmann, M. Mudrich, C.P. Schulz, F. Stienkemeier, Eur. Phys. J. D 52, 67 (2009)
S. Müller, S. Krapf, T. Koslowski, M. Mudrich, F. Stienkemeier, Phys. Rev. Lett. 102, 183401 (2009)
M. Dvorak, M. Müller, O. Bünermann, F. Stienkemeier, J. Chem. Phys. 140, 144301 (2014)
A. Boatwright, C. Feng, D. Spence, E. Latimer, C. Binns, A.M. Ellis, S. Yang, Faraday discussions 162, 113 (2013)
G. Haberfehlner, P. Thaler, D. Knez, A. Volk, F. Hofer, W.E. Ernst, G. Kothleitner, Nat. commun. 6, 1 (2015)
H. Schmidt, J. von Vangerow, F. Stienkemeier, A.S. Bogomolov, A.V. Baklanov, D.M. Reich, W. Skomorowski, C.P. Koch, M. Mudrich, J. Chem. Phy. 142, 044303 (2015)
R. Monni, G. Auböck, D. Kinschel, K.M. Aziz-Lange, H.B. Gray, A. Vlcek, M. Chergui, Chem. Phy. Lett. 683, 112 (2017)
M. Lundwall, M. Tchaplyguine, G. Öhrwall, A. Lindblad, S. Peredkov, T. Rander, S. Svensson, O. Björneholm, Surface science 594, 12 (2005)
A. Masson, L. Poisson, M.-A. Gaveau, B. Soep, J.-M. Mestdagh, V. Mazet, F. Spiegelman, J. Chem. Phys. 133, 054307 (2010)
A.V. Benderskii, J. Eloranta, R. Zadoyan, V.A. Apkarian, J. Chem. Phys. 117, 1201 (2002)
H. Buchenau, E.L. Knuth, J. Northby, J.P. Toennies, C. Winkler, J. Chem. Phys. 92, 6875 (1990)
A. Mauracher, M. Daxner, J. Postler, S.E. Huber, S. Denifl, P. Scheier, J.P. Toennies, J. Phys. Chem. Lett. 5, 2444 (2014)
A. Mauracher, O. Echt, A. Ellis, S. Yang, D. Bohme, J. Postler, A. Kaiser, S. Denifl, P. Scheier, Phys. Rep. 751, 1 (2018)
T. González-Lezana, O. Echt, M. Gatchell, M. Bartolomei, J. Campos-Martínez, P. Scheier, International Reviews in Physical Chemistry 39, 465 (2020)
R. Fröchtenicht, J.P. Toennies, A. Vilesov, Chem. Phys. Lett. 229, 1 (1994)
N. Halberstadt, K.C. Janda, Chem. Phys. Lett. 282, 409 (1998)
D. Buchta et al., J. Chem. Phys. 139, 084301 (2013)
T. Ruchti, B.E. Callicoatt, K.C. Janda, Phys. Chem. Chem. Phys. 2, 4075 (2000)
W. Lewis, C. Lindsay, R. Bemish, R. Miller, J. Am. Chem. Soc. 127, 7235 (2005)
N. Saito et al., Chem. Phys. Lett. 441, 16 (2007)
J. Zobeley, R. Santra, L.S. Cederbaum, J. Chem. Phys. 115, 5076 (2001)
A. LaForge et al., Phy. Rev. Lett. 116, 203001 (2016)
D.S. Peterka, A. Lindinger, L. Poisson, M. Ahmed, D.M. Neumark, Phys. Rev. Lett. 91, 043401 (2003)
D.S. Peterka, J.H. Kim, C.C. Wang, L. Poisson, D.M. Neumark, J. Phys. Chem. A 111, 7449 (2007)
C.C. Wang, O. Kornilov, O. Gessner, J.H. Kim, D.S. Peterka, D.M. Neumark, J. Phys. Chem. 112, 9356 (2008)
S. Mandal et al., Phys. Chem. Chem. Phys. 22, 10149 (2020)
L.B. Ltaief, M. Shcherbinin, S. Mandal, S.R. Krishnan, R. Richter, T. Pfeifer, M. Mudrich, J. Phy. B: Atomic. Molecul. Optic. Phys. 53, 204001 (2020)
L.B. Ltaief, M. Shcherbinin, S. Mandal, S. Krishnan, R. Richter, S. Turchini, N. Zema, M. Mudrich, J. Low. Temp. Phys. (2021)
M. Shcherbinin, F.V. Westergaard, M. Hanif, S.R. Krishnan, A. LaForge, R. Richter, T. Pfeifer, M. Mudrich, J. Chem. Phys. 150, 044304 (2019)
K. von Haeften, A.R. B.d. Castro, M. Joppien, L. Moussavizadeh, R.V. Pietrowski, T.Möller, Phys. Rev. Lett. 78, 4371 (1997)
K. von Haeften, T. Laarmann, H. Wabnitz, T. Möller, Phys. Rev. Lett. 87, 153403 (2001)
K. von Haeften, T. Laarmann, H. Wabnitz, T. Möller, Phys. Rev. Lett. 88, 233401 (2002)
K. von Haeften, T. Laarmann, H. Wabnitz, T. Möller, J. Phys. B 38, 373 (2005)
O. Bünermann, O. Kornilov, S.R. Leone, D.M. Neumark, O. Gessner, IEEE. J. Sel. Top. Quantum Electron. 18, 308 (2012)
O. Bünermann, O. Kornilov, D.J. Haxton, S.R. Leone, D.M. Neumark, O. Gessner, J. Chem. Phys. 137, 214302 (2012)
L.F. Gomez et al., Science 345, 906 (2014)
B. Langbehn et al., Phys. Rev. Lett. 121, 255301 (2018)
O. Gessner, A.F. Vilesov, Ann. Rev. Phy. Chem. 70, 173 (2019)
J.D. Asmussen et al., Phys. Chem. Chem. Phys. 23, 15138 (2021)
V. Lyamayev et al., J. Phys. B 46, 164007 (2013)
M. Joppien, R. Karnbach, T. Möller, Phys. Rev. Lett. 71, 2654 (1993)
R. Michiels et al., Phys. Rev. Lett. 127, 093201 (2021)
K.D. Closser, O. Gessner, M. Head-Gordon, J. Chem. Phys. 140, 134306 (2014)
C.-Z. Gao, P.M. Dinh, P.-G. Reinhard, E. Suraud, Phys. Chem. Chem. Phys. 19, 19784 (2017)
U. Hergenhahn, J. Electron. Spectrosc. Relat. Phenom. 184, 78 (2011)
T. Jahnke, J. Phys. B 48, 082001 (2015)
T. Jahnke et al., Chem. Rev. 120, 11295 (2020)
L.S. Cederbaum, J. Zobeley, F. Tarantelli, Phys. Rev. Lett. 79, 4778 (1997)
M. Mucke et al., Nat. Phys. 6, 143 (2010)
V. Stumpf, K. Gokhberg, L.S. Cederbaum, Nat. Chem. 8, 237 (2016)
X. Ren, E. Wang, A.D. Skitnevskaya, A.B. Trofimov, K. Gokhberg, A. Dorn, Nat. Phys. 14, 1062–1066 (2018)
F. Wiegandt et al., Phys. Rev. A 100, 022707 (2019)
Y. Ovcharenko et al., Phys. Rev. Lett. 112, 073401 (2014)
A.C. LaForge et al., Sci. Rep. 4, 3621 (2014)
R. Katzy, M. Singer, S. Izadnia, A.C. LaForge, F. Stienkemeier, Rev. Sci. Instr. 87, 013105 (2016)
Y. Ovcharenko et al., New J. Phys. 22, 083043 (2020)
N.V. Kryzhevoi, V. Averbukh, L.S. Cederbaum, Phys. Rev. B 76, 094513 (2007)
N.V. Kryzhevoi, D. Mateo, M. Pi, M. Barranco, L.S. Cederbaum, Phy. Chem. Chem. Phy. 15, 18167 (2013)
S. Kazandjian et al., Phys. Rev. A 98, 050701 (2018)
A.I. Kuleff, K. Gokhberg, S. Kopelke, L.S. Cederbaum, Phys. Rev. Lett. 105, 043004 (2010)
S. Augst, D. Strickland, D.D. Meyerhofer, S.L. Chin, J.H. Eberly, Phys. Rev. Lett. 63, 2212 (1989)
A. Heidenreich, B. Grüner, M. Rometsch, S. Krishnan, F. Stienkemeier, M. Mudrich, New J. Phys. 18, 073046 (2016)
A. Heidenreich, B. Grüner, D. Schomas, F. Stienkemeier, S.R. Krishnan, M. Mudrich, J. Modern Opt. 64, 1061 (2017)
B. Schütte, M. Arbeiter, A. Mermillod-Blondin, M.J. Vrakking, A. Rouzée, T. Fennel, Phys. Rev. Lett. 116, 033001 (2016)
U. Saalmann, C. Siedschlag, J.M. Rost, J. Phys. B 39, R39 (2006)
T. Fennel, K.-H. Meiwes-Broer, J. Tiggesbäumker, P.-G. Reinhard, P.M. Dinh, E. Suraud, Rev. Mod. Phys. 82, 1793 (2010)
T. Döppner, S. Teuber, T. Diederich, T. Fennel, P. Radcliffe, J. Tiggesbäumker, K.-H. Meiwes-Broer, Eur. Phys. J. D 24, 157 (2003)
B. Schütte, M. Arbeiter, T. Fennel, G. Jabbari, A. Kuleff, M. Vrakking, A. Rouzée, Nat. Phys. 6, 8596 (2015)
T. Oelze et al., Sci. Rep. 7, 40736 (2017)
C. Medina et al., New J. Phys. 23, 053011 (2021)
D. Schomas et al., arXiv preprint arXiv:2005.02944 (2020)
C. Gnodtke, U. Saalmann, J.M. Rost, Phys. Rev. A 79, 041201 (2009)
C. Bacellar et al., submitted (2021)
B. Schütte et al., Sci. Rep. 6, 39664 (2016)
J. Passig et al., Nat. commun. 8, 1 (2017)
J. Miao, T. Ishikawa, I.K. Robinson, M.M. Murnane, Science 348, 530 (2015)
M.J. Bogan et al., Nano Lett. 8, 310 (2008)
M. Dantus, V.V. Lozovoy, Chem. Rev. 104, 1813 (2004), publisher: American Chemical Society
K. Ohmori, Annu. Rev. Phys. Chem. 60, 487 (2009)
M. Shapiro, P. Brumer, Phy. Rep. 425, 195 (2006)
L. Bruder, A. Eisfeld, U. Bangert, M. Binz, M. Jakob, D. Uhl, M. Schulz-Weiling, E.R. Grant, F. Stienkemeier, Phys. Chem. Chem. Phys. 21, 2276 (2019)
K. Lazonder, M.S. Pshenichnikov, D.A. Wiersma, Opt. Lett. 31, 3354 (2006)
M. Fushitani, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem. 104, 272 (2008)
R.M. Hochstrasser, PNAS 104, 14190 (2007)
M. Cho, Chem. Rev. 108, 1331 (2008)
P. Nuernberger, S. Ruetzel, T. Brixner, Angew. Chem. Int. Ed. 54, 11368 (2015)
F.D. Fuller, J.P. Ogilvie, Annu. Rev. Phys. Chem. 66, 667 (2015)
L. Bruder, U. Bangert, M. Binz, D. Uhl, F. Stienkemeier, J. Phys. B: At. Mol. Opt. Phys. 52, 183501 (2019)
D.M. Jonas, Annu. Rev. Phys. Chem. 54, 425 (2003)
P. Malý, T. Mančal, J. Phys. Chem. Lett. 9, 5654 (2018)
O. Kühn, T. Mančal, T. Pullerits, J. Phys. Chem. Lett. 11, 838 (2020)
M. Maiuri, J. Brazard, Top Curr Chem (Z) 376, 10 (2018)
T. Brixner, J. Stenger, H.M. Vaswani, M. Cho, R.E. Blankenship, G.R. Fleming, Nature 434, 625 (2005)
J. Dostál, J. Pšenčík, D. Zigmantas, Nat. Chem. 8, 705 (2016)
K.L.M. Lewis, J.P. Ogilvie, J. Phys. Chem. Lett. 3, 503 (2012)
G. Moody, S.T. Cundiff, Adv. Phys. 2, 641 (2017)
M.K. Petti, J.P. Lomont, M. Maj, M.T. Zanni, J. Phys. Chem. B 122, 1771 (2018)
P. Hamm, M. Zanni, Concepts and Methods of 2D Infrared Spectroscopy (Cambridge University Press, 2011)
P.F. Tekavec, T.R. Dyke, A.H. Marcus, J. Chem. Phys. 125, 194303 (2006)
P.F. Tekavec, G.A. Lott, A.H. Marcus, J. Chem. Phys. 127, 214307 (2007)
B. Lomsadze, B.C. Smith, S.T. Cundiff, Nat. Photon. 12, 676 (2018)
K. Hirano, K. Enomoto, M. Kumakura, Y. Takahashi, T. Yabuzaki, Phys. Rev. A 68, (2003)
M. Zbiri, C. Daul, J. Chem. Phys. 121, 11625 (2004)
M. Binz, Ph.D. thesis, University of Freiburg, 2021
J. Nagl, G. Auböck, A.W. Hauser, O. Allard, C. Callegari, W.E. Ernst, Phy. Rev. Lett. 100, 063001 (2008)
O. Allard, J. Nagl, G. Auböck, C. Callegari, W.E. Ernst, Journal of Physics B-Atomic Molecular And Optical. Physics 39, S1169 (2006)
L. Bruder, U. Bangert, M. Binz, D. Uhl, R. Vexiau, N. Bouloufa-Maafa, O. Dulieu, F. Stienkemeier, Nat. Commun. 9, 4823 (2018)
M. Wewer, F. Stienkemeier, J. Chem. Phys. 120, 1239 (2004)
R. Lehnig, A. Slenczka, J. Chem. Phys. 118, 8256 (2003)
U. Bangert, F. Stienkemeier, L. Bruder, arXiv:2112.05418 (2021)
D. Mani et al., Sci. Adv. 5, eaav8179 (2019)
A. Gutberlet, G. Schwaab, O. Birer, M. Masia, A. Kaczmarek, H. Forbert, M. Havenith, D. Marx, Science 324, 1545 (2009)
K. Nauta, R.E. Miller, Science 287, 293 (2000)
G. Haberfehlner, P. Thaler, D. Knez, A. Volk, F. Hofer, W.E. Ernst, G. Kothleitner, Nat. Commun. 6, 8779 (2015)
K. Nauta, R.E. Miller, Phys. Rev. Lett. 82, 4480 (1999)
J. Küpper, J.M. Merritt, Int. Rev. Phys. Chem. 26, 249 (2007)
J. Roden, A. Eisfeld, M. Dvořák, O. Bünermann, F. Stienkemeier, J. Chem. Phys. 134, 054907 (2011)
G.E. Douberly, J.M. Merritt, R.E. Miller, Phys. Chem. Chem. Phys. 7, 463 (2005)
A. Wituschek et al., Nat. Commun. 11, 1 (2020)
F. Bencivenga et al., Sci. Adv. 5, eaaw5805 (2019)
C. Svetina et al., Opt. Lett. 44, 574 (2019)
R. Mincigrucci et al., arXiv preprint arXiv:2010.04860 (2020)
D. Rupp et al., Nat. Commun. 8, 1 (2017)
L. Flückiger et al., New J. Phys. 18, 043017 (2016)
Acknowledgements
M.K. acknowledges financial support by the Austrian Science Fund (FWF) under Grants P29369-N36 and P33166-N, as well as support from NAWI Graz. M.M. gratefully acknowledges funding from the Carlsberg Foundation and from the SPARC Programme, MHRD, India. F.S. and L.B. gratefully acknowledge financial support by the European Research Council (ERC) with the Advanced Grant “COCONIS” (694965) and by the Deutsche Forschungsgemeinschaft (DFG) IRTG CoCo (2079). F.S. acknowledges funding within the DFG Priority Program QUTIF (STI 125/22).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Bruder, L., Koch, M., Mudrich, M., Stienkemeier, F. (2022). Ultrafast Dynamics in Helium Droplets. In: Slenczka, A., Toennies, J.P. (eds) Molecules in Superfluid Helium Nanodroplets. Topics in Applied Physics, vol 145. Springer, Cham. https://doi.org/10.1007/978-3-030-94896-2_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-94896-2_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-94895-5
Online ISBN: 978-3-030-94896-2
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)