
Abstract
The anatomy and physiology of the nasal cavity provide unique advantages for accessing targets for local, systemic, and potentially central nervous system drug delivery. This chapter discusses these advantages and the challenges that must be overcome to reach these targets. The chapter then comprehensively reviews nasal dosage forms, analytical testing, and regulatory requirements in the context of existing nasal spray products. Since nasal sprays are moving towards being preservative-free, the chapter covers specialized methods of achieving a sterile product, namely, formulation strategies, manufacturing strategies, and the device landscape that support this upcoming platform. Finally, the chapter reviews various pathways for regulatory approval around the world, for brand and generic, with particular emphasis on the growing acceptance of in vitro data for locally acting nasal spray products.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Preservative-free nasal spray drug products represent a small portion of the overall drug delivery market. However, the desire to remove preservatives from formulations driven by concerns over potential damage from long-term use coupled with innovations in device technology has allowed Pharma companies to consider preservative-free nasal sprays as a viable option. In this chapter, an overview of nasal cavity physiology will be presented along with a review of locally and systemically acting drug products. Current formulation and manufacturing strategies are discussed along with the device landscape that enables preservative-free formulations. Finally, the pathway for global regulatory approval will be outlined including considerations for in vitro analytical test requirements.
2 Nasal Physiology
A schematic of a human nasal cavity is shown in Fig. 5.1. Two nostrils, also referred to as the nasal vestibule, mark the entrance into the nasal cavities. At the end of the nasal vestibule, the diameter of each cavity decreases at a point called the nasal ostium (Newman 1993). The septum separates the two cavities, which extend, on average, 12–14 cm from the nostrils to the junction between the nose and pharynx (Vidgren and Kublik 1998; Marom et al. 1984). This junction is called the nasopharynx. The nasal-associated lymphoid tissue (NALT), an area that may be associated with inducing mucosal immunity, is located in the nasopharynx. Within the nose itself, the main nasal passage is further divided by three projections from the septum called turbinates (Pontiroli et al. 1989). The inferior, middle, and superior turbinates increase the total surface area of the nasal cavity to 150 cm2 (Pontiroli et al. 1989). The total volume of each cavity is 7.5 mL.

Human nasal cavity anatomy (courtesy of Aptar Pharma)
The nasal mucosa is lined with stratified squamous, pseudostratified columnar, and transitional epithelia cells (Adams 1986). The stratified squamous and transitional types are mainly found in the anterior third of each cavity. Cells in this region are neither ciliated nor well vascularized. The columnar type, also known as respiratory epithelium, is located in the posterior two thirds. The respiratory region contains ciliated cells, mucous secreting goblet cells, and basal cells (Petruson et al. 1984). The respiratory epithelium is also highly vascularized, innervated, and drained by an extensive lymphatic network (Pontiroli et al. 1989; Schipper et al. 1991). The olfactory epithelium, which contains cells that provide a sense of smell, is located near the superior turbinate and adjacent to the nasal septum (Schipper et al. 1991). The main function of the nose is to warm and humidify inspired air and to filter inhaled, potentially toxic or infectious, particles from the airstream (Pontiroli et al. 1989). Thus, the nasal cavity primarily acts as a defense mechanism by protecting the lower respiratory tract (Andersen and Proctor 1983).
Inhaled particles or droplets are thought to deposit in the nose by three mechanisms: inertial impaction, gravitational sedimentation, and Brownian diffusion (Brain and Valberg 1979; Newman et al. 1982; Gonda and Gipps 1990). Of these, inertial impaction is the most predominant for two main reasons. First, the air passageway constricts sharply approximately 1.5 cm into the nose at the nasal ostium (Mygind 1985). This constriction accelerates the inhaled air and increases turbulence (Yu et al. 1998). Secondly, the air stream must change direction at this constriction to enter the turbinate region. Particles that are large or moving at high velocity cannot follow the air stream as it changes direction due to their high momentum. Such particles continue in their original direction of travel and impact the airway walls, particularly at the leading edge of the turbinates. Because the drug-laden droplets for most aqueous nasal sprays are so large (30–60 μm) (Chien et al. 1989), a high percentage of the spray impacts in the anterior third of the nasal cavity (Hardy et al. 1985). However, droplets that are smaller than 10 μm may bypass the nasal cavity and deposit in the lower respiratory tract, which may be deemed as a risk by regulatory agencies.
A particle that deposits on the nasal mucosa may exert a local effect and/or be absorbed into the blood stream. Absorption is facilitated by a highly vascularized, large surface area with relatively low enzymatic activity. Since blood leaving the nasal cavity bypasses the liver, first pass hepatic metabolism can be avoided, making the nose a suitable target for drugs with low oral bioavailability. However, cytochrome P-450-dependent monooxygenase has been reported to metabolize compounds in the nasal mucosa such as cocaine and progesterone (Dahl and Hadley 1983; Brittebo 1982).
Nasal absorption can be rapid. Concentration vs. time profiles similar to intravenous administration have been reported for nicotine and butorphanol (Henningfield and Keenan 1993; Bristol Myers Squibb Company 1999). Absorption is thought to take place primarily in the respiratory zone (posterior, ciliated two thirds) of the nasal cavity. However, the absorption rate at specific deposition sites has not been clearly defined (Vidgren and Kublik 1998). Animal studies have shown that drugs can be absorbed through transcellular and paracellular passive absorption, carrier-mediated transport, and by transcytosis (Bjork 1993; McMartin et al. 1987).
Caution should be exercised when extrapolating results from animal models to man, according to some published literature (Illum 2000). Rats, rabbits, sheep, pigs, dogs, and monkeys have all been used as models for nasal drug absorption. In man, the surface area/body weight ratio is 2.5 cm2/kg (Illum 2000). The surface area/body weight ratios for the animals above range from 7.7 to 46 cm2/kg except for sheep that have a ratio of 0.2 cm2/kg (Illum 2000). In addition, animal’s nasal cavities are structurally different than man because they lack a third turbinate. To deliver nasal sprays into the nose of many of these animals, the animal needs to be anesthetized or sedated, which also can affect drug absorption. In short, animal models produce absorption results that fail to accurately predict the results in man (Illum 2000).
The nose filters undesirable chemicals and bacterial and viral particles from the inhaled airstream. Particles depositing in the anterior regions are physically removed from the nose by wiping, blowing, or sneezing. Although these regions (nasal vestibule and leading edge of the turbinates) are non-ciliated, some of the surfaces are covered with mucus. Here mucus flow is slow, 1–2 mm/h, and occurs mainly due to its connection to the mucus layer in the posterior nose (Hilding 1963).
Unabsorbable particles that adhere to the mucus layer that lines the respiratory epithelium are swept towards the nasopharynx by ciliated cells through a process called mucociliary clearance. They are ultimately swallowed.
The mucus layer is predominately aqueous (90–95 %). However, glycoproteins in mucus give it a gel-like structure. The velocity of mucus transport in ciliated regions is about 6 mm/min (Andersen and Proctor 1983). Particles that partition into mucus or deposit on its surface are typically removed from the nasal cavity in 20 min (Andersen and Proctor 1983). Obviously, physical removal of particles either by wiping the nose or by mucociliary clearance is a major component of the nose’s defense mechanism. For drug delivery, these processes can oppose local drug activity or absorption.
The rate of mucociliary clearance can be altered by pathophysiology such as a common cold or cystic fibrosis, environmental conditions that affect the mucus content, by drug-induced side effects, or potentially by excipients found in nasal spray formulations. A controversial example of such an excipient is benzalkonium chloride (BAC) which is used to prevent microbial growth. A review of BAC (Marple et al. 2004) studies suggest that BAC may cause changes to ciliary beat frequency, ciliary morphology, mucociliary clearance or may potentially damage the epithelial lining. However, after assessing all the literature, the reviewers concluded that BAC is safe to use in nasal spray formulations. A more thorough discussion of use of BAC in formulations is presented later in this chapter (Sect. 5.4).
When delivering drugs to the nose, one must consider the interplay between the formulation, device, and the patient. These three factors greatly affect where the drug-laden droplets or drug particles deposit within the nasal cavity. The site of deposition in the nose is recognized as one of the keys to success or failure of nasal drug therapy. Although this concept is widely recognized (Vidgren and Kublik 1998), only one study actually relates deposition pattern to biologic response (Harris et al. 1986).
This detailed study related deposition pattern, clearance, absorption, and response for desmopressin admixed with radiolabeled HSA delivered by sprays and drops (Harris et al. 1986). The spray formulation deposited in the front of the nose (anteriorly) while the drops covered more surface area. Since the drops covered a larger surface area, it seems logical that the drops would have elicited a greater response. In fact, the opposite was true. The drops were cleared faster by mucociliary clearance since they deposited in posterior regions of the nasal cavity (where cilia move the mucus layer faster). The spray was retained longer, allowing more time for absorption of desmopressin to occur (Table 5.1). The levels of factor VIII in the blood in response to delivery of desmopressin were significantly greater after administration with the spray compared to the drops.
Today’s generations of nasal devices typically deposit droplets in the anterior portions of the nasal cavity due to inertial impaction and the size and/or velocity of the droplets. For example, the deposition patterns from two commonly used nasal spray pumps (Suman et al. 2002) were compared in human volunteers. A radiolabeled nasal nicotine solution was administered in a crossover study. Deposition pattern was determined by gamma scintigraphy. The mean droplet sizes for each of the pumps were 47 and 53 μm for Pump A and Pump B, respectively. The results, Fig. 5.2, indicated that both pumps produced similar deposition patterns and that the droplets were deposited primarily in the anterior regions of the nose and along the floor of the nasal cavity. In this case, the size of the droplets determined the primary site of deposition.

This figure shows gamma scintigraphs following use of Pump A and Pump B in the same volunteer. The nasal cavity was divided into a nine region grid. Deposition in the upper, lower, inner, and outer regions of the grid was calculated as described previously (Suman 1999). The outer region represents the anterior portion of the nasal cavity including the nostrils
While nasal nebulizers have been shown to cover more surface area in the nasal cavity by decreasing droplet size (Suman et al. 1999), a simple reduction in droplet size alone does not guarantee an increase in the deposition pattern beyond the anterior nose. Nasal aerosols (Newman et al. 1987b) that utilize propellants to generate the spray have been shown to have smaller droplets compared to conventional nasal sprays. However, the deposition pattern is even more localized because of the exit velocity of the plume. The droplets cannot make the bend in the nasal airway and deposit in the front of the nose. This also leads to slower clearance from the nasal cavity for the pressurized formulation as the droplets deposit on non-ciliated regions of the nose.
Despite the challenges of delivery and maintaining contact with the nasal epithelium, the nose is a very attractive site for administration for both locally and systemically acting drugs.
3 Local vs. Systemic Action
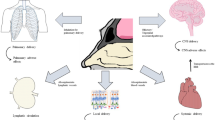
The easy access to the middle meatus and turbinates gives nasal drug delivery a unique advantage for local pharmacological action, systemic delivery, and potential for nose to brain delivery. The turbinates are richly vascularized and have a large surface area, which makes them an ideal target for systemic drug delivery. In addition, both the olfactory nerve and trigeminal nerve innervate the nasal cavity, which makes them a potential target for nose to brain delivery (Dhuria et al. 2009). Drugs reaching these targets can be rapidly absorbed across the thin membranes and can achieve potentially faster onset of action at lower doses while avoiding the disadvantages of oral dosage forms, namely, first pass metabolism and side effects from drug interactions with other organs (Dhuria et al. 2009; Laube 2007).
By delivering directly to sites of action, nasal drug delivery offers greater convenience and safety. It is a noninvasive and a painless method of drug administration, encouraging greater compliance compared to other routes of administration. Another advantage of nasal drug delivery for patients taking multiple drugs is that a nasally delivered drug may act as an adjunct to another drug given orally or intravenously (Behl et al. 1998a; Costantino et al. 2007).
3.1 Local Targets for Allergies
For the treatment of allergies, nasal drug delivery can place therapeutic agents within close proximity of the middle meatus and turbinates, the sites of inflammation. Thus sufficiently high levels of potent corticosteroids, antihistamines, or decongestants (Newman et al. 2004) can reach receptor sites at the target tissue, while systemic blood levels of these drugs are minimized.
Reducing this systemic exposure minimizes well documented side effects (Trangsrud et al. 2002; van Drunen et al. 2005). For example, antihistamines are known to sedate and interfere with psychomotor abilities. Delivered intranasally, these symptoms are absent (Costantino et al. 2007) because the drug does not reach the blood. Locally acting drugs have minimal or low bioavailability, and any blood levels that are detected have no correlation to efficacy because the drugs act locally. Table 5.2 summarizes commercially available prescription treatments for locally acting drugs approved in the United States and EU.
3.2 Systemic Delivery
In addition to topical treatments, the vascular-rich turbinates lend themselves to systemic drug delivery. Absorption in the nose can be rapid, and allows some molecules to achieve a greater bioavailability compared to oral administration. The turbinates have a large surface area and thin membranes. When drug contacts these membranes, rapid absorption into the blood occurs (Laube 2007; Newman et al. 2004). Unlike oral dosing, this absorption into the blood happens without first undergoing enzymatic degradation in the gastrointestinal (GI) tract nor first pass metabolism in the liver (other than the small amount that may be swallowed). Bypassing these metabolic pathways for poorly absorbed drugs allows comparable or greater blood levels, faster onset, and at a lower dose. These advantages (e.g., improved bioavailability, faster onset of action, lower dose) are particularly beneficial for drugs with potential toxic effects on the liver. When delivered through the nasal cavity, only a fraction of dose that may be swallowed could potentially reach the liver, instead of the entire dose when orally administered. When given orally, all drugs that clear the gastrointestinal tract are then available for the liver. Systemically acting drugs could therefore be more effective and safer when delivered intranasally directly to the blood supply within the turbinates.
Several marketed products use the intranasal route of administration to systemically deliver drugs for conditions such as pain and osteoporosis. MedImmue’s FluMist®, approved in 2003, delivers an annual influenza vaccine intranasally (see Product Profile) while Novartis’ Miacalcin® and Unigne Laboratories’ Fortical® are indicated for osteoporosis. Other systemically acting nasal products include pain medications for migraines: Imitrex® (sumtriptan nasal spray) marketed by GlaxoSmithKline, Migranal® (marketed by Valeant), and Zomig® (marketed AstraZeneca) and examples for pain management indications include Sprix® (marketed by Daiichi Sankyo) and Instanyl® (marketed by Takeda). Refer to Table 5.3 for a summary of the current commercial prescription landscape for systemically delivered nasal products in the United States and EU. Several areas of research and development are ongoing for nasal delivery routes of administration including the delivery of insulin for treatment of Type 1 diabetes (including Nasulin® under development by Cpex Pharmaceuticals) and the treatment of infectious diseases (including hepatitis C, HRV/SARS).
3.2.1 Product Profile: MedImmune’s FluMist® (Influenza Vaccine Live, Intranasal)
FluMist® is an annual influenza vaccine that is delivered intranasally (see Fig. 5.3). It is a live attenuated influenza vaccination (LAIV, trivalent, types A and B) that is preservative-free and contains three live attenuated influenza virus reassortants recommended by the US Centers for Disease Control and Prevention (CDC) (identified for the Northern Hemisphere 2011–2012 flu season as an A/California/7/2009 (H1N1)-like virus; an A/Perth/16/2009 (H3N2)-like virus; and a B/Brisbane/60/2008-like virus) (Fiore et al. 2010; MedImmune, online 2003), the same three CDC-recommended influenza strains in the traditional flu shot (a needle injection which builds up the body’s immunity to the flu through antibody production carried in the bloodstream—using inactivated (dead) virus (TIV)).

FluMist influenza vaccine live, intranasal (courtesy of MedImmune)
Once dosed intranasally (one 0.1 mL spray per nostril), the formulation stimulates an immune response by producing antibodies in the lining of the nose where the flu virus typically enters the body. FluMist is termed cold-adapted since the virus is engineered to replicate efficiently at temperatures below that of the body (25 °C) as is the case in the nasal passages (2003). Protective immunity is built up in the nasopharynx by the antigenic properties from the ca, ts, and att phenotypes derived from master donor virus (MDV) influenza strains (MedImmune, online 2003).
It was first approved by the FDA in June 2003 and is currently approved in five countries including Canada and EU (marketed by AstraZeneca as Fluenz® in select European counties). The original BLA for FluMist was submitted to the FDA for approval in 1998 and was subsequently rejected due to a lack of manufacturing validation and stability data (Food and Drug Administration 2003). MedImmune (formerly Aviron) was able to win US regulatory backing approximately 5 years later. Since FluMist contains a live virus, it is recommended for use by children, adolescents, and adults ages 2–49 years old.
In a placebo-controlled study in adults 18–49 years of age (study AV009), FluMist showed a decrease in any febrile illness of 10.9 % (95 % CI: −5.1, 24.4) and febrile upper respiratory illness of 23.7 % (95 % CI: 6.7, 37.5) (MedImmune, online). In comparative efficacy data between FluMist and an active control (study MI-CP111 using an injectable influenza vaccine made by Sanofi Pasteur, Inc.) FluMist demonstrated a 44.5 % (95 % CI: 22.4, 60.6) reduction in influenza rate in children <5 years of age as measured by culture-confirmed modified CDC-ILI (MedImmune, online). Given the comparative efficacy and safety of FluMist (Ambrose et al. 2011), US regulatory approval was received by MedImmune with four post-marketing clinical commitments (including a 60,000 patient safety trial, adverse event monitoring in patient subsets, an investigation of vaccine virus shedding and immune response, along with providing additional revaccination data) and one nonclinical commitment (to complete additional reproductive toxicology studies) (U.S. Food and Drug Administration, online). FluMist 2011 revenue totaled $161 MM and $174 MM for full year 2010 (Astrazeneca, online).
3.3 Nose to Brain
Nasal delivery also offers the opportunity to bypass the blood–brain barrier and deliver drugs directly to the central nervous system. This barrier prevents systemically delivered drugs, whether delivered orally, intravenously, or by other routes, from reaching significant concentrations in the brain. Two cranial nerves, the olfactory nerve and the trigeminal nerve, pass through the nasal cavity. An intranasally delivered drug could use these pathways to reach tissue in the central nervous system and achieve levels necessary to be of therapeutic benefit. Additionally, there are other potential vascular, cerebrospinal, or lymphatic pathways as routes to the central nervous system (Dhuria et al. 2009).
Currently, no marketed drug products exist that act via nose to brain. One challenge is targeting deposition of sprayed droplets in the regions where olfactory neurons are located. However, there are research programs to treat Alzheimer’s and Parkinson’s diseases, some of which have shown some success (Dhuria et al. 2009). Given the overall difficulties with treating central nervous disease, nose to brain delivery could offer a promising way to achieve efficacy while minimizing side effects of drugs.
3.4 Challenges of Nasal Drug Delivery
Nasally delivering drugs to therapeutic areas of interest can make them more effective for local action, systemic action, and central nervous system action, at lower doses with minimum side effects. However, delivering drug to the specific regions of interest is challenging. As mentioned previously, these challenges arise because the winding and narrow geometry of the nasal airways filter most droplets into the anterior third of the cavity (Kimbell et al. 2007; Laube 2007; Hardy et al. 1985; Newman et al. 1987a; Suman et al. 1999; Vidgren and Kublik 1998). Most targets, though, are located in the posterior nasal cavity. Even less reach the access points for the nerves to the brain in the olfactory region. To overcome these challenges, new devices are in development to target drugs specifically to these regions (Djupesland et al. 2006). Also with these new devices come challenges to accurately assess how well they deposit within specific areas of the nasal cavity.
Another challenge with nasal drug delivery is mucociliary clearance. Most droplets landing within the therapeutically beneficial posterior nasal cavity are removed by mucociliary clearance within 20 min (Hochhaus et al. 2002). The drug, therefore, must absorb and/or act quickly. Formulation changes, such as using absorption enhancers (Behl et al. 1998b; Costantino et al. 2007; Na et al. 2010) and using mucoadhesives to increase residence time (Ugwoke et al. 2005), are actively being researched in order to take advantage of benefits of nasal drug delivery.
4 Formulation Strategies
Until recently, nasal formulations were primarily prepared in the form of either solutions or suspensions and frequently required the use of preservatives (such as BAC) to prevent microbial contamination and microbial growth. Due to potential adverse events associated with the use of these preservatives, regulatory agencies from several countries, including Germany, requested that the manufacturers avoid the use of preservatives in the nasal formulations. These limitations necessitated the development of preservative-free formulations and thereby led to adoption and implementation of various strategies to circumvent the use of preservatives.
In order to develop preservative-free nasal formulations, novel approaches including the use of preservative-free devices and various sterilization techniques have gained widespread attention. Since the aforementioned approaches tend to rely heavily on the use of sterile techniques for manufacturing, compliance with the procedures related to the use of sterile techniques, as outlined in USP <797> Pharmaceutical Compounding-Sterile Preparation, is critical.
The following summarizes the current landscape of nasal formulation development, the limitations of using preservatives, and describes USP <797> regulations as they apply to manufacturing of nasal preparations under sterile conditions.
4.1 Current Landscape
The majority of commercially available nasal formulations are active pharmaceutical ingredient(s) (APIs) mixed with excipients such as preservatives, suspending agents, emulsifiers, or buffering agents. Microbial growth can occur in the nasal formulation preparations either during manufacture or while in use by the patient. During manufacture, the most commonly occurring sources of microbial contamination include the handling process and the use of contaminated excipients (Groves and Murty 1990).
These sources of contamination, either alone or in conjunction, can negatively impact the quality of the finished product and shelf life. Once the nasal product is used by the patient, factors such as unhygienic handling or the contact between the tip of the nasal delivery device and nasal cavity can further introduce contamination via migration into the nasal spray tip. Further, the conventional design of the nasal delivery device may allow microbial contamination to enter the formulation by the intake of unfiltered air.
4.1.1 Currently Adopted Approaches to Address Microbial Contamination
To avoid contamination and prevent microbial growth, manufacturers use some of the following approaches:
-
Adding preservatives to the nasal formulations: This is the most commonly used approach and there are a variety of commercially available preservatives that are routinely employed.
-
Preventing the entry of microorganisms through sterile manufacturing of the nasal formulation: This approach can be applied to unit-, bi-, as well as multi-dose products. The drug formulation is prepared under sterile conditions where no preservative is added or the product can be terminally sterilized. If the product is not sterile, then the finished product is generally subjected to radiation to ensure inactivation of microbial contamination (if any) after filling. A detailed description of sterile manufacture is discussed in Sect. 5.6.
-
Selection of a preservative-free device: After manufacturing the formulation under sterile conditions, these devices (discussed in Sect. 5.5) require no preservatives. Several companies also manufacture preservative-free pumps for multi-dose formulations. The special tip seal and filter in these pumps reduce microbial growth upon repeated use. Another type of device platform, called “Bag-on-Valve” (BOV), also supports preservative-free formulations, as discussed in Sect. 5.5.1.3.
4.1.2 Use of Preservatives
Adding preservatives is a simple, robust, and cost-effective method of controlling microorganisms. The FDA guidance states that if preservatives are used in the nasal formulation, the minimum content limit should be demonstrated as microbiologically effective by performing a microbial challenge assay of the drug formulated with an amount of preservative equal to or less than the minimum amount specified. Although BAC is by far the most widely used preservative, other preservatives such as thiomersal, chlorhexidine, chlorobutanol and phenylethanol, potassium sorbate, and parabens are also routinely employed in the formulation of nasal drops and cosmetics. Table 5.4 includes the list of preservatives and the ranges of concentration used.
4.1.3 Limitations Associated with Preservatives
Although preservatives have been used for decades, and they are simple, they do have limitations. These include adverse effects on the nasal mucosa—particularly in children, and the potential of preservatives to cause discomfort, irritation, and other side effects after long-term use. In certain cases, preservatives affected the cilia in the nasal cavity by altering the elimination of the nasal mucus (in cases of nasal infection) and slowing down or even stopping mucociliary clearance, an essential natural mechanism for protecting the upper airways. Several reviews have examined adverse events associated with the use of preservatives (Lebe et al. 2004; Mallants et al. 2007; Bernstein 2000; Merkus et al. 2001; van de Donk et al. 1980, 1982).
Preservatives also introduce formulation challenges due to drug stability/drug-device compatibility issues, and/or by modifying the smell and/or taste of the nasal drug products. For example, phenylethylalcohol can be perceived by some patients as causing an unpleasant odor, potentially reducing patient compliance.
Quite recently, several countries have expressed concern about the risk associated with the use of BAC. Therefore, manufacturers in Europe, Latin America, and more recently Japan have started to consider eliminating the use of preservatives and reformulating their nasal products. Although the FDA still allows the use of preservatives, the FDA has started to encourage manufacturers to actively adopt the use of preservative-free techniques.
4.2 Development of Preservative-Free Nasal Products
Since the goal is to avoid adding preservatives while ensuring that the formulation is sterile during the manufacturing and use period, it is important that the formulation is prepared and processed under aseptic conditions before and during transfer of the formulation into the nasal delivery device. Alternatively, terminal sterilization may be employed if suitable for the formulation and device.
Recently, several US manufacturers have ventured into the arena of aseptic nasal formulation processes for manufacturing nasal formulations. Since the processes related to sterile manufacturing techniques, as outlined in the United States Pharmacopoeia USP <797>, are considered the “gold standard,” these processes have also been adopted by manufacturers of nasal products. USP <797> provides information on procedures and practices that may be adopted to prevent microbial contamination. The chapter discusses minimum quality standards based on state-of-the-art scientific information and the best sterile compounding practices. It is important to note that the goal of USP <797> is to provide a global view of the various practices that can be adopted to prepare sterile formulations across the manufacturing spectrum, rather than describe the approaches that can be adopted for a particular formulation. Hence, although USP <797> does not specifically describe the application of various sterilization techniques in the context of manufacturing nasal formulations, the general principles outlined in USP <797> still apply to the manufacturing of nasal formulations.
Once the formulation is prepared under aseptic conditions using the principles outlined in USP <797>, the next step is to ensure that the nasal formulation delivered to the patient is free of microbial contamination. Therefore, it is critical that the device used for delivering the nasal formulation provides a sterile environment to the nasal preparation. For unit- and bi-dose formulations, a preservative-free pump is not needed because the formulations are designed for a single use. In some cases, the conventional pump used for multi-dose preservative-free formulations can be subjected to gamma radiation to ensure that the pump is free from microbial contamination. This could be performed before or after the filling process.
4.2.1 Ideal Design Characteristics of Preservative-Free Pump
Throughout the use of product life, the conventional nasal delivery devices can introduce microbial contamination by the following routes: the orifice, the venting air which replaces the dispensed liquid, or due to insufficient container/dispenser fit (Brouet and Grosjean 2003).
In order to ensure that nasal delivery devices are free of microbial contamination, it is vital that the device can be sterilized before or after the filling process. Therefore, specific polymeric materials such as high density polyethylene are a good choice for manufacturing of devices as they resist gamma irradiation and maintain their physical properties.
For unit-dose and bi-dose devices, creating a preservative-free environment for the device is not a major concern since the disposable devices are capable of delivering one or two shots only. However, for multi-dose devices, the following additional considerations apply to ensure that the formulation remains protected inside the container:
-
Pump as a closed system: Metering spray pump should work as a closed system (full seal system). Unlike conventional metering nasal spray pumps, the closed system does not allow air to enter into the container and come into contact with the nasal drug product, thus preventing contamination from airborne germs.
-
Using a filter: When metering spray pumps are equipped with a filter, the venting air is sucked through a filter assembled inside the pump, which eliminates the airborne germs and keeps them out of the container.
-
BOV technology: Unpreserved product is stored in a pouch and dispensed through a valve. The content of the pouch is not in contact with the outside atmosphere.
-
Using bacteriostatic agent: The agent such as silver ions could be added to device components so that liquid that comes in contact with them gets protected. Silver ions have a large antibacterial spectrum and low toxicity to humans.
4.2.2 Case Study on Preservative-Free Systems: Mechanical Spray Pumps
The orifice of any container is a contamination risk because it contacts the mucosa and/or skin, areas populated by microorganisms and body fluids. Some marketed systems use the oligodynamic activity of a silver wire in the tip of the actuator, a silver-coated spring, and ball (Groß 2000). These components control release of silver ions into the formulation over time. The system minimizes microorganisms between long dosing intervals, even when the tip is immersed into bacterial-contaminated fluid (Bagel and Wiedemann 2004). Silver ions are widely used for their antiseptic properties and are even used for wound dressings. They are safe and have no adverse effects. One must ensure, however, that the silver ions do not react with the formulation, e.g., chloride ions forming micro-precipitations. This effect may be overlooked because it is most relevant for spans of 6–12 h between individual actuations, intervals not usually evaluated during development.
Consequently, the most recent preservative-free systems follow a purely mechanical approach to minimize interactions between device parts and formulation. One way to prevent contamination via the orifice is “tip seal technology.” Both spray pumps and ophthalmic droppers use this technology. A spring-loaded valve is located directly below the opening of the tip orifice, not allowing any microbes to migrate from any surfaces or contacted liquids into the system, sealing the orifice under resting conditions. The tip seal keeps the system closed until a defined pressure (for sprays it is more than 3 bar) is reached by actuating the system. Once a defined pressure is reached, the system opens and formulation is forced through the orifice at a higher pressure than needed to open the valve. When the pressure drops at the end of the actuation, the tip seal immediately closes the orifice with an outward movement. Therefore, no backflow of potentially contaminated medication or other liquid is possible. Depending on the pump system, the fluid path may even be “metal-free,” which means the springs needed for the device operation do not come in contact with the formulation.
At any time when a liquid is dispensed out of a container, the pressure inside such container decreases gradually. To avoid contamination of the formulation via venting air, different technical solutions are used. The simplest way is sterile filtration of the venting air via separate filters or filter gaskets. For oxygen-sensitive formulations, the so-called collapsing bags or depressed systems are used. The formulation is filled in a special, microbial tight bag which is protected by a surrounding bottle. When dispensing the product, the bag collapses with the content not coming in contact with the ambient air. Some pumps are constructed in such a way that the entire system is air-tight and during use some vacuum (up to −300 mbar) is generated within the bottle. Those systems allow even a purging with inert gases to reduce oxygen content in the container headspace.
While appearing complex, these approaches to avoid the use of preservatives for multi-dose devices are well established and matured technologies. Though not commercially available in the United States yet (as of publication), unpreserved multi-dose nasal sprays have gained substantial interest and market share in places like Europe and Latin America (Fig. 5.4).

Examples of commercially available products from Brazil (top left), Australia (bottom left), France (top right), and Austria (bottom right)
As the development paradigm for nasal formulations shifts from preservative-based formulations to preservative-free formulations, in particular for Latin American countries, the information outlined in USP <797> will continue to provide the roadmap for manufacturing preservative-free nasal formulations prepared under a sterile environment which will ultimately benefit the entire healthcare community.
5 Device Landscape for Nasal Drug Delivery
5.1 Nasal Spray Devices: Liquid Formulations
Nasal spray devices for liquid formulations come in various dose and container volume sizes. The devices include unit-dose, bi-dose, and multi-dose delivery systems for both preserved and preservative-free. Fill volumes range from 125 μL (for unit-dose) to 30 mL or larger (for multi-dose) and spray volumes range from 25 to 140 μL. The selection of the spray volume is driven by the therapeutic dose. The selection of the spray pump is driven by the volume of formulation that is required to support that dose. The selection of the fill volume is generally driven by the intended frequency of use of the drug product—for a chronic-use product (for example, for nasal allergies), a multi-dose device containing 1 month’s supply might be selected; for an acute-use product (for example, for controlling seizures or pain management), a unit-dose or bi-dose device might be chosen.
5.1.1 Case Study for Characterization of Multi-dose Nasal Spray Devices
This case study reviews the steps typically taken when selecting a multi-dose nasal spray device. The selection procedure is based on the spray characteristics of the product formulation from the device in question.
A minimum of 12 devices from one lot were taken and filled with the product formulation for the study. The amount of dose delivered on n = 6 actuations was determined by hand after priming. The number of doses delivered per bottle was then determined on n = 6 devices. At this time a visual evaluation of the plume shape was made (a nicely formed plume should be evident rather than a liquid stream).
Once the formulation “sprayability” had been demonstrated, the device was loaded into a computer-controlled device actuator (supplied by Proveris Scientific) to determine stroke length, which is the distance moved when the spray pump is compressed. Using this value, and default velocity and acceleration parameters, the dose weight was determined. The droplet size distribution at 3 cm from the orifice at both the beginning and end of the container life was determined using the stroke length and default velocity and acceleration parameters.
Selection criteria:
-
The number of doses delivered per bottle must meet the label claim.
-
The dose weights obtained must meet the label claim, and the variation (% RSD) in the dose weight data (both hand-actuated and computer-actuated data) must be within the acceptable range.
-
The droplet size distribution at the 10th, 50th, and 90th percentiles must be within the acceptable range at both the beginning and the end of the container life.
If droplets are too large, the formulation may deposit in the front of the nose and tend to drip out of the nose; on the other hand, droplets smaller than 10 μm may travel deeper into the nasal cavity and reach the lungs—which are not the intended delivery site. Ideally, the percentage of droplet smaller than 10 μm should be kept to a minimum. At the upper end of the size range (90th percentile), the majority of droplets should be less than 150–200 μm.
5.1.2 Container/Closure Systems
Suppliers of nasal spray container/closure systems include: Becton Dickinson, Coster, MeadWestvaco, Rexam, and Aptar Pharma. Examples of the various systems currently available for use with liquid nasal sprays are shown in Figs. 5.5, 5.6, 5.7, and 5.8.

Classic pump (multi-dose pump that is sterilized for a low preservative nasal spray application) (courtesy of Aptar Pharma)

Cartridge pump system, CPS (multi-dose nasal spray pump with microfilter air filtration system to protect non-preserved formulation) (courtesy of Aptar Pharma)

Unit-dose, UDS liquid (single dose liquid nasal spray device) (courtesy of Aptar Pharma)

Bi-dose, BDS liquid (two spray single dose liquid nasal device) (courtesy of Aptar Pharma)
Classic spray pumps are widely used for local and systemic nasal drug delivery, and are used for preserved formulations. The extensive range of closures, actuators, and accessories available make this spray pump highly adaptable to fit customers’ specific requirements. Classic pumps are incorporated into a number of drug products marketed in Asia, Europe, Latin America, and the United States.
The cartridge pump system (CPS) is a highly versatile spray pump. It is designed for the multi-dose delivery of preserved or non-preserved drug formulations. CPS can be used for a wide range of therapeutic applications including allergy, pain, and intranasal mass vaccination. CPS can be terminally sterilized by gamma irradiation.
Unit-dose liquid delivery systems are available for delivery of sterile or preserved single dose medicines. For unit-dose (UDS) and bi-dose (BDS) devices, a coated rubber stopper is placed in the device vial. This stopper contains “fins,” which create a good seal to prevent evaporation of the formulation during storage, and prevent the ingress of microorganisms. During the insertion of the stoppers, the “fins” are compressed to allow air within the vial to escape and prevent a build-up of pressure within the sealed unit.
The pumps used with multi-dose devices contain a gasket which is compressed during application to give an air-tight seal. These pumps can be a screw-on, snap-on, or crimp-on design.
After manufacture, the sterility of drug products is maintained by the container/closure system. For single use or unit-dose devices, sterility is assured by the integrity of the container/closure system itself until the time of use. For multi-dose devices, however, the situation is not as straight forward, and sterility can be compromised when the device is sprayed for the first time, and on each subsequent use. One method used to maintain the sterility of the product in-use is by incorporating a 0.2 μm filter into the dispensing tip/actuator (see Fig. 5.6). With these devices, the return air that is introduced into the container after the dose is expelled is filtered through the 0.2 μm filter—thereby maintaining the sterility of the product throughout its in-use lifetime.
Alternatively, antimicrobial preservatives, as discussed in Sect. 5.4, may be included in the formulation to kill or to inhibit the growth of microorganisms inadvertently introduced during use. Single preservatives, and more often combinations of preservatives, are commonly used in pharmaceutical formulations (including some sterile formulations, for example, eye drops and multi-dose injections) to prevent the growth of bacteria.
5.1.3 Alternative Preservative-Free Nasal Product: The “Bag-on-Valve”
In addition to maintaining a sterile environment, engineering of the physical device also helps ensure that the preservative-free formulation remains free from microbial contamination. One example of how optimizing the design of the device can help prevent microbial contamination for saline nasal sprays is the BOV technology.
Briefly, BOV technology can potentially be used whenever it is important to separate the drug product from the propellant, thereby ensuring product purity. The major benefits of using the BOV technology include cost-effectiveness, better preservation of the drug product, and environmental safety. Figure 5.9 shows key components of the BOV system.

An overview of “bag-on-valve” (BOV) technology. The bag and valve are inserted into the canister during assembly. The canister is pressurized with the bag then filled with drug product (courtesy of Aptar Pharma)
The product is sealed inside a pressurized container (generally an aluminum can) and is released by compressed air or nitrogen. The BOV technology offers several benefits to the consumer such as longer shelf use without the use of preservatives and ability to use at all angles because the spray is driven by compression of the bag by the propellant.
5.1.4 Nasal Spray Characterization Testing and FDA Nasal Spray Guidance (Food and Drug Administration 2003)
In order to support a regulatory filing for a nasal spray product, the current FDA guidance documents make recommendations as to the characterization and test data. These test recommendations are summarized in Table 5.5. To date, the FDA has not issued guidances specific to unpreserved nasal spray formulations. Unpreserved formulations are generally manufactured sterile, and the testing in Table 5.5 applicable to that type of formulation is undertaken.
5.2 Nasal Spray Devices: Dry Powder Formulations
Following the success of liquid formulation nasal sprays, research and marketing interest has expanded to include dry powder nasal devices. Dry powder inhaler (DPI) systems were undertaken as an alternative to the pressurized metered dose inhalers (pMDI) that use ozone depleting propellants. Dry powder systems (both DPI and nasal) generally comprise a micronized active drug and suitable powdered excipients within an apparatus that is designed to aerosolize the formulation. Some of the advantages of dry powder drug delivery systems include formulation stability, a system that is propellant-free, and that less coordination between actuation and inhalation is required (Telko and Hickey 2005; Serra-Batlles et al. 2002). Also, because of the absence of moisture in the dry powder system, microbial growth is minimized or eliminated, and the use of preservatives is less critical than in the case of liquid formulations. Deposition efficiency, dose uniformity, complexity of manufacturing, and device to device performance remain as concerns for dry powder technology (Chan 2006; Islam and Gladki 2008).
Dry powder devices come in unit-dose, bi-dose, and multi-dose systems. Some of the container/closure systems currently available for use with dry powder nasal sprays are shown in Fig. 5.10.

Unit-dose powder (UDP) and bi-dose powder (BDP) devices (courtesy of Aptar Pharma)
6 Manufacturing and Filling Nasal Delivery Systems
There are several different methods to achieving a sterile product that cover both aseptic manufacture and terminal sterilization. In aseptic manufacture, the drug product, container, and closure are first subjected to sterilization separately, and then brought together in an extremely high-quality environment. Terminal sterilization, on the other hand, involves manufacturing a low bioburden product in an environment designed to minimize microbial and particulate contamination, and then subjecting the final container to a sterilization process such as heat (e.g., autoclaving), chemical sterilant (e.g., ethylene oxide), or ionizing radiation (e.g., gamma or electron beam). Each of these methods of producing a sterile product has its own technical challenges.
6.1 Aseptic Manufacture
Various sterilization processes are employed for both the container/closure system (for example, glass containers are subjected to dry heat; suitable plastic containers such as high density polyethylene are subjected to ionizing radiation; rubber closures are subjected to moist heat) and the dosage form. Some options for producing a sterile dosage form are presented below.
6.1.1 Sterile Filtration
If the dosage form is a liquid solution, or a very low viscosity emulsion, sterilization can be affected by passing the solution through a filter with a pore size small enough to trap out any microbial contamination (0.2 μm). The filtered dosage form is then kept sterile until it is enclosed in the final container/closure system (see Fig. 5.11).

Sterile filtered dosage form
During process development, an assessment of the filter and filtration process must be carried out—including:
-
Retention of the drug substance by the filter/loss of potency—by testing the dosage form pre- and post-filtration
-
Testing the filter for potential extractables and leachables
-
Testing the filter for microbial retention
The bioburden of the pre-filtered solution must be evaluated as part of the in-process testing to ensure that the filter does not become overloaded with contaminants. The integrity of the filter must also be checked following the filtration process.
Filtration cannot be used as the sole means of achieving a sterile bulk formulation in the case where the dosage form is viscous or contains suspended particles (for viscous or suspension formulations, see Sect. 5.6.1.2). The following case study summarizes work carried out to validate a sterilizing filter.
6.1.1.1 Case Study: Sterilizing Filter Validation
6.1.1.1.1 Purpose
The purpose of this study was to demonstrate that the sterilizing filter was acceptable and capable for the sterile filtration of the product base. This was achieved through a review of the supplied filter documentation, and by carrying out various verification activities.
6.1.1.1.2 Verification Requirements and Results
The critical operating parameters associated with the sterile filtration of the product base (temperature, flow rate, and pressure) that could potentially impact the performance and integrity of the filtration process must be within the design capability of the filter cartridge/membrane. Table 5.6 lists the verification requirements for the sterilizing filter, the acceptance criteria, and the results obtained.
6.1.2 Combination Processes
In cases where filtration cannot be used as the sole means of sterilization (for example, for viscous or suspension formulations), there are several combination processes available.

Presterilized isolator attached to the mixing vessel
6.1.2.1 Dry Heat or Ionizing Radiation of Powders Followed by Aseptic Addition to Pre-filtered Base
Here, the powders are first packed into a suitable container, and then subjected to sterilization by dry heat (e.g., 170 °C for 1 h) or ionizing radiation (gamma or electron beam). The liquid formulation base is sterilized by filtration. The sterile powder is then added to the sterile formulation base by aseptic addition—for example, via a presterilized isolator attached to the mixing vessel (Fig. 5.12). Factors to consider when presterilizing powdered active ingredients include heat stability of the active ingredient; stability to ionizing radiation; packaging of the active ingredient; compatibility between the active ingredient and the packaging; and extractables and leachables from the packaging.
6.1.2.2 Aseptically Combining Phases Sterilized by Different Methods
Here, the bulk formulation is split into two distinct phases—for example, the oil and aqueous phases of an emulsion. The drug substance is dissolved in one of the phases. The oil phase is then sterilized by passing it from a phase tank into the final mixing tank, via a 0.2 μm filter; the aqueous phase is autoclaved in a second phase tank. These two phases are then combined in a final mixing tank, and held sterile until packaging (Fig. 5.13).

Aseptic combination of sterilized phases
Once the bulk formulation has been produced sterile, the manufacturing environment for the downstream processes (filling and closing the nasal delivery system) needs to be kept and monitored at a very high quality (low bioburden and particulate levels). The whole manufacturing process needs to be validated at regular intervals (usually every 6 months) to demonstrate that the aseptic handling techniques and manufacturing operations do not compromise the sterility of the final product. This validation exercise involves carrying out media simulations, processing microbiological growth media through the entire process train and into the final container/closure system—and needs to include all anticipated process interventions, manual and mechanical manipulations, and machine downtime. The media is then incubated to determine if the process is contamination-free. If contamination is found, the contaminants need to be identified, and causes assigned to the failure of the aseptic operation.
In an aseptic operation, controlling the sterility of the drug product and container/closure system is relatively straight forward; it is the human interface that provides the biggest challenge and the most likely cause of contamination.
6.2 Terminal Sterilization: Heat
The use of heat (dry heat or autoclaving) to terminally sterilize the drug product can lead to challenges to the thermal stability of the formulation and formulation ingredients. Dry heat sterilization involves taking the product up to 170 °C for a set period of time; autoclaving (or wet heat sterilization) involves heating to 121 °C. Whereas dry heat uses the heat itself to bring about sterilization, autoclaving uses the water contained within the formulation to achieve this. As the temperature of the product increases during autoclaving, the vapor pressure within the container/closure also increases. To prevent package rupturing, the pressure within the autoclave chamber must be controlled to match that within the container/closure.
Many drug substances will degrade or denature at high temperature, leading to loss of potency and the generation of degradation products. Also, many formulation bases will fail when exposed to such high temperatures—for example, ointment bases will lose viscosity and lead to sedimentation of any suspended solids; emulsions will exhibit phase separation. Aqueous gels are typically the most tolerant formulation type to the effects of heat sterilization—usually being sterilized by autoclaving.
6.3 Terminal Sterilization: Chemical Sterilant
Chemical sterilants are highly reactive and affect sterilization by oxidation. Approved chemical sterilants are ethylene oxide, hydrogen peroxide, and ozone. The technical challenges with the use of chemical sterilants are (a) getting the chemical sterilant into the pack so that it can interact with any microbial contaminants, (b) ensuring that the sterilant doesn’t affect the potency of the drug substance, and (c) getting the sterilant (and any degradation products) back out of the pack after the sterilization process is complete.
Semipermeable packaging (to enable gas transfer) is used to allow penetration and removal of the sterilant. As a result, this method of sterilization is unsuitable for liquid and semisolid formulations.
Desorption studies are carried out on the sterilized product to ensure that the chemical sterilant and any degradation products are reduced to acceptable levels before the product can be distributed and used. In the case of ethylene oxide, the degradation products are ethylene glycol and ethylene chlorhydrin. These degradation products have toxic effects and their acceptable level is controlled.
Because of the challenges with the use of chemical sterilants, they are mainly used for the sterilization of device components rather than the finished nasal spray product.
6.4 Terminal Sterilization: Ionizing Radiation
The use of radiation to bring about terminal sterilization is very effective, but poses technical challenges regarding the stability of the drug substance and drug product to its ionizing effects. Aqueous-based formulations are unsuitable to sterilization by this route due to the formation of hydroxyl radicals, which then react with other chemicals within the formulation. Although the majority of nasal spray formulations are currently aqueous-based, a few nonaqueous-based liquid formulations exist in the development phase.
Radiation can also affect polymers causing either cross-linking or chain scission. This can manifest itself in many ways; for example, some plastics can become brittle; some plastics can discolor; gels can lose viscosity; adhesives can become hard and less sticky; or, conversely, adhesives can become stringy and more sticky. The effect of ionizing radiation needs to be monitored on a product by product basis, and over an extended period of time—as these effects are not always apparent immediately after processing.
The following two case studies summarize work carried out to validate a gamma irradiation cycle for nasal spray device components, and to determine the effects of gamma radiation on the extractable and leachable profile of a gamma-irradiated delivery device.
6.4.1 Case Study: Radiation Sterilization of Nasal Spray Device Components
6.4.1.1 Purpose
The purpose of this study was to qualify radiation sterilization as an acceptable means of sterilization for nasal spray device components. The ANSI/AAMI/ISO 11137-2: 2006 (VDmax25) guideline was followed to achieve a sterility assurance level of 10−6.
6.4.1.2 Definitions
-
Bioburden: Population of viable microorganisms on a material (e.g., product, package, or component).
-
Dosimeter: Device or system having a reproducible and measureable response to radiation, which can be used to measure dose exposure.
-
Sterility assurance level (SAL): Probability of a viable microorganism being present after sterilization (normally expressed as 10−n).
-
Sterility testing: Test performed to determine if viable microorganisms are present.
-
Verification dose: A radiation dose estimated to produce a sublethal SAL for a material. Verification doses are used in dose setting to establish or confirm the sterilization dose.
6.4.1.3 Dose Setting
First, the mean bioburden of the components was determined by evaluating ten unirradiated samples randomly selected from each of three separate production lots. Aerobic and fungal bioburden counts were performed on each sample. A verification dose (SAL 10−1) was selected based upon the average bioburden results, adjusted for recovery efficiency, and referencing Table 9 in ANSI/AAMI/ISO 11137-2: 2006. The closest number greater than or equal to the average adjusted bioburden was selected for dose determination.
Next, a sublethal dose verification experiment was carried out. Samples were exposed to the verification target dose, ±10 %. Calibrated dosimeters were used to verify the dose. After exposure, each sample was visually checked for damage and/or compromised packaging prior to sterility testing. Sterility testing was performed by adding the test sample to Soybean Casein Digest Broth and Fluid Thioglycollate Medium, and incubating for 14 days at 20–25 and 30–35 °C, respectively. Bacteriostasis and fungistasis testing was also carried out.
6.4.1.4 Acceptance/Rejection Criteria
If, after completion of the verification dosing, the results of the sterility test showed that one or fewer positives were observed, the sterilization dose of 25 kGy minimum would be considered valid. If, however, the results of the sterility test showed more than two positives, and if after repeat verification dosing, the results of sterility testing still showed positives, the adequacy of the 25 kGy sterilization dose might not be acceptable.
6.4.1.5 Establishing Sterilization Specifications and Revalidation
Factors to consider when establishing sterilization specifications include a description of the material to be sterilized and its packaging, carrier loading configuration, dose mapping, minimum dose (to give acceptable sterilization), maximum dose (for materials compatibility), and placement of dosimeters.
Once established, routine dose auditing exercises are carried out (e.g., every 3 months) to assess the ongoing material bioburden and continued effectiveness of the sterilization cycle. Any changes in the material or manufacturing location must be evaluated for their possible influence on the sterility validation.
6.4.2 Case Study: Extractable and Leachable Study on a Gamma-Irradiated Delivery Device
6.4.2.1 Purpose
The purpose of this study was to identify any extractable and leachable materials present in a delivery device that was to be used in contact with a sterile product. The device in question was to be pre-irradiated using gamma irradiation, and then aseptically filled with the product.
6.4.2.2 Method Development: Volatile and Semi-volatile Materials
Initially, a headspace GC/MS method was developed to analyze the device for the presence of any volatile and semi-volatile components prior to irradiation.
Sections of material were placed into a 20 mL headspace vial and analyzed using GC/MS at a range of oven temperatures between 80 and 230 °C. The results demonstrated that the temperature which yielded the maximum number of extractable peaks was 230 °C.
Next, a headspace vial equilibration time study was conducted to determine the optimum time at which peak areas were maximized. The peak areas of four randomly chosen peaks were monitored at six different vial equilibration times ranging from 10 to 120 min. For the four peaks studied, a common trend of the peak area plateauing after 30 min was exhibited. This 30 min vial equilibration time was then used in an attempt to characterize all unknown peaks.
The identities of the peaks were confirmed by injecting pure standards of the components proposed by the GC/MS NIST library, and matching the R t values and mass spectral fragments. In total, nine peaks were identified and confirmed by MS.
6.4.2.3 Method Development: Nonvolatile Materials
In order to characterize the nonirradiated device material, a solvent extraction procedure was developed. GC/MS and LC/MS methods were also developed for the analysis of any possible nonvolatile species.
Initially, an 8 % ethanol in water solution was used in contact with the device housed in a stoppered graduated cylinder. This was placed into a water bath and incubated for 2 h at 65 °C. The cylinder was then cooled, and 15 mL of the extract was pipetted into a conical glass vial and evaporated to dryness using N2 gas. After complete evaporation, the remaining extract was reconstituted with 0.5 mL of solvent, and then analyzed using the GC/MS conditions previously developed. As no extractable peaks were observed, the extract procedure was repeated using a solution of 3 % acetic acid in water, and incubated for 2 h at 100 °C. Again, no peaks were observed.
Since the last two approaches failed to yield any extractable peaks, a more aggressive solvent (n-heptane) was chosen. Initially, a blank of n-heptane was analyzed by GC/MS. Many peaks were observed in the n-heptane solvent that could possibly interfere with any extractable peaks, so n-hexane was chosen as an alternative. The change in solvent from n-heptane to n-hexane resulted in fewer solvent peaks and a cleaner baseline. Therefore, n-hexane was implemented as the extraction solvent.
Initially, n-hexane was used with an incubation time of 2 h at 50 °C. After evaporating to dryness with N2 and reconstituting the remaining extract with 0.5 mL of n-hexane, analysis by GC/MS showed no additional peaks other than those present in the solvent. As a result, longer incubation periods of 6 and 24 h were implemented. Even with the increased incubation period at 50 °C, no components were extracted. Additionally, no components were extracted when solvent studies were carried out at room temperature and incubation periods up to 168 h.
Overall, the GC/MS solvent extractable studies conducted on the nonirradiated device material showed that only solvent peaks were present and no extractables were observed.
An LC/MS method was then developed and used to analyze the n-hexane solvent extract which had been incubated for 2 h at 50 °C. In this case, 0.5 mL of acetonitrile was used to reconstitute the extract. An electrospray positive (ES(+)) mode of ionization was initially employed. Comparing the extract chromatogram to that of blank acetonitrile, a single peak at R t = 25 min was observed. Upon further investigation, the mass spectral pattern of the unknown peak was also observed in a control sample where the n-hexane solvent had been evaporated to dryness using N2 gas, and then reconstituted with acetonitrile. This suggested that the peak was related to the solvent, and not an extractable peak. Even when the incubation period was increased up to 24 h at 50 °C, or 72 h at room temperature, no other peaks were observed other than those present in the solvent control or the blank. Furthermore, no additional peaks were observed on changing the mode of ionization from ES(+) to Atmospheric Pressure Chemical Ionization positive (APCI(+)), ES(−), or APCI (−).
6.4.2.4 Evaluation of Irradiated Devices
Devices that had been exposed to gamma radiation at both a nominal dose (26.0–26.5 kGy) and a higher dosage (51.7–53.3 kGy) were analyzed using the same conditions listed above. The results from this analysis showed that the irradiation performed on the devices removed the volatile components previously observed in the headspace analysis of the nonirradiated material. Likewise, no peaks were observed using the solvent extraction procedure.
Finally, a leachable study was performed on an aged product sample to determine if any extractables previously detected were present. Product that had been stored in an irradiated device at 40 °C for 6 months was analyzed and compared against the formulation base stored in a glass jar, not exposed to the device material, to observe if any leachable peaks were present. GC/MS and LC/MS analysis showed no peaks were present in the stability sample other than those present in the placebo.
7 Analytical Techniques and Drug Product Characterization Studies for Nasal Spray and Nasal Aerosols
Analytical tests that are used to characterize the performance include methods that measure the size of emitted droplet, the shape of the spray, as well as critical formulation components such as viscosity and content uniformity. A list of these tests is shown in Table 5.5 and is described in more detail as this section progresses. These tests can be used to characterize the reproducibility of performance and make decisions regarding device selection and formulation optimization. Of the in vitro tests that will be discussed, droplet size is likely to be the most important parameter to predict where droplets may deposit in the nasal cavity. It should be noted that these tests can be used to facilitate development and can also be used as quality control tests. One should be careful to denote the differences between the two applications. To date, a significant correlation between in vitro analytical tests such as spray pattern and in vivo outcomes has not been established.
Because of the importance of deposition, many researchers (Shah et al. 2011; Shah et al. 2013; Suman et al. 2006; Newman et al. 2004; Laube 2007; Aggarwal et al. 2004; Schroeter, et al. 2006; Djupesland et al. 2006; Djupesland and Skretting 2012; Cheng et al. 2001; Foo et al. 2007; Guo et al. 2005; Hughes et al. 2008; Kundoor and Dalby 2010) have turned to nasal casts and computational fluid dynamic (CFD) models to assess the deposition patterns of new nasal devices and/or formulations. Often used in early development, nasal cast studies have become easier to perform with increasing ease in the creation of nasal casts from MRI and CT scans (Fig. 5.14). With rapid prototyping techniques, nasal casts can be machined for use in a lab setting. These casts are typically coated with a material to simulate the mucus layer and to prevent particle bounce. These casts can provide both a qualitative and quantitative picture of the sites of drug deposition, and can be combined with impaction-based techniques to quantify the mass of drug exiting the nasal cavity.

Silicon model of a nasal cast
CFD modeling can also be used to simulate changes in airflow, angle of insertion, disease state, or patient geometry as a mechanism to predict nasal deposition. Several studies (Chen et al. 2010; Segal et al. 2008) have been cited in the literature that have simulated nasal hypertrophy and to assess potential patient to patient variability. With the availability of software from Mimic and Fluent, the end user can perform analysis of many different simulations.
Reverting back to the traditional tools to characterize performance, spray pattern (Fig. 5.15) and plume geometry (Fig. 5.15) are in vitro tests used to define the shape of the emitted spray and to confirm that the molding process of the pump components was successful. These tests are performed from the analysis of a two-dimensional image of the emitted plume. Traditionally spray pattern and plume geometry have been performed with impaction systems such as TLC plates and fast speed cameras. Nowadays, spray pattern and plume geometry analyses are mostly performed using non-impaction laser sheet-based instruments. Spray pattern is characterized by the D max, D min, ovality ratio, and area. D max is the longest diameter measured on the resulting spray pattern image. D min is the shortest diameter measured on the resulting spray pattern image. Ovality ratio is the ratio of D max to D min. This ratio provides a quantitative value for the overall shape of the spray. The spray pattern area is automatically detected by the software. Percent area is the ratio of the spray pattern area to the entire image area. Plume geometry is characterized by the plume height, spray angle, and plume width. Spray angle is the angle of the emitted plume measured from the vertex of the spray cone and spray nozzle. Plume width is the width of the plume at a given distance (e.g., 3 cm) from the spray nozzle. Plume height is the height of the emitted plume measured from the tip of the device. While specifications may be set for all spray pattern parameters, FDA recommends using area and ovality ratio for statistical comparison (Food and Drug Administration 2003) to establish bioequivalence between test and reference nasal drug products. In case of plume geometry, FDA recommends using spray angle and plume width for statistical comparison (Food and Drug Administration 2003).

Spray pattern (left) and plume geometry (right) images (courtesy of Proveris Scientific)
Droplet size distribution is an important in vitro test based on laser diffraction principle to characterize droplet size distributions from nasal sprays. The droplet size distribution is characterized by the volume distribution (Dv10, Dv50, and Dv90), span, and percentage (%) less than 10 μm. Dv50 is the volume median diameter. It indicates that 50 % of the distribution is contained in droplets that are smaller than this value while the other half is contained in droplets that are larger than this value. Similarly the Dv10 and Dv90 values indicate that 10 % and 90 %, respectively, of the distribution are contained in droplets that are smaller than these values. Span is calculated by the following equation: (Dv90 − Dv10/Dv50) and quantifies the spread of the droplet size distribution. Percentage (%) less than 10 μm is the cumulative volume of the particles with size less than 10 μm. This cumulative fraction provides a risk estimate of particles from nasal spray that may be inhaled into lung. For bioequivalence assessment, FDA recommends using Dv50 and span for statistical comparison (Food and Drug Administration 2003) to establish bioequivalence between test and reference nasal drug products. Droplet size is also a quality control test.
Single actuation content (used for in vitro bioequivalence) or spray content uniformity (SCU) through container life and pump delivery (PD) through container life testing are used to characterize the delivery of drug discharged from the actuator of an aerosol or nasal spray against the label claim through container life. This test ensures that the product delivers the label claim over the labeled number of actuations. This test is also used to confirm the number of priming and repriming shots under different storage conditions and orientations. Typically the spray from the nasal unit is collected in a collection tube or glass bottle and the mass of drug is quantified by HPLC. Pump delivery is calculated from the weight difference of the collection tube or the glass bottle before and after shot collection. Single actuation content/SCU and pump delivery are performed at the beginning and end of the unit life for multi-dose drug products. Drug mass per single actuation is recommended by FDA (2003) for bioequivalence assessment.
For suspension products, drug particle size distribution by microscopy can estimate the rate of dissolution. Drug particle size distribution and extent of agglomerates are characterized in the spray or aerosol formulation prior to actuation, and in the spray following actuation. A sample from a nasal spray unit is sprayed onto a substrate (e.g., a microscope slide or a gridded filter paper). A polarized light microscope is used to analyze the size of the primary drug particle present in the sample. A count-based particle size histogram and a cumulative particle size graph are reported. Optical microscopy coupled with Raman spectroscopy (Kippax et al. 2011) imaging techniques (Klueva et al. 2008) can provide an improved method to establish equivalent particle size distribution between Test and Reference products that can be in accordance with FDA’s critical path initiative (Food and Drug Administration 2003). While current optical microscopy relies on the morphology of the drug particle, Raman spectroscopy or imaging techniques can provide chemical information and hence can improve the specificity and accuracy of the method through ingredient-specific particle size analysis.
Aerodynamic particle size distribution by cascade impaction is intended to determine the amount of drug in small particles/droplets. Small droplets defined as droplets smaller in size than the nominal effective cutoff diameter of the top stage of the cascade impactor may potentially be delivered to regions of the airways beyond the nose which may be a safety issue. The amount of drug in small particles is typically measured by an Andersen Cascade Impactor (ACI) operated by drawing the sample laden air through the ACI at 28.3 L/min. ACI is made up of classification stages consisting of a series of jets and impaction surfaces. At each stage, an aerosol stream passes through the jets and impacts upon the surface. Particles in the aerosol stream with significant inertia will settle upon the impaction plate. Smaller particles pass as aerosols on to the next jet stage. By designing the following consecutive stages with higher aerosol jet velocities, smaller diameter particles are collected at each subsequent stage giving the cascade affect of separation. The ACI is assembled to a 2 L glass nasal induction port and a pre-separator. Aerosol collected in the induction port, pre-separator, and the impactor is analyzed using HPLC to quantify the mass of drug. The amount and % of drug less than 9 μm and the mass balance are reported. Deposition profile (i.e., distribution of mass deposited on various components of the ACI and associated accessories) is recommended by FDA (Food and Drug Administration 2003) for bioequivalence assessment.
8 Global Regulatory Perspective
The regulatory landscape for nasal spray drug products is well established in the Western world. However, as utilization of nasal sprays, particularly generics, gains momentum in the BRIC countries (Brazil, Russia, India, China), regulatory bodies such as ANVISA in Brazil and CFDA in China are looking to adopt regulatory strategies similar to FDA. The following sections will discuss analytical regulatory expectations from both a new drug and generic drug approval perspective.
8.1 New Drug Approvals
From a Chemistry, Manufacturing, and Controls (CMC) perspective (Food and Drug Administration 1999), nasal spray product performance depends on the interaction between the formulation and delivery device. Hence, analytical requirements for the approval of the drug product consist of techniques that assess the chemical and physical stability of the formulation and the functionality of the device. While the relationship between certain spray characteristics and the efficacy of a product is still under investigation, FDA currently requires 12 different techniques for characterizing the spray and device for nasal spray product New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs) (refer Table 5.5 in Sect. 5.5). These methods are used to support stability, batch release, and drug product characterization for NDAs. The extensive nature of analytical requirements puts nasal drug products in a category of most highly tested dosage forms when compared to, for example, oral solid dosage forms.
Developers are cautioned to perform these tests even if they are not planning to market in the United States. Neither Health Canada nor EMA (Health Canada 2006; European Medicines Agencies 2006) require spray pattern and plume geometry analyses (Table 5.7), and specifications on pump delivery, SCU, and droplet size distribution vary between regulatory bodies. However, if later there is interest in launching the product in the United States, and these tests had not been performed, extensive reformulation or device design may be required for FDA approval, requiring new clinical studies. It should also be noted that both ANVISA in Brazil and the CFDA in China are beginning to incorporate analytical requirements into their expectations for these drug products. In India, the World Health Organization (WHO) requires analytical testing, such as droplet size, for devices used for nasally administered vaccines.
Both preserved and preservative-free nasal spray drug products will be required to complete the series of testing outlined above. If the drug product is manufactured in a sterile environment, then sterility testing will be required. An antimicrobial active may be self-preserving and, therefore, may not need routine preservative effectiveness testing. As previously discussed, BAC, phenylethyl alcohol, EDTA, and potassium sorbate have a history of use as preservatives in nasal spray formulations. If an alternate or novel preservative is used that does not have a history of use in the nasal cavity or respiratory tract, regulatory bodies may require additional toxicological studies on the excipient.
Nasal powders and pressurized nasal aerosols can be considered alternates to preservative-free systems. For both of these, particle size characterization by cascade impaction to quantify the mass of drug less than 9 μm will be required. This will be used to address the potential for lung deposition via the nasal cavity, which is a FDA and EMA safety concern. Cascade impaction may also be required on a routine basis by FDA for nasal powders and aerosols. HFA-based nasal aerosols may also need to follow some of the analytical tests outlined in the Inhalation CMC Guidance (Food and Drug Administration 1999; European Medicines Agencies 2006). For passive nasal powders, where the patient’s inspiratory effort aerosolizes the powder, spray pattern and plume geometry would not be required.
Recently, FDA has requested that sponsors submit additional CMC data with the Investigational Drug Application (IND), and spray performance measurements can provide some of that data. Spray characterization data appropriate for this stage might include any or all of the following: pump delivery (PD), SCU, droplet size distribution, spray pattern, and plume geometry.
In the case of a solution formulation, pump delivery (PD) may serve as a surrogate for SCU to conserve resources at this phase of development, since PD takes only minutes to complete compared to hours for SCU. However, confirming first that the correlation between the PD and SCU exists is prudent. Since the distribution of API in suspensions might result in differing amounts of API in each actuation, PD might not equate to SCU, therefore suspension formulations always require SCU.
FDA requires evaluation of potential leachables on stability. Leachables, which may be seen as a potential contaminate and harmful to public health, are also on the radar of the CFDA. To address, an extractable study is required to determine if potential components from the device may leach into the drug product. If the extractable profile reveals entities above the analytical evaluation threshold (AET) that require monitoring, a leachable study is necessary. For practical and financial reasons this study should take place concurrently with your registration stability batches because units can be stored for both studies at the same time under the same conditions. In order to have sufficient planning time for the leachable study, you will need to complete the extractable profile at least 6 months prior to the scheduled start of registration stability studies.
FDA requires testing of three registration batches prior to submission of an NDA. In addition to analysis of physical characteristics and microbiological testing over the course of the stability study, most sponsors also choose to include spray pattern, although spray pattern and plume geometry are not required. These registration stability study designs (Table 5.8) typically involve the analysis in excess of 10,000 units over a 2- to 3-year period. As a result, poor planning, such as failing to place a sufficient number of units in the stability chambers, can result in a disastrous loss of time and money.
Drug product characterization studies on samples from three registration batches should also take place along with clinical batch release testing. One-time drug product characterization studies performed at this stage include (where appropriate) photostability, temperature cycling device robustness, profiling, effect of dosing orientation, prime/reprime, and cascade impaction for nasal sprays to determine the percentage of droplets less than 10 μm.
8.2 Generic Drug Approvals
The global interpretation of qualitative and quantitative (Q and Q) sameness, as required for generic drug products, may actually vary from country to country. In the United States, FDA’s interpretation of Q and Q is well defined in that the active must be the same and the inactive excipients must be with 5 % of the reference label drug (RLD) for nasal sprays. Health Canada and EMA have a similar approach. However, the similarities may end there. In the United States, the FDA expects that the patient has the same experience when using the device. In other words, there is a need to have the same type of device, e.g., CPS pump to CPS pump, used for the RLD and generic. In Brazil, the regulatory bodies allow omission or alternate excipients for generic drug products. For example, Budecort (budesonide, RLD) is available on the market as a preserved multi-dose nasal spray and a generic budesonide formulation that is non-preserved is also on the market. This Brazilian example would not meet FDA’s expectations of a generic drug product because it is not Q and Q from a formulation and device standpoint.
Other regulatory differences are the bioequivalence guidances that request in vitro analytical testing (FDA and ANVISA) or deposition studies (EMA). There is a draft FDA Bioequivalence Guidance (Food and Drug Administration 2003; European Medicines Agencies 2006) that outlines a series of analytical tests that can be used to determine equivalence. These tests include droplet size by laser diffraction, drug in small particles/droplets as determined by cascade impaction, spray pattern, plume geometry, single actuation content uniformity, microscopy for suspensions, and priming and repriming. A combination of statistical approaches is used to determine equivalence (Table 5.9). For a locally acting nasal spray solution, equivalence of these six tests (no particle size by microscopy) may allow the generic sponsor to avoid performing pharmacokinetic, pharmacodynamic, or clinical endpoint studies.
In Europe, the EMA has defined a stepwise approach for approval of generic drug products (European Medicines Agencies 2006). While this model is meant for inhaled drug products, in theory, this could also be considered relevant for nasal sprays. The stepwise approach relies on similarity of in vitro tests as the starting point. The question for nasal sprays is that the exact in vitro tests are not defined. However, if in vitro tests are not equivalent, the next step may be a deposition study with a technique like gamma scintigraphy and demonstrating similar systemic exposure. Unlike FDA, deposition studies could be used as a tool for bioequivalence in the EU.
In October 2010, ANVISA issued a guidance (Brazil National Health Surveillance Agency 2008) similar in design to the FDA draft bioequivalence guidance. ANISA has also reissued a more detailed guidance in March 2012 (Brazil National Health Surveillance Agency 2013). The tests required for generic approval in Brazil are spray pattern, droplet/particle size by laser diffraction, uniformity of delivered dose, number of actuations, priming and repriming, and general assays for the drug product found in pharmacopoeias (Brazil, USP, EP, etc.). The statistical approach is not defined in this guidance; however, it is believed that an approach defined by FDA will be utilized to determine equivalence.
9 Conclusion
In the future, there is likely to be an increase in preservative-free formulations, especially in certain countries, both in the prescription and over-the-counter markets. Preservative-free nasal sprays are made possible by the device platforms that allow for sterilization before or after manufacture of the drug product. In addition, preservative-free devices add another barrier by preventing microbial ingress during use by the patient. Sterile manufacturing technology is adaptable to preservative-free nasal sprays and the regulatory pathway is similar to that of traditional nasal spray drug products.
References
Adams DR (1986) Transitional epithelial zone of the bovine nasal mucosa. Am J Anat 176:159–170
Aggarwal R, Cardozo A, Homer JJ (2004) The assessment of topical nasal drug distribution. Clin Otolaryngol Allied Sci 29:201–205
Ambrose CS, Levin MJ, Belshe RB (2011) The relative efficacy of trivalent live attenuated and inactivated influenza vaccines in children and adults. Influenza Other Respir Viruses 5:67–75
Andersen I, Proctor DF (1983) Measurement of nasal mucociliary clearance. Eur J Respir Dis Suppl 127:37–40
Anon (2003) Influenza virus vaccine live intranasal—MedImmune vaccines: CAIV-T, influenza vaccine live intranasal. Drugs R D 4:312–319
Astrazeneca (2012) Fourth quarter and full year results 2011. http://www.astrazeneca.com/Investors/financial-information/Financial-results. Accessed 2 Feb 2012
Bagel S, Wiedemann B (2004) Extension of in-use stability of preservative-free nasalia. Eur J Pharm Biopharm 57:353–358
Behl CR, Pimplaskar HK, Sileno AP, deMeireles J, Romeo VD (1998a) Effects of physicochemical properties and other factors on systemic nasal drug delivery. Adv Drug Deliv Rev 29:89–116
Behl CR, Pimplaskar HK, Sileno AP, Xia WJ, Gries WJ, deMeireles JC, Romeo VD (1998b) Optimization of systemic nasal drug delivery with pharmaceutical excipients. Adv Drug Deliv Rev 29:117–133
Bernstein IL (2000) Is the use of benzalkonium chloride as a preservative for nasal formulations a safety concern? A cautionary note based on compromised mucociliary transport. J Allergy Clin Immunol 105:39–44
Bjork E (1993) Starch microspheres as a nasal delivery system for drugs. Uppsala University, Uppsala
Boukarim C, Abou JS, Bahnam R, Barada R, Kyriacos S (2009) Preservatives in liquid pharmaceutical preparations. Drug Test Anal 1:146–148
Brain JD, Valberg PA (1979) Deposition of aerosol in the respiratory tract. Am Rev Respir Dis 120:1325–1373
Brazil National Health Surveillance Agency (2008) Guidance for pharmaceutical equivalence and the bioequivalence of nasal sprays and aerosols. Agência Nacional de Vigilância Sanitária, Brazil
Brazil National Health Surveillance Agency (2013) Technical standard No. 001/2013/CEFAR/GTFAR/GGMED/ANVISA Bristol Myers Squibb Company (1999) Butorphanol (Stadol Nasal Spray) package insert. Bristol Myers Squibb Company, Princeton, PA
Brittebo EB (1982) Metabolism of progesterone by the nasal mucosa in mice and rats. Acta Pharmacol Toxicol (Copenh) 51:441–445
Brouet G, Grosjean B (2003) Development of preservative free nasal spray products. Drug Deliv Technol 3
Chan HK (2006) Dry powder aerosol delivery systems: current and future research directions. J Aerosol Med 19:21–27
Chen XB, Lee HP, Chong VF, de Wang Y (2010) A computational fluid dynamics model for drug delivery in a nasal cavity with inferior turbinate hypertrophy. J Aerosol Med Pulm Drug Deliv 23:329–338
Cheng YS, Holmes TD, Gao J, Guilmette RA, Li S, Surakitbanharn Y, Rowlings C (2001) Characterization of nasal spray pumps and deposition pattern in a replica of the human nasal airway. J Aerosol Med 14:267–280
Chien YK, Yu K, Chang S (1989) Physical, biopharmaceutical, and toxicophysiological considerations. In: Nasal systemic drug delivery. Marcel Dekker, New York, pp 39–88
Costantino HR, Illum L, Brandt G, Johnson PH, Quay SC (2007) Intranasal delivery: physicochemical and therapeutic aspects. Int J Pharm 337:1–24
Dahl AR, Hadley WM (1983) Formaldehyde production promoted by rat nasal cytochrome P-450-dependent monooxygenases with nasal decongestants, essences, solvents, air pollutants, nicotine, and cocaine as substrates. Toxicol Appl Pharmacol 67:200–205
Dhuria SV, Hanson LR, Frey WH (2010) Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J Pharm Sci 99(4):1654–1673
Djupesland PG, Skretting A (2012) Nasal deposition and clearance in man: comparison of a bidirectional powder device and a traditional liquid spray pump. J Aerosol Med Pulm Drug Deliv 25(5):280–289
Djupesland PG, Skretting A, Winderen M, Holand T (2006) Breath actuated device improves delivery to target sites beyond the nasal valve. Laryngoscope 116:466–472
Food and Drug Administration (2003) Approval letter to MedImmune. U.S. Food and Drug Administration, Washington, DC. http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm123753.htm. Accessed 1 Feb 2012
European Medicines Agencies (2006) Guideline on the pharmaceutical quality of inhalation and nasal products. European Medicines Agencies, London
Fiore AE, Uyeki TM, Broder K, Finelli L, Euler GL, Singleton JA et al (2010) Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm Rep 59:1–62
Foo MY, Cheng YS, Su WC, Donovan MD (2007) The influence of spray properties on intranasal deposition. J Aerosol Med 20:495–508
Food and Drug Administration (1999) Nasal spray and inhalation solution, suspension, and spray drug products: chemistry, manufacturing, and controls documentation. US Food and Drug Administration, Washington, DC
Food and Drug Administration (2003) Bioavailability and bioequivalence studies for nasal aerosols and nasal sprays for local action. US Food and Drug Administration, Washington, DC
Gonda I, Gipps E (1990) Model of disposition of drugs administered into the human nasal cavity. Pharm Res 7:69–75
Groß D (2000) The COMOD-System—a preservative free drug therapy against glaucoma. In: Orgül S, Flammer J (eds) Pharmacotherapie in glaucoma. Hans Huber, Bern, pp 321–328
Groves MJ, Murty R (1990) Aseptic pharmaceutical manufacturing II: applications for the 1990s. Interpharm Press, Buffalo Grove
Guo Y, Laube B, Dalby R (2005) The effect of formulation variables and breathing patterns on the site of nasal deposition in an anatomically correct model. Pharm Res 22:1871–1878
Hardy JG, Lee SW, Wilson CG (1985) Intranasal drug delivery by spray and drops. J Pharm Pharmacol 37:294–297
Harris AS, Nilsson IM, Wagner ZG, Alkner U (1986) Intranasal administration of peptides: nasal deposition, biological response, and absorption of desmopressin. J Pharm Sci 75:1085–1088
Health Canada (2006) Pharmaceutical quality of inhalation and nasal products. Health Canada, Ottowa, ON
Henningfield JE, Keenan RM (1993) Nicotine delivery kinetics and abuse liability. J Consult Clin Psychol 61:743–750
Hilding AC (1963) Phagocytosis, mucous flow, and ciliary action. Arch Environ Health 6:61–71
Hochhaus G, Sahasranaman S, Derendorf H, Mollmann H (2002) Intranasal glucocorticoid delivery: competition between local and systemic effects. STP Pharma Sci 12:23–31
Hughes R, Watterson J, Dickens C, Ward D, Banaszek A (2008) Development of a nasal cast model to test medicinal nasal devices. Proc Inst Mech Eng H 222:1013–1022
Illum L (2000) The significance of animal models in the investigation of respiratory therapies, practical approaches to nasal and pulmonary drug delivery. Paris, 24–25th January, 2002
Islam N, Gladki E (2008) Dry powder inhalers (DPIs)—a review of device reliability and innovation. Int J Pharm 360:1–11
Kimbell JS, Segal RA, Asgharian B, Wong BA, Schroeter JD, Southall JP et al (2007) Characterization of deposition from nasal spray devices using a computational fluid dynamics model of the human nasal passages. J Aerosol Med 20:59–74
Kippax P, Huck D, Levoguer C, Virden A, Suman J (2011) Characterizing a nasal spray formulation from droplet to API particle size. Pharm Technol Eur 23:34–39
Klueva O, Nelson M, Treado P, Su G, Suman J (2008) Size distribution of active ingredient in generic and brand nasal sprays based on Raman chemical imaging. Respiratory Drug Delivery pp 479–482
Kundoor V, Dalby RN (2010) Assessment of nasal spray deposition pattern in a silicone human nose model using a color-based method. Pharm Res 27:30–36
Laube BL (2007) Devices for aerosol delivery to treat sinusitis. J Aerosol Med 20:S5–S17
Lebe E, Baka M, Yavasoglu A, Aktug H, Ates U, Uyanikgil Y (2004) Effects of preservatives in nasal formulations on the mucosal integrity: an electron microscopic study. Pharmacology 72:113–120
Mallants R, Jorissen M, Augustijns P (2007) Effect of preservatives on ciliary beat frequency in human nasal epithelial cell culture: single versus multiple exposure. Int J Pharm 338:64–69
Marom Z, Shelhamer J, Kaliner M (1984) Nasal mucus secretion. Ear Nose Throat J 63:36–37, 44
Marple B, Roland P, Benninger M (2004) Safety review of benzalkonium chloride used as a preservative in intranasal solutions: an overview of conflicting data and opinions. Otolaryngol Head Neck Surg 130:131–141
McMartin C, Hutchinson LE, Hyde R, Peters GE (1987) Analysis of structural requirements for the absorption of drugs and macromolecules from the nasal cavity. J Pharm Sci 76:535–540
MedImmune (2011) FluMist [package insert]. MedImmune, Philadelphia, PA. http://www.medimmune.com/pdf/products/flumist_pi.pdf. Accessed 1 Feb 2012
Merkus P, Romeijn SG, Verhoef JC, Merkus FW, Schouwenburg PF (2001) Classification of cilio-inhibiting effects of nasal drugs. Laryngoscope 111:595–602
Mygind N (1985) Upper airway: structure, function and therapy. In: Aerosols in medicine. Principles, diagnosis, and therapy. Elsevier BV, Amsterdam, pp 1–26
Na L, Mao S, Wang J, Sun W (2010) Comparison of different absorption enhancers on the intranasal absorption of isosorbide dinitrate in rats. Int J Pharm 397:59–66
Newman SP, Agnew JE, Pavia D, Clarke SW (1982) Inhaled aerosols: lung deposition and clinical applications. Clin Phys Physiol Meas 3:1–20
Newman SP, Moren F, Clarke SW (1987a) Deposition pattern from a nasal pump spray. Rhinology 25:77–82
Newman SP, Moren PF, Clarke SW (1987b) The nasal distribution of metered dose inhalers. J Laryngol Otol 101:127–132
Newman SP (1993) Scintigraphic assessment of therapeutic aerosols. Crit Rev Ther Drug Carrier System 10(1):65–109
Newman SP, Pitcairn GR, Dalby RN (2004) Drug delivery to the nasal cavity: In vitro and in vivo assessment. Crit Rev Ther Drug Carrier Syst 21:21–66
Petruson B, Hansson HA, Karlsson G (1984) Structural and functional aspects of cells in the nasal mucociliary system. Arch Otolaryngol 110:576–581
Pontiroli AE, Calderara A, Pozza G (1989) Intranasal drug delivery. Potential advantages and limitations from a clinical pharmacokinetic perspective. Clin Pharmacokinet 17:299–307
Schipper NG, Verhoef JC, Merkus FW (1991) The nasal mucociliary clearance: relevance to nasal drug delivery. Pharm Res 8:807–814
Schroeter JD, Kimbell JS, Asgharian B (2006) Analysis of particle deposition in the turbinate and olfactory regions using a human nasal computational fluid dynamics model. J Aerosol Med 19:301–313
Segal RA, Kepler GM, Kimbell JS (2008) Effects of differences in nasal anatomy on airflow distribution: a comparison of four individuals at rest. Ann Biomed Eng 36:1870–1882
Serra-Batlles J, Plaza V, Badiola C, Morejon E (2002) Patient perception and acceptability of multidose dry powder inhalers: a randomized crossover comparison of Diskus/Accuhaler with Turbuhaler. J Aerosol Med 15:59–64
Shah SA, Berger RL, Gupta P, Wan J, Monteith D, Connor A, McDermott J, Lin W (2011) In vivo nasal deposition from different delivery devices and formulations. Int’l Pharmaceutical Aerosol Consortium on Regulation and Science
Shah SA, Dickens CJ, Ward DJ, Banaszek AA, George C, Horodnik W (2013) Design of experiments to optimize an in vitro cast to predict human nasal drug deposition. J Aerosol Med Pulm Drug Deliv. doi:10.1089/jamp.2012.1011
Suman JD, Laube BL, Dalby R (1999) Comparison of nasal deposition and clearance of aerosol generated by nebulizer and an aqueous spray pump. Pharm Res 16:1648–1652
Suman JD, Laube BL, Lin TC, Brouet G, Dalby R (2002) Validity of in vitro tests on aqueous spray pumps as surrogates for nasal deposition. Pharm Res 19:1–6
Suman JD, Laube BL, Dalby R (2006) Validity of in vitro tests on aqueous spray pumps as surrogates for nasal deposition, absorption, and biologic response. J Aerosol Med 19:510–521
Telko MJ, Hickey AJ (2005) Dry powder inhaler formulation. Respir Care 50:1209–1227
Trangsrud AJ, Whitaker AL, Small RE (2002) Intranasal corticosteroids for allergic rhinitis. Pharmacotherapy 22:1458–1467
Ugwoke MI, Agu RU, Verbeke N, Kinget R (2005) Nasal mucoadhesive drug delivery: background, applications, trends and future perspectives. Adv Drug Deliv Rev 57:1640–1665
van de Donk HJ, Muller-Plantema IP, Zuidema J, Merkus FW (1980) The effects of preservatives on the ciliary beat frequency of chicken embryo tracheas. Rhinology 18:119–133
van de Donk HJ, van den Heuvel AG, Zuidema J, Merkus FW (1982) The effects of nasal drops and their additives on human nasal mucociliary clearance. Rhinology 20:127–137
van Drunen C, Meltzer EO, Bachert C, Bousquet J, Fokkens WJ (2005) Nasal allergies and beyond: a clinical review of the pharmacology, efficacy, and safety of mometasone furoate. Allergy 60(suppl 80):5–19
Vidgren MT, Kublik H (1998) Nasal delivery systems and their effect on deposition and absorption. Adv Drug Deliv Rev 29:157–177
Yu G, Zhang Z, Lessmann R (1998) Fluid flow and particle diffusion in the human upper respiratory system. Aerosol Sci Tech 28:146–158
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 American Association of Pharmaceutical Scientists
About this chapter
Cite this chapter
Ehrick, J.D. et al. (2013). Considerations for the Development of Nasal Dosage Forms. In: Kolhe, P., Shah, M., Rathore, N. (eds) Sterile Product Development. AAPS Advances in the Pharmaceutical Sciences Series, vol 6. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7978-9_5
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7978-9_5
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7977-2
Online ISBN: 978-1-4614-7978-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)