
Abstract
The process of eukaryotic ribosome assembly stretches across the nucleolus, the nucleoplasm and the cytoplasm, and therefore relies on efficient nucleocytoplasmic transport. In yeast, the import machinery delivers ~140,000 ribosomal proteins every minute to the nucleus for ribosome assembly. At the same time, the export machinery facilitates translocation of ~2000 pre-ribosomal particles every minute through ~200 nuclear pore complexes (NPC) into the cytoplasm. Eukaryotic ribosome assembly also requires >200 conserved assembly factors, which transiently associate with pre-ribosomal particles. Their site(s) of action on maturing pre-ribosomes are beginning to be elucidated. In this chapter, we outline protocols that enable rapid biochemical isolation of pre-ribosomal particles for single particle cryo-electron microscopy (cryo-EM) and in vitro reconstitution of nuclear transport processes. We discuss cell-biological and genetic approaches to investigate how the ribosome assembly and the nucleocytoplasmic transport machineries collaborate to produce functional ribosomes.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Eukaryotic ribosome assembly takes place across multiple cellular compartments : the nucleolus , the nucleoplasm and the cytoplasm (Fig. 1). This dynamic and energy consuming process requires the coordination of three RNA polymerases (I, II, and III), the RNA splicing, and nucleocytoplasmic transport machineries [1]. Despite this complexity, ribosome biogenesis is an incredibly efficient process, with yeast producing up to 60 ribosomes every second [2].

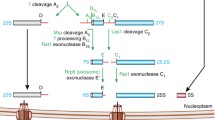
Current model for eukaryotic ribosome assembly . Transcription of primary 35S rRNA transcript, the common precursor of 18S, 5.8S, and 25S rRNAs , by Pol I from rDNA repeats together with cotranscriptional joining of U3 snoRNP , r-proteins and 40S assembly factors form the 90S ribosomal precursor. 5S rRNA is transcribed separately by Pol III for assembly of 5S RNP before joining the 60S pre-ribosome. Cotranscriptional cleavage of 35S rRNA at A2 site separates the 60S and 40S r-subunit maturation pathways. Pre-ribosomal particles undergo a cascade of maturation steps in the nucleoplasm by transient association with assembly factors until reaching nuclear export competency. Once exported to the cytoplasm, last maturation events and quality control steps can occur resulting into functional r-subunits production. (Adapted from Peña et al. 2017)
The small subunit (SSU) processome is the first ribosome precursor assembled cotranscriptionally in the nucleolus through stepwise association with UTP-A, UTP-B, and UTP-C complexes and U3 snoRNP [1, 3]. Cryo-electron microscopy (cryo-EM) studies are revealing how UTP-A, UTP-B complexes, U3 snoRNP and additional biogenesis factors encapsulate and guide pre-rRNA folding in a hierarchical 5′-to-3′-oriented manner [4,5,6,7]. During nucleolar maturation, the 5′ domain of the 18S rRNA achieves a mature conformation, and the central domain is correctly positioned relative to the 5′ domain. In contrast, the 3′ major domain is buried within the SSU processome core, and its conformation is distinct as in the mature 40S subunit. Release of the 40S pre-ribosome requires endonucleolytic cleavages within the pre-rRNA [8,9,10,11], the release of U3 snoRNA and associated proteins catalyzed by the RNA helicase Dhr1 and its cofactor Utp14 [12, 13]. The RNA exosome mediates degradation of 5′-ETS (external transcribed spacer) rRNA within the 5′-ETS rRNA –UTP complex to disassemble and recycle the UTPs for a new round of 40S assembly [14].
Following 40S preribosome release the growing 27S pre-rRNA associates with 60S-specific r-proteins and maturation factors to initiate 60S pre-ribosome assembly . Cryo-EM structures of early states of the 60S pre-ribosome suggest a sequence of events during nucleolar 60S assembly. Early states the nucleolar 60S pre-ribosome show a characteristic arch-like morphology, whereas the later states adopt a compact shape closer to a mature 60S subunit. The 5′ domains (I and II) within 27S pre-rRNA adopt a near mature conformation, and connect with the 3′ terminal domain VI, thus forming the solvent-exposed backside of the 60S subunit. In all states, the pre-rRNA spacer ITS2, located in the 27S pre-rRNA between 5.8S and 25S rRNA , and the associated ITS2 factors form the “foot” structure. The central domains III, IV and V, which form the subunit interface, are not visible in the early states. Although cotranscriptional assembly of the 60S pre-ribosome occurs in a sequential manner, it undergoes nonlinear compaction of domains I, II, and VI, which then allows domains III, IV, and V to fall onto the arch-like structure, thus preparing the pre-ribosomal cargo for nuclear export [15,16,17].
In yeast , before every cell division, ~200,000 pre-ribosomes are transported to the cytoplasm through ~200 NPCs (nuclear pore complexes) by the exportin Crm1 that recognizes nuclear export sequences (NESs) on cargos and interacts with the FG-rich (phenylalanine/glycine-rich) meshwork of the NPC transport channel [18,19,20,21,22]. Nmd3 is the only identified essential adaptor for Crm1-mediated 60S pre-ribosome export [23, 24]. In contrast, no essential NES-containing export adaptor has been identified for 40S pre-ribosome export. 40S pre-ribosome bound shuttling assembly factors Ltv1 and Rio2 can recruit Crm1 in the presence of RanGTP through a leucine-rich NES . Pre-ribosomes also employ multiple export factors (Mex67-Mtr2 , Arx1 , Ecm1 , Bud20 ) that directly interact with the FG-meshwork of the transport channel [1, 21, 22]. In addition, a non-FG pathway involving the mRNA export factor Gle2 also facilitates 60S pre-ribosome export [25].
Exported pre-ribosomes undergo final maturation and proofreading before initiating translation [26]. Cytoplasmic proofreading of 60S pre-ribosomes involves release of assembly factors that block binding of r-proteins , interactions with translation factors, or pairing with the 40S . Tif6 prevents binding of immature 60S pre-ribosomes to mature 40S subunits, ensuring that only properly assembled subunits engage in translation . Quality control of the 40S pre-ribosome relies on assembly factors that prevent the premature binding of initiation factors, mRNA , tRNA, and the 60S subunit. Ltv1 and Enp1 directly bind uS3 on its solvent side, thereby blocking the mRNA channel opening. Rio2 , Tsr1 , and Dim1 bind the subunit interface, thus preventing joining of the mature 60S subunit and translation initiation factor eIF1A. Nob1 and Pno1 block the binding of eIF3, thereby interfering with translation initiation [27]. After release of Rio2 , Tsr1 , and Dim1 initiated by the reorganization of the beak structure, the 40S pre-ribosome becomes competent to interact with a mature 60S subunit. This translation-like interaction is thought to test the ability of a 40S pre-ribosome to engage with a mature 60S subunit, and only then triggers Nob1 to cleave 20S pre-RNA to mature 18S rRNA in vitro [28, 29]. Cryo-EM studies are revealing how these late factors interact with the 40S pre-ribosome and have provided a structural framework for the ordering of cytoplasmic maturation events [30,31,32].
In addition to >200 assembly factors , ribosome assembly relies on efficient nucleocytoplasmic transport [1]. All r-proteins and assembly factors need to be imported into the nucleus, and correctly assembled pre-ribosomal particles need to be transported through nuclear pore complexes into the cytoplasm. How the ribosome assembly machinery collaborates with the cellular trafficking pathways is currently under intense investigation. Here, we outline biochemical approaches that permit rapid screening and analyses of pre-ribosomal particles for single particle cryo-EM studies, and reconstitution of nucleocytoplasmic transport processes. We discuss genetic and cell-biological approaches that will enable the unveiling of the functional interface between the ribosome assembly and nucleocytoplasmic machineries.
2 Materials, Reagents, and Yeast Media
2.1 Material (Listed in Alphabetical Order)
-
1.
15 mL Centrifuge tube (Greiner Bio-One, 188271)
-
2.
20 mL Luer Solo, Inject (Braun, 4606205V)
-
3.
50 mL Centrifuge tube (Greiner Bio-One, 227261)
-
4.
50 mL Magnetic Separation Rack (New BioLabs, S1507S)
-
5.
Centrifuge 5804 R (Eppendorf, 5805 000.327).
-
6.
Centrifuge Sorvall RC3BP with H-6000A Rotor (Thermo-Scientific, 75007530, 11250).
-
7.
Cover glasses (VWR, 631-0137).
-
8.
DynaMag™—Spin Magnet (Invitrogen, 12320D).
-
9.
Fluorescence microscope Leica DM6000 B (Leica).
-
10.
Incubator IFE 600 (Memmert).
-
11.
Incubator shaker ISF1-X (Kuhner).
-
12.
Inoculation loop (Greiner, 731101).
-
13.
Membrane filters 0.45 mm (Millipore, HAWP04700).
-
14.
Microcentrifuge 5415R (Eppendorf, 022621408).
-
15.
Microscope slides (VWR, 631-1550).
-
16.
Planetary mill Pulverisette 6 (Fritsch, 06.2000.00).
-
17.
Spectrophotometer Ultrospec 2100 pro (GE Healthcare, 80-2112-21).
-
18.
Stainless Steel Grinding Balls (Fritsch, 55.0200.10).
-
19.
Stainless Steel Grinding Bowls, 80 mL (Fritsch, 50.4100.00).
-
20.
Sterifil Aseptic System (Millipore, XX1104700).
-
21.
Syringe filter, pore size: 2.7 μm (GE Healthcare, 6888-2527).
-
22.
Syringe filter pore size: 1.6 μm (GE Healthcare, 6882-2516).
-
23.
Thermomixer comfort (Eppendorf).
2.2 Reagents (Listed in Alphabetical Order)
-
1.
Acetone (Merck, 1.00014.1000).
-
2.
AcTEV protease (Invitrogen, 12575015).
-
3.
Beta-mercaptoethanol (CalBioChem, 444203).
-
4.
CaCl2(Sigma-Aldrich, 22,350-6).
-
5.
cOmplete protease inhibitor cocktail tablets, EDTA free (Roche, 11873580001),
-
6.
Coomassie Brilliant Blue R-250 (ThermoScientific, 20,278).
-
7.
Difco Agar (BD, 214530).
-
8.
Dithiothreitol (DTT) (AppliChem, A1101).
-
9.
Dynabeads™ M-270 Epoxy (Invitrogen, 14301).
-
10.
Glucose (Sigma-Aldrich, G8270).
-
11.
Glutathione Sepharose 4 Fast Flow (GE Healthcare, 17-5132-01).
-
12.
Glycerol (AppliChem, A1123).
-
13.
HEPES (Sigma-Aldrich, H4034).
-
14.
IgG from rabbit serum (Sigma-Aldrich, I5006).
-
15.
KOAc (Sigma-Aldrich, P1147).
-
16.
LDS sample buffer (Invitrogen, NP0008).
-
17.
MgCl2 (Fluka, 63072).
-
18.
Mg(OAc)2 (Sigma-Aldrich, M5661).
-
19.
NaCl (Merck, 1064045000).
-
20.
NH4Cl (Sigma-Aldrich, A9434).
-
21.
NuPAGE 4–12% Bis–Tris (Novex, NP0321BOX).
-
22.
Phenylmethanesulfonyl fluoride (PMSF) (AppliChem, A0999).
-
23.
Polyvinylpyrrolidone (K90) (AppliChem, A6393).
-
24.
Sodium phosphate (Sigma-Aldrich, 342483).
-
25.
TCA (Sigma-Aldrich, T6399).
-
26.
Tris–HCl (Sigma-Aldrich, T3253).
-
27.
Triton X-100 (Merck, 1086431000).
-
28.
Tween 20 (Sigma-Aldrich, P9416).
2.3 Yeast Media
-
1.
Synthetic complete (SC) media: 0.69% yeast nitrogen base without amino acids (ForMedium, CYN0410) 2% glucose (Sigma-Aldrich, G8270) 0.6–0.8% appropriate drop-out supplements complete mixtures (ForMedium).
-
2.
SC plates (SC, 2% agar).
-
3.
Yeast-extract peptone dextrose (YPD) media: 1% yeast extract, (ForMedium, YEM03), 2% peptone (ForMedium, PEP03), 2% glucose (Sigma-Aldrich, G8270).
-
4.
YPD plates (YPD , 2% agar).
3 Pre-ribosome Isolation for Single Particle Cryo-Electron Microscopy
During the last decade, affinity-purification protocols combined with sensitive mass spectrometry have dramatically altered our understanding of eukaryotic ribosome assembly [33]. By employing the powerful “tandem affinity-purification ” (TAP ), several groups have unravelled the compositions of the 90S, 60S , and 40S pre-ribosomes and thus expanded the inventory of the assembly machinery and ordered the 40S and 60S maturation pathways [34,35,36,37,38,39,40]. Recently, improvement in these biochemical approaches together with advances of cryo-EM have driven structural studies of pre-ribosomal particles, thus providing high-resolution snapshots of the process of eukaryotic ribosome assembly [4,5,6, 15,16,17, 30,31,32, 41, 42] (Fig. 2a, b).

Purification and structure of a late 40S pre-ribosome. (a) Protein composition of 80S ribosomes Nob1-D15N particles, purified via ProteinA (pA)-tag. Proteins were separated by SDS-PAGE and visualized by silver staining. Labelled protein bands were characterized by mass spectrometry . (b) Front and back view of cryo-EM structure of a cytoplasmic 40S preribosome (PDB: 6FAI). The 20S rRNA is shown in light gray, r-proteins in dark gray—except of uS3 in yellow. Assembly factors are shown in color: Tsr1 in orange, Rio2 in blue, Pno1 in green, Ltv1 in purple, and Enp1 in red. (Adapted from Scaiola et al. 2018)
3.1 Preparing Yeast Cells for Cryogenic Lysis (Modified from Rout Lab Protocol: Harvesting Cells and Making Yeast Noodles [43])
3.1.1 Buffers and Solutions
Resuspension Buffer: 1.2% PVP-40 (polyvinylpyrrolidone), 20 mM HEPES pH 7.4, Supplemented with 1:100 complete protease inhibitor cocktail tablet (Roche), 2 mM PMSF, 1 mM DTT (see Note 1 ).
3.1.2 Yeast Cell Preparation
-
1.
Grow cell culture to OD600 = 2.5–3.5 in complete media/ 1.5–2.0 in synthetic media.
-
2.
Spin cultures down (4500 × g, 15 min, 4 °C).
-
3.
Resuspend pellet in 50 mL ddH2O on ice. Put resuspended solution into 50 mL Falcon tube(s) and spin down (4500 × g, 5 min, 4 °C). Repeat this step.
-
4.
Resuspend pellet on ice in a volume of resuspension buffer equal to the volume of the pellet. Spin down (4500 × g, 15 min, 4 °C). Aspirate all liquid from the pellet.
-
5.
Spin just the pellet down again (4500 × g, 2 min, 4 °C) to remove the residual buffer (repeat if needed).
-
6.
Pellet should be fairly dry and resemble a thick paste.
-
7.
Fill up a Styrofoam box with a liquid nitrogen and prepare a metal holder next to it.
-
8.
Use a metal clamp to precool and fill up completely a 50 mL Falcon tube with liquid nitrogen using and fix the tube to a metal holder.
-
9.
With a spatula, scoop out cell paste and place into a 20 mL syringe. Press out the cell paste into the liquid nitrogen in the Falcon tube. Avoid touching the liquid nitrogen directly with the tip of the syringe to prevent syringe clogging.
-
10.
When all cell paste is gone, remove liquid nitrogen from the tube (poke holes into cap of Falcon tube, screw on the cap and turn tube upside down to pour out the liquid nitrogen).
-
11.
Do not tighten the tube completely, in order to allow liquid nitrogen vapor to escape. Store tubes at −80 °C.
3.1.3 Cryogenic Lysis of Yeast Cells (See Also Note 2 ) (Modified from Rout Lab Protocol: Cryogenic Lyses of yeast Cells [43])
-
1.
Fill a rectangular ice bucket with liquid nitrogen.
-
2.
Pre-chill everything. Immerse the 80 mL stainless steel grinding jars, the stainless steel lid, the grinding balls, steel spoon and the storage tube, with the frozen yeast noodles, in the liquid nitrogen.
-
3.
Pre-cooling is finished when nitrogen bath is no longer bubbling vigorously.
-
4.
Once everything is chilled pour the noodles into the grinding jar.
-
5.
Weigh the grinding jar with noodles and then adjust the counterbalance weight.
-
6.
Use 7–8 of the 20 mm stainless steel balls based on the amount of noodles.
-
7.
Be sure no liquid nitrogen is in the grinding jar prior to grinding to avoid an explosion.
-
8.
Grinding is done in 4 cycles, each cycle composed of 500 rpm, 3 min and in reverse rotation 500 rpm, 2 min (see also Note 3 ).
-
9.
Between each cycle the jars are removed and cooled in liquid nitrogen. Do not remove the lid (removal of lid may result in cell loss). To ensure lid is chilled use an empty Falcon tube to pour liquid nitrogen over the top of the grinding jar while the bowl of the grinding jar cools in the liquid nitrogen bath. Do not submerge the jar completely as this will allow liquid nitrogen into the grinding bowl and may also result in cell loss.
-
10.
When 4 cycles are complete, carefully remove powder from the balls with a pre-cooled spatula and transfer the powder into a new prechilled 50 mL Falcon tube (if there is powder stuck to the side of the jar repeat 1 grinding cycle) (see Note 4 ).
-
11.
Jars and balls are cleaned with warm water.
-
12.
Frozen ground cells are stored at −80 °C.
3.1.4 Isolating Pre-ribosomes Using Magnetic Beads (Modified from Oeffinger et al. 2007 [43])
3.1.4.1 Buffers and Solutions
M-IgG Buffer: 20 mM HEPES pH 7.4, KOAc, 40 mM NaCl, 0.5% Triton-X, 0,1% Tween 20.
Supplemented with (see Note 1 ) 2 mM PMSF, 1 mM DTT, 2–10 mM MgCl2 (optional).
3.1.4.2 Method (See Also Notes 5–8)
-
1.
Transfer 2–5 g of ground yeast cells, to a pre-cooled 50 mL falcon, and put it into liquid nitrogen.
-
2.
Pour 40 mL M-IgG Buffer + Detergents into cooled sterile glass beaker with sterile magnet fish and start mixing.
-
3.
Cool down and sterilize metal sieve in liquid nitrogen.
-
4.
Add yeast powder slowly through the sieve into mixing buffer (avoid freezing of clumps).
-
5.
Allow powder to resuspend completely in buffer (5–10 min).
-
6.
Transfer yeast lysate to 50 mL Falcon tube. Spin down (4500 × g, 5 min, 4 °C) to get rid of big chunks.
-
7.
Meanwhile take 100 μL bead slurry and wash 3× with 1 mL M-IgG Buffer + Detergents using Magnet rack to wash away buffer and resuspend in 1 mL M-IgG Buffer + Detergents.
-
8.
Filter lysate with 60 mL syringe through 2.7 μM Whatman filter slowly into new 50 mL Falcon (do not push too hard, rather exchange the filter for new one).
-
9.
Repeat with 60 mL syringe through 1.6 μM Whatman filter into new 50 mL Falcon (stop as soon as white foam comes out).
-
10.
Add beads to clarified yeast lysate, seal with parafilm and incubate 30 min at 4 °C on rotating wheel.
-
11.
Collect beads with magnetic rack, wait for complete bead binding (solution clears up).
-
12.
Collect flow-through in a new Falcon tube and repeat bead collection to avoid loss of the beads.
-
13.
Resuspend all collected beads in 1 mL M-IgG Buffer + Detergents and transfer it into 1.5 mL tube.
-
14.
Wash the beads 3× with 1 mL M-IgG Buffer + Detergents using magnetic rack.
-
15.
Wash the beads further 3–5× with 1 mL M-IgG Buffer without Detergents (to get rid of foaming).
-
16.
Resuspend beads in 50–75 μL M-IgG buffer without Detergents.
-
17.
Add 0.5 μL (10 U) TEV protease (Sigma, 20 U/L), seal with parafilm and incubate overnight at 4 °C on rotating wheel.
-
18.
Collect flow-through (purified sample) in new tube using magnetic rack.
-
19.
Use 5 μL sample per EM Copper Grid.
-
20.
To analyze samples, proceed with TCA precipitation, RNA extraction , Silver gel, or RNA gel.
-
21.
Boil beads in 25–50 μL 2× LDS and collect LDS in new tube (using magnetic rack) for TEV cleavage control.
3.2 Nuclear Import Assays for Ribosomal Proteins
It is assumed that like a typical import cargo, RanGTP dissociates the r-protein from the importin after arriving in the nuclear compartment. However, r-proteins contain disordered regions, which make them prone to non-specific interactions with other nucleic acids, aggregation and degradation in their non-assembled state. During a single 90 min generation time ~14 million r-proteins synthesized in the cytoplasm, need to be targeted to the yeast nucleus and handed over to the ribosome assembly machinery [1, 44]. Thus, safe and efficient transport of r-proteins to their rRNA binding site is a logistical challenge.
A number of groups have uncovered dedicated chaperones that interact with newly synthesized r-proteins in the cytoplasm [44,45,46,47,48,49,50,51,52,53,54,55]. The dedicated chaperone–r-protein complexes recruit the import machinery and are transported to the nucleus where they are released by the action of RanGTP .
A different mechanism is employed by the r-protein eS26 to reach the 90S pre-ribosome. Like a typical import cargo protein, eS26 uses importins for targeting to the nucleus. However, after reaching the nuclear compartment, eS26 is removed from the importin by an unloading factor, the escortin Tsr2 , without the aid of RanGTP . Tsr2 shields eS26 from proteolysis and enables its safe transfer to the 90S pre-ribosome [56]. Below, we outline a step-by step protocol to investigate the canonical RanGTP dependent disassembly of a Pse1–Slx9 complex (Fig. 3a), and non-canonical Tsr2 dependent disassembly of a Kap123–eS26 complex (Fig. 3b). Slx9 is a predominantly nuclear localized protein, yet it shuttles between the nucleus and the cytoplasm [57, 58]. The import receptor transports Slx9 to the nuclear compartment, where Slx9 facilitates assembly of a Crm1-export complex and efficient export of 40S pre-ribosomes from the nucleus [57,58,59,60].

In vitro binding assays for nuclear import . (a) RanGTP-dependent release of Slx9 from Pse1 importin. Left panel: Workflow overview of the importin binding assay. POI = protein of interest. Right panel: GST–Pse1 complex formation with Slx9 dissociated in the RanGFP-dependent manner. GST–Pse1 (lanes 1–6) or GST alone (lanes 7–8) was immobilized on Glutathione Sepharose, washed with PBS-KMT and incubated with 4 μM Slx9 for 1 h at 4 °C (lanes 2–4 and 6). Pse1-Slx9 complex was subsequently incubated with 1.5 μM RanGTP under same conditions (lane 5). After washing with PBS-KMT, bound proteins were eluted in SDS sample buffer, separated by SDS-PAGE and visualized by Coomassie Blue staining and Western analyses using α-Slx9 antibodies. L = input. (b) RanGTP-independent release of eS26 from Kap123 importin. Immobilized GST-Kap123 on Glutathione Sepharose was washed and incubated with 4 μM eS26FLAG for 1 h at 4 °C. GST-Kap123:eS26FLAG complex was washed and incubated with either buffer alone (lane 2 and 8), 0.375 μM His6-RanQLGTP (left panel lanes 3–6) or 0.375 μM His6-Tsr2 (right panel lanes 9–12). Samples were withdrawn at the indicated time points and washed with PBS-KMT. Bound proteins were eluted in SDS sample buffer, separated by SDS-PAGE and visualized by Coomassie Blue staining and Western analyses using α-eS26 and α-RanGTP antibodies. L = input. GST-Kap123 is indicated by asterisk. (Adapted from Schütz et al. 2014)
3.2.1 A. RanGTP Mediated Disassembly of a Pse1–Slx9 Complex
3.2.1.1 Buffers and Solutions
PBS-KMT buffer (pH 7.3): 150 mM NaCl, 25 mM Sodium phosphate, 3 mM KCl, 1 mM MgCl2, 0,1% Tween 20.
3.2.1.2 Method
-
1.
Pipet 50 μL of GSH-Sepharose slurry (per reaction) into a 1.5 mL reaction tube and wash 3 times with 1 mL PBS-KMT buffer, spin down in between for 30 s at 850 x g . Wash GSH-Sepharose beads for all reactions together.
-
2.
Meanwhile thaw recombinant GST-Pse1 and GST-alone sample on ice.
-
3.
Centrifuge the GST-Pse1 and GST-alone or lysates for 10 min at 4 °C at 16,000 x g to pellet aggregates and store the tubes on ice until use.
-
4.
Add 12 μg GST-Pse1 or GST-alone (per reaction) to washed GSH-Sepharose beads suspended in 500 μL of PBS-KMT buffer.
-
5.
Incubate on a rotating platform for 1 h at 4 °C to immobilize GST-Pse1 or GST-alone on GSH-Sepharose beads.
-
6.
Wash samples as indicated in step 1 to remove unbound protein. Keep samples on ice between each washing step.
-
7.
To assess the amount of GST-Pse1 or GST-alone bound to the GST-Sepharose beads remove supernatant completely. Add 30 μL 2× LDS sample buffer, mix and heat up at 70 °C for 10 min to elute the bound proteins. Separate the eluted denatured proteins on an 12–15% SDS-PAGE and analyze by Coomassie Blue staining.
-
8.
To form an importin–protein complex, add 3–6 μM Slx9 to the immobilized GST-Pse1 or GST-alone on GSH-Sepharose beads suspended in 500 μL PBS-KMT buffer (from step 6).
-
9.
Incubate on a rotating platform for 1 h at 4 °C.
-
10.
Wash samples as indicated in step 1 to remove unbound protein. Samples are kept on ice between each washing step.
-
11.
To assess the amount of GST-Pse1–Slx9 complex bound to the GST-Sepharose beads remove supernatant completely. Add 30 μL 2× LDS sample buffer, mix and heat up at 70 °C for 10 min to elute the bound proteins. Separate the eluted denatured proteins on a 12–15% SDS-PAGE and analyze by Western blotting using antibodies directed against Slx9 .
-
12.
To investigate RanGTP sensitivity, incubate the GST-Pse1–Slx9 complex with 500 μL PBS-KMT containing 1.5 μM RanQLGTP for 1 h at 4 °C on a rotating wheel. In parallel incubate GST-Pse1–Slx9 in 500 μL PBS-KMT buffer without RanQLGTP to assess whether Slx9 is released by buffer alone.
-
13.
Wash samples as indicated in step 1 to remove released protein. Keep samples on ice between each washing step.
-
14.
Remove supernatant completely, and add 30 μL 2× LDS sample buffer, mix and heat up at 70 °C for 10 min to elute the bound proteins. Separate the eluted denatured proteins on a 12–15% SDS-PAGE and analyze by Coomassie Blue staining and Western blotting using antibodies directed against Slx9 .
3.2.2 Tsr2 Mediated Disassembly of a Kap123–eS26FLAG Complex (See Also Notes 9–14)
-
1.
Pipet 50 μL of GSH-Sepharose slurry (per reaction) into a 1.5 mL reaction tube and wash 3 times with 1 mL PBS-KMT buffer, spin down in between for 30 s at 850 × g. Wash GSH-Sepharose beads for all reactions together.
-
2.
Meanwhile thaw recombinant GST-Kap123 and GST-alone sample on ice.
-
3.
Centrifuge the GST-Kap123 and GST-alone or lysates for 10 min at 4 °C at 16,000 × g to pellet aggregates and store the tubes on ice until use.
-
4.
Add 12 μg GST-Kap123 or GST-alone to washed GSH-Sepharose beads suspended in 500 μL of PBS-KMT buffer.
-
5.
Incubate on a rotating platform for 1 h at 4 °C to immobilize GST-Kap123 or GST-alone on GSH-Sepharose beads.
-
6.
Wash samples as indicated in step 1 to remove unbound protein. Keep samples on ice between each washing step.
-
7.
The amount of GST-Kap123 or GST-alone bound to the GST-Sepharose beads can be assessed at this stage. For this, remove supernatant completely. Add 30 μL 2× LDS sample buffer, mix and heat up at 70 °C for 10 min to elute the bound proteins. Separate the eluted denatured proteins on a 12–15% SDS-PAGE and analyze by Coomassie Blue staining.
-
8.
To form importin–protein incubate the immobilized GST-Pse1 or GST-alone on GSH-Sepharose beads with 3–6 μM eS26FLAG in 500 μL PBS-KMT buffer from step 6.
-
9.
Incubate on a rotating platform for 1 h at 4 °C.
-
10.
Wash samples as indicated in step 1 to remove unbound protein. Samples are kept on ice between each washing step.
-
11.
To assess the amount of GST-Kap123–eS26FLAG complex bound to the GST-Sepharose beads, remove supernatant completely. Add 30 μL 2× LDS sample buffer, mix and heat up at 70 °C for 10 min to elute the bound proteins. Separate the eluted denatured proteins on a 12–15% SDS-PAGE and analyze by Western blotting using antibodies directed against FLAG tag.
-
12.
To investigate Tsr2 dependent disassembly of the complex from step 10, incubate the GST-Kap123–eS26FLAG complex with 500 μL PBS-KMT containing 1.5 μM Tsr2 . In parallel, incubate GST-Kap123–eS26FLAG with 500 μL of PBS-KMT buffer as a control to assess whether eS26FLAG, is released by buffer itself.
-
13.
Wash samples as indicated in step 1 to remove released protein. Keep samples on ice between each washing step.
-
14.
Remove supernatant completely, and add 30 μL 2× LDS sample buffer, mix and heat up at 70 °C for 10 min to elute and denature the bound proteins. Separate the eluted denatured proteins on a 12–15% SDS-PAGE and analyze by Coomassie Blue staining and Western blotting using antibodies directed against FLAG tag.
3.2.3 Cell Biological Assays for r-protein Nuclear Import
It is complicated to monitor the nuclear targeting of r-proteins in vivo since at steady state they reside in the cytoplasm. Therefore, one strategy is to uncouple r-protein nuclear import from their export by selectively impairing recruitment to the pre-ribosome. Fusing GFP to either the N- or C- terminus can prevent r-protein incorporation into the pre-ribosome. These non-functional proxy constructs can be employed to determine nuclear localization signals within r-proteins [56]. Further, by monitoring their mislocalization to the cytoplasm in different importin-deficient (Table 1) and/or mutant strains, these non-functional r-protein fusions (listed in Table 3) can pinpoint their nuclear pathway(s).
3.3 Nuclear Export Assays for Pre-ribosomes
3.3.1 Monitoring Nuclear Export of Pre-ribosomes (See Also Notes 15 and 16)
Transport of 60S and 40S pre-ribosomes from the nucleus into the cytoplasm can be investigated by monitoring the localization of r-protein fusions with GFP (uL23-GFP, uL5-GFP, uL18-GFP for the 60S subunit and uS5-GFP for the 40S subunit) in distinct mutant strains [72,73,74,75]. These r-protein fusions are stably incorporated into pre-ribosomes, and their steady-state localization in wild type (WT) cells is cytoplasmic. Nuclear accumulation of these reporters indicates either an early pre-ribosome assembly and/or export defect. Next to the r-protein GFP reporters, nuclear accumulation of GFP fused export adaptor factors serve as reporters for impairment in late assembly steps and/or of export of 60S (Nmd3-GFP) and 40S (Rrp12-GFP and Rio2-GFP) pre-ribosomes, respectively (Fig. 4).

Monitoring ribosome export defect by fluorescence microscopy. GFP-reporters for large r-subunit (uL18-GFP or Nmd3-GFP) and small r-subunit (uS5-GFP or Rrp12-GFP) were transformed into WT or indicated mutant strains and they localization was analyzed by fluorescence microscopy. Scale bar = 5 μm. Output of the experiment is indicated on the left scheme: accumulation of the reporter in the nucleus in case of export defect as seen for bud20Δ and yrb2Δ
3.3.1.1 Method
-
1.
Transform yeast strains with the reporter plasmids (uL18-GFP, uS5-GFP) and plate on appropriate media.
-
2.
The transformants expressing reporters are grown in appropriate liquid media (for mutants at permissive temperature) until saturation (overnight).
-
3.
The next day, dilute cells into 10 mL fresh media and grown until mid-log phase for at least 2–3 divisions. (In the case of cold or temperature sensitive mutants, shift the cultures to the appropriate restrictive temperature after 2–3 divisions of growth at permissive temperature).
-
4.
Pellet cells by centrifugation in a 15 mL Falcon tube ( 450 × g, 3 min) and wash once with 10 mL dH2O.
-
5.
Pour off the supernatant and resuspend the cell pellet in remaining liquid.
-
6.
Place 2 μL of cell suspension on a slide, press a coverslip over before inspection under a fluorescence microscope.
3.3.2 Reconstitution of Crm1-Nuclear Export Complexes In Vitro
In actively growing yeast cells , every minute ~25 pre-ribosomal particles are exported into the cytoplasm [2]. The exportin Crm1 (yeast Xpo1) plays an essential role in transporting pre-ribosomes into the cytoplasm. To initiate export, Crm1 , in the presence of RanGTP recognizes a nuclear export signal (NES) on adaptor proteins bound to pre-ribosomes, and cooperatively forms a Crm1-export complex [1, 23, 24, 76, 77]. In vitro binding assay with recombinant Crm1 and RanQL are used to analyze potential NES containing export adaptors of preribosomal subunits [58]. Here, using Ltv1 :Crm1 :RanQLGTP as an example to outline a protocol for assembly of a trimeric Crm1-export complex in vitro (Fig. 5a).

In vitro binding assays for nuclear export. (A) Ltv1 NES-dependent export complex formation. Left panel: Workflow overview of the binding assay. POI = protein of interest. Right panel: GST-Ltv1 or GST-Ltv1 lacking NES sequence (450-463aa) was immobilized on the Glutathione Sepharose, washed with PBS-KMT and incubated with buffer alone (lanes 1 and 5), buffer with 2μM His6-RanQLGTP (lanes 2 and 6), 50nM Crm1-His6 (lanes 3 and 7) or 50nM Crm1-His6 and 2μM His6-RanQLGTP simultaneously (lane 4 and 8) for 1h at 4°C. Bound proteins were washed with PBS-KMT, eluted in 2XLDS sample buffer, separated by SDS-PAGE and visualized by Coomassie Blue staining and Western analyses using α-RanGTP and α-Crm1 antibodies. L = input. Adapted from Fischer et al., 2015. (B) Heterodimeric transport receptor Mex67-Mtr2 interacts with FG repeat sequences of different nucleoporins. Left panel: Workflow overview of the binding assay. POI = protein of interest. Right panel: Indicated GST-Nucleoporin fusion proteins were immobilized on Glutathione Sepharose and washed with PBS-KMT before 1 h incubation with 3.6 μg His6-Mex67-Mtr2 at 4°C. Bound proteins were washed, eluted with 2X LDS buffer and analyzed by SDS-PAGE followed by Coomassie staining or Western blotting using α-Mex67/Mtr2 antibody. L = Load; Asterisks indicate Mex67 bands. Adapted from Altvater et al., 2012
3.3.2.1 Buffers and Solutions
PBS-KMT buffer (pH 7.3): 150 mM NaCl, 25 mM sodium phosphate, 3 mMKCl, 1 mM MgCl2 Tween-20.
3.3.2.2 Method (See Also Notes 17–21)
-
1.
Pipet 50 μL of GSH-Sepharose slurry (per reaction) into a 1.5 mL reaction tube and wash 3 times with 1 mL PBS-KMT buffer; spin down in between for 30 s at 850 × g. Wash GSH-Sepharose beads for all reactions together.
-
2.
In meanwhile thaw GST-fusion proteins (GST-Ltv1 , GST-Ltv1 ΔNES ) and GST sample on ice.
-
3.
Centrifuge the GST-Ltv1 and GST-alone or lysates for 10 min at 4 °C at 16,000 × g to pellet aggregated proteins and store the tubes on ice until use.
-
4.
Add 12 μg GST-Ltv1 protein or GST-alone to washed GSH-Sepharose beads suspended in 500 μL PBS-KMT buffer.
-
5.
Incubate on a rotating platform for 1 h at 4 °C to immobilize GST-Ltv1 and GST-alone on GSH-Sepharose beads.
-
6.
Wash samples as indicated in step 1 to remove unbound protein. Keep samples on ice between each washing step.
-
7.
The amount of GST-Ltv1 or GST-alone bound to the GST-Sepharose beads can be assessed at this stage. For this, remove supernatant completely, add 30 μL 2× LDS sample buffer, mix and heat up at 70 °C for 10 min to elute the bound proteins. Separate the eluted denatured proteins on a 12–15% SDS-PAGE and analyze by Coomassie Blue staining.
-
8.
Thaw on ice and centrifuge substrate proteins (RanQLGTP and Crm1 ) at 10 min at 4 °C at 16,000 × g to pellet aggregated proteins.
-
9.
Add simultaneously 2 μM RanQLGTP and 50 nM Crm1 to GSH-Sepharose beads immobilized GST-Ltv1 or GST-alone and suspended in 500 μL PBS-KMT buffer. In parallel, as controls, incubate immobilized GST-Ltv1 with Crm1 and RanQLGTP separately.
-
10.
Incubate on a rotating platform for 1 h at 4 °C.
-
11.
Wash samples as indicated in step 1 to remove unbound protein. Keep samples on ice between each washing step.
-
12.
Remove supernatant completely. Add 30 μL 2× LDS sample buffer, mix, and heat up at 70 °C for 10 min to elute and denature the bound proteins.
-
13.
Separate proteins on a 12–15% SDS-PAGE and subject them to Coomassie Blue staining and Western blot analysis using directed antibodies against Ran and Crm1 .
3.3.3 FG-Repeat Binding Assay
40S and 60S pre-ribosomes are transported through NPC into the cytoplasm by export factors that interact transiently with FG-repeat lining nucleoporins that line the transport channel. In addition to RanGTP-dependent exportins such as Crm1 and Exp5 [23, 24, 78] several shuttling factors such as Mex67-Mtr2 , Arx1 , Ecm1 , Bud20 , and Rrp12 promote nuclear export of pre-ribosomes by directly interacting with FG-rich meshwork of NPC [1, 57, 71, 79,80,81,82]. Here, we outline a protocol that demonstrates interactions between the heterodimeric transport receptor Mex67-Mtr2 with FG-rich nucleoporins (Fig. 5b).
3.3.3.1 Buffers and Solutions
Universal buffer (Künzler and Hurt, 1998 [83]): 20 mM HEPES–KOH, pH 7.0, 100 mM KOAc, 2 mM Mg(OAc, 0,1% Tween 20, 10% glycerol. Supplemented with (see Note 1 ) 1 complete protease inhibitor cocktail tablet (Roche) per 100 mL 5 mM ß-mercaptoethanol.
3.3.3.2 Method (See Also Notes 22–28)
-
1.
Wash 80 μL of GSH-Sepharose slurry (per reaction) into a 1.5 mL reaction tube and wash 3 times with 1 mL universal buffer; spin down in between for 30 s at 3000 rpm at 4 °C. Wash GSH-Sepharose beads for all reactions together.
-
2.
Meanwhile thaw lysates containing GST-nucleoporins (Nup1, Nup100, Nup116, Nup42) or GST alone on ice.
-
3.
Centrifuge the lysates for 10 min at 4 °C at 16,000 × g to pellet aggregated proteins and store the tubes on ice until use.
-
4.
Add 1 mL of each lysate to washed GSH-Sepharose beads (combine GSH-Sepharose beads for reactions with same GST-fusion protein) (see Note 22 ).
-
5.
Incubate tubes on a rotating platform for 1 h at 4 °C to immobilize GST and GST fusion protein on GSH-Sepharose beads.
-
6.
Wash samples 3× (850 × g, 30 s, 4 °C) with 1.5 mL universal buffer and resuspended in 1 mL buffer each.
-
7.
Split beads, by pipetting 500 μL, into two new 1.5 mL eppendorf tubes.
-
8.
Add 1–5 μg of recombinant affinity-purified His6-tagged protein (3.6 μg for Mex67-Mtr2 ) or respective volume of buffer as negative control to the GST-/GST-nucleoporin-coated beads and fill up to 1 mL with universal buffer.
-
9.
Incubate tubes on a rotating platform for 1 h at 4 °C.
-
10.
Wash beads 3× (850 × g, 30s , 4 °C) with 1 mL universal buffer, remove supernatant completely. Elute the bound proteins by adding 80 μL 2× LDS buffer and heating at 70 °C for 10 min.
-
11.
Spin down beads and analyze the supernatant by SDS-PAGE, Coomassie Blue staining and Western analysis using directed antibodies against Mex67-Mtr2 (see Note 28) .
3.4 Genetic Analyses of Nuclear Export
Pre-ribosomal particles are among the largest and most abundant cargoes that need to cross the permeability barrier of the NPC. Rapid transit through the NPC is especially important, as any delay within the channel may hinder transport of other cargoes. Therefore, pre-ribosomal particles deploy a set of multiple redundant export receptors for their transport to the cytoplasm [1, 21]. High-copy suppressor screens and synthetic lethal interactions and in budding yeast have uncovered ancillary factors that also mediate efficient pre-ribosome nuclear export . These approaches have proved to be powerful tools to either discover new components of the ribosome export machinery or to directly test functional overlap of a factor of interest with the process of nuclear export .
The essential export adaptor Nmd3 was identified as a high copy suppressor of the bud20 Δ mutant slow growth and 60S pre-ribosome export defect [72]. Further, the bud20 Δ strain exhibits synthetic lethal or synthetic enhanced growth defects when combined with mutants of established 60S pre-ribosome export factors (nmd3ΔNES1 , xpo1-1 , arx1 Δ , gle2-1, and ecm1 Δ ), included mex67 and mtr2 mutant alleles ( mex67kraa and mtr2-33 ) that are specifically impaired in 60S subunit [72]. These analyses culminated in the discovery of new FG-repeat binding proteins that functions directly to the process of nuclear export of 60S pre-ribosomes.
Similarly, a high-copy suppressor screen aimed at identifying genes that suppress the impaired growth of the slx9Δ mutant, led to the discovery of the mRNA and 60S export receptor Mex67-Mtr2 as an auxiliary export receptor for in 40S pre-ribosome export [57]. Mex67-Mtr2 mutants that fail to bind to the 40S pre-ribosome were synthetically lethal when combined with the slx9Δ mutant [57]. Further, the slx9Δ mutant displayed a synthetic growth defect when combined with a strain expressing Rrp12-GFP. Rrp12 is a 40S pre-ribosome export factor that directly interacts with FG-rich nucleoporins [79]. Finally, Yrb2 that stimulate the assembly of Crm1-export complexes on certain NES-containing cargos by cooperatively binding Crm1 and RanGTP also showed a strong genetic interaction with Slx9 [57]. This genetic link culminated in the identification of a new type of RanGTP binding protein that binds to a specific NES-containing cargo and RanGTP and promotes Crm1 recruitment.
Tables 2 and 3 provide a comprehensive list of mutants and their sources to investigate 60S and 40S preribosome export using standard yeast genetic approaches.
4 Conclusions
Diverse approaches in budding yeast have greatly advanced our understanding of ribosome assembly and transport. Genetic perturbation of energy-consuming enzymes can now be combined with biochemical affinity purifications to obtain high-resolution snapshots of the ribosome assembly pathway. We also anticipate that traditional genetic, biochemical, and cell-biological approaches will provide eagerly awaited integration of the ribosome pathway with other biological pathways including nucleocytoplasmic transport , cellular quality control and signaling.
5 Notes
-
1.
Add just before use.
-
2.
Outlined below is a step-by-step protocol to lyse frozen yeast noodles using a planetary ball mill (Pulverisette 6, Fritsch).
-
3.
You MUST hear the balls rattling around in the jar!
If there is no rattling: add or remove balls to the jar until you hear it rattle. It is not considered a grinding cycle unless there is rattling.
-
4.
Typically, ~90% of yeast cells can be disrupted in such procedure.
-
5.
Use cryoprotective gloves for harvesting cells.
-
6.
Work on ice or cold room as much as possible.
-
7.
Prepare powder stock from big culture, store at −80 °C and weight only necessary amount for an experiment.
-
8.
The M-IgG beads are prepared by conjugation of Dynabeads (Invitrogen) with rabbit IgG (Sigma) according to the standard protocol.
-
9.
Plasmids encoding GST-Kap123 and GST-Pse1 [86], RanQLGTP mutant [87] originate from Schlenstedt laboratory. Slx9 [58] and eS26Flag (His6-eS26FLAG; [56]) can be requested from Panse laboratory.
-
10.
GST recombinant proteins were expressed in E.coli and lysed in PBS-KMT (150 mM NaCl, 25 mM sodium phosphate, 3 mM KCl, 1 mM MgCl2, 0.1% Tween, pH 7.3) buffer. His-tagged proteins were purified in buffer containing 50 mM HEPES pH 7.5, 50 mM NaCl, 10% glycerol. It is possible to use directly E.coli lysates containing recombinant proteins without purification steps [56, 86]. His6-tagged importins and RanQLGTP (His6-Gsp1Q71L-GTP) were expressed and purified as previously described [86,87,88].
-
11.
To prepare E. coli lysate BL21 cells was grown overnight in 100 mL LB media at 37 °C and 220 rpm. Diluted in 1 l of LB media to OD600 0.2 and again incubated at the same condition. Cells were harvest at logarithmic growth phase and resuspended in PBS-KMT. After cell lysis and centrifugation supernatant is used for the experiment.
-
12.
To prevent unspecific interactions with GST-tagged proteins or GST it is possible to add E. coli lysate to the incubation reaction (100 μL is usually sufficient).
-
13.
The Ran protein mutant used in this assay, RanQL (Gsp1Q71L), is a GTPase deficient mutant originating from Schlenstedt laboratory. His6-RanQL is stored in KPi buffer (24.8 mM KH2PO4, 25 mM, K2HPO4, 20 mM KCl, 5 mM MgCl2, 2 mM imidazole, 5 mM β-mercaptoethanol, pH 6.8).
-
14.
Antibodies against FLAG tag are commercially available reagent from Sigma (F1804).
-
15.
This assay can be performed with strains containing the reporters growing on solid selective media. In this case, strains typically in stationary phase, are spread on fresh YPD plates to induce ribosome production. After growing the cells for 4–8 h at the appropriate temperature, cells can be scraped from plate using an inoculation loop, mixed with 2 μL H2O placed on a glass slide and analyzed under a fluorescence microscope.
-
16.
The uL23-GFP and uS5-GFP reporter plasmids with different selection markers (LEU2, URA3, TRP1, and HIS3) originate from the Hurt laboratory [73, 74], the uL5-GFP reporter (LEU2) originates from the Silver laboratory [75], and the uL18-GFP reporter plasmids (LEU2, URA3, TRP1 and HIS3) can be requested from the Panse laboratory [72]. All the above reporters are expressed under the endogenous promoters. The ribosomal protein uL18 can be C-terminally tagged with GFP at the genomic locus for monitoring the nuclear export of the large pre-ribosomal subunit.
-
17.
Plasmids for yeast Crm1 (Xpo1-His6) and RanQL mutant (His6-Gsp1Q71L) originate from Schlenstedt lab [87], GST-Ltv1 and GST-Ltv1 ΔNES can be requested from Panse lab [58].
-
18.
Recombinant proteins were expressed in E. coli and either purified in PBS-KMT or used as lysate.
-
19.
GST-tagged proteins and His6-Gsp1Q71L was expressed in E. coli as described in [87, 88]. Crm1-His6 was expressed in E. coli as described in [58]. GST, GST-Ltv1 and GST-Ltv1 ΔNES , and Crm1-His6 proteins are stored in PBS-KMT buffer. His6-Gsp1Q71L is stored in KPi buffer (24.8 mM KH2PO4, 25 mM, K2HPO4, 20 mM KCl, 5 mM MgCl2, 2 mM imidazole, 5 mM β-mercaptoethanol, pH 6.8).
-
20.
To prevent unspecific interactions with GST-tagged proteins or GST it is possible to add E.coli lysate to the incubation reaction (100 μL is usually sufficient).
-
21.
Antibodies against Ran and Crm1 can be requested in Panse laboratory.
-
22.
For the load: Respective amount of affinity-purified His6-tagged Mex67-Mtr2 is added to final 100 μL 2× LDS, heated at 70 °C for 10 min.
-
23.
All the steps should be carried out at 4 °C or on ice.
-
24.
Cut pipette tip when pipetting beads.
-
25.
Recombinant His6-tagged protein (Mex67-Mtr2 ) was expressed in BL21 E. coli strain, lysed by sonication in lysis buffer (500 mM NaCl, 20 mM Tris–HCl pH 8.0, 0.1% Triton X-100, 1 mM PMSF) and purified using Ni-Sepharose (GE Healthcare) following a standard protocol.
-
26.
The lysates containing GST-tagged nucleoporins (Nup1, Nup100, Nup116, Nup42) or GST alone were expressed in BL21 E. coli strain lysed by sonication in universal buffer.
-
27.
Due to different expression levels, different volumes of GST-NUPs lysates may need to be used to uniformly load GSH-Sepharose beads.
-
28.
The antibody directed against Mex67-Mtr2 can be requested from the Panse laboratory [72].
References
Pena C, Hurt E, Panse VG (2017) Eukaryotic ribosome assembly, transport and quality control. Nat Struct Mol Biol 24:689–699
Warner JR (1999) The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24:437–440
Kressler D, Hurt E, Bassler J (2017) A puzzle of life: crafting ribosomal subunits. Trends Biochem Sci 42:640–654
Kornprobst M, Turk M, Kellner N, Cheng J, Flemming D, Kos-Braun I, Kos M, Thoms M, Berninghausen O, Beckmann R, Hurt E (2016) Architecture of the 90S pre-ribosome: a structural view on the birth of the eukaryotic ribosome. Cell 166:380–393
Cheng J, Kellner N, Berninghausen O, Hurt E, Beckmann R (2017) 3.2-A-resolution structure of the 90S preribosome before A1 pre-rRNA cleavage. Nat Struct Mol Biol 24:954–964
Sun Q, Zhu X, Qi J, An W, Lan P, Tan D, Chen R, Wang B, Zheng S, Zhang C, Chen X, Zhang W, Chen J, Dong MQ, Ye K (2017) Molecular architecture of the 90S small subunit pre-ribosome. elife 6:e22086
Chaker-Margot M, Barandun J, Hunziker M, Klinge S (2017) Architecture of the yeast small subunit processome. Science 355:eaal1880
Osheim YN, French SL, Keck KM, Champion EA, Spasov K, Dragon F, Baserga SJ, Beyer AL (2004) Pre-18S ribosomal RNA is structurally compacted into the SSU processome prior to being cleaved from nascent transcripts in Saccharomyces cerevisiae. Mol Cell 16:943–954
Kos M, Tollervey D (2010) Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol Cell 37:809–820
Billy E, Wegierski T, Nasr F, Filipowicz W (2000) Rcl1p, the yeast protein similar to the RNA 3′-phosphate cyclase, associates with U3 snoRNP and is required for 18S rRNA biogenesis. EMBO J 19:2115–2126
Horn DM, Mason SL, Karbstein K (2011) Rcl1 protein, a novel nuclease for 18 S ribosomal RNA production. J Biol Chem 286:34082–34087
Sardana R, Liu X, Granneman S, Zhu J, Gill M, Papoulas O, Marcotte EM, Tollervey D, Correll CC, Johnson AW (2015) The DEAH-box helicase Dhr1 dissociates U3 from the pre-rRNA to promote formation of the central pseudoknot. PLoS Biol 13:e1002083
Zhu J, Liu X, Anjos M, Correll CC, Johnson AW (2016) Utp14 recruits and activates the RNA helicase Dhr1 to undock U3 snoRNA from the Preribosome. Mol Cell Biol 36:965–978
Thoms M, Thomson E, Bassler J, Gnadig M, Griesel S, Hurt E (2015) The exosome is recruited to RNA substrates through specific adaptor proteins. Cell 162:1029–1038
Kater L, Thoms M, Barrio-Garcia C, Cheng J, Ismail S, Ahmed YL, Bange G, Kressler D, Berninghausen O, Sinning I, Hurt E, Beckmann R (2017) Visualizing the assembly pathway of nucleolar pre-60S ribosomes. Cell 171(1599–1610):e14
Sanghai ZA, Miller L, Molloy KR, Barandun J, Hunziker M, Chaker-Margot M, Wang J, Chait BT, Klinge S (2018) Modular assembly of the nucleolar pre-60S ribosomal subunit. Nature 556:126–129
Zhou D, Zhu X, Zheng S, Tan D, Dong MQ, Ye K (2019) Cryo-EM structure of an early precursor of large ribosomal subunit reveals a half-assembled intermediate. Protein Cell 10:120–130
Winey M, Yarar D, Giddings TH Jr, Mastronarde DN (1997) Nuclear pore complex number and distribution throughout the Saccharomyces cerevisiae cell cycle by three-dimensional reconstruction from electron micrographs of nuclear envelopes. Mol Biol Cell 8:2119–2132
Stade K, Ford CS, Guthrie C, Weis K (1997) Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 90:1041–1050
Neville M, Rosbash M (1999) The NES-Crm1p export pathway is not a major mRNA export route in Saccharomyces cerevisiae. EMBO J 18:3746–3756
Nerurkar P, Altvater M, Gerhardy S, Schutz S, Fischer U, Weirich C, Panse VG (2015) Eukaryotic ribosome assembly and nuclear export. Int Rev Cell Mol Biol 319:107–140
Gerhardy S, Menet AM, Pena C, Petkowski JJ, Panse VG (2014) Assembly and nuclear export of pre-ribosomal particles in budding yeast. Chromosoma 123:327–344
Ho JH, Kallstrom G, Johnson AW (2000) Nmd3p is a Crm1p-dependent adapter protein for nuclear export of the large ribosomal subunit. J Cell Biol 151:1057–1066
Gadal O, Strauss D, Kessl J, Trumpower B, Tollervey D, Hurt E (2001) Nuclear export of 60s ribosomal subunits depends on Xpo1p and requires a nuclear export sequence-containing factor, Nmd3p, that associates with the large subunit protein Rpl10p. Mol Cell Biol 21:3405–3415
Occhipinti L, Chang Y, Altvater M, Menet AM, Kemmler S, Panse VG (2013) Non-FG mediated transport of the large pre-ribosomal subunit through the nuclear pore complex by the mRNA export factor Gle2. Nucleic Acids Res 41:8266–8279
Panse VG, Johnson AW (2010) Maturation of eukaryotic ribosomes: acquisition of functionality. Trends Biochem Sci 35:260–266
Strunk BS, Loucks CR, Su M, Vashisth H, Cheng S, Schilling J, Brooks CL 3rd, Karbstein K, Skiniotis G (2011) Ribosome assembly factors prevent premature translation initiation by 40S assembly intermediates. Science 333:1449–1453
Strunk BS, Novak MN, Young CL, Karbstein K (2012) A translation-like cycle is a quality control checkpoint for maturing 40S ribosome subunits. Cell 150:111–121
Lebaron S, Schneider C, van Nues RW, Swiatkowska A, Walsh D, Bottcher B, Granneman S, Watkins NJ, Tollervey D (2012) Proofreading of pre-40S ribosome maturation by a translation initiation factor and 60S subunits. Nat Struct Mol Biol 19:744–753
Scaiola A, Pena C, Weisser M, Bohringer D, Leibundgut M, Klingauf-Nerurkar P, Gerhardy S, Panse VG, Ban N (2018) Structure of a eukaryotic cytoplasmic pre-40S ribosomal subunit. EMBO J 37:e98499
Heuer A, Thomson E, Schmidt C, Berninghausen O, Becker T, Hurt E, Beckmann R (2017) Cryo-EM structure of a late pre-40S ribosomal subunit from Saccharomyces cerevisiae. elife 6:e30189
Zhou Y, Musalgaonkar S, Johnson AW, Taylor DW (2019) Tightly-orchestrated rearrangements govern catalytic center assembly of the ribosome. Nat Commun 10:958
Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17:1030–1032
Bassler J, Grandi P, Gadal O, Lessmann T, Petfalski E, Tollervey D, Lechner J, Hurt E (2001) Identification of a 60S preribosomal particle that is closely linked to nuclear export. Mol Cell 8:517–529
Harnpicharnchai P, Jakovljevic J, Horsey E, Miles T, Roman J, Rout M, Meagher D, Imai B, Guo Y, Brame CJ, Shabanowitz J, Hunt DF, Woolford JL Jr (2001) Composition and functional characterization of yeast 66S ribosome assembly intermediates. Mol Cell 8:505–515
Grandi P, Rybin V, Bassler J, Petfalski E, Strauss D, Marzioch M, Schafer T, Kuster B, Tschochner H, Tollervey D, Gavin AC, Hurt E (2002) 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol Cell 10:105–115
Dragon F, Gallagher JE, Compagnone-Post PA, Mitchell BM, Porwancher KA, Wehner KA, Wormsley S, Settlage RE, Shabanowitz J, Osheim Y, Beyer AL, Hunt DF, Baserga SJ (2002) A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417:967–970
Fatica A, Cronshaw AD, Dlakic M, Tollervey D (2002) Ssf1p prevents premature processing of an early pre-60S ribosomal particle. Mol Cell 9:341–351
Nissan TA, Bassler J, Petfalski E, Tollervey D, Hurt E (2002) 60S pre-ribosome formation viewed from assembly in the nucleolus until export to the cytoplasm. EMBO J 21:5539–5547
Schafer T, Strauss D, Petfalski E, Tollervey D, Hurt E (2003) The path from nucleolar 90S to cytoplasmic 40S pre-ribosomes. EMBO J 22:1370–1380
Greber BJ, Gerhardy S, Leitner A, Leibundgut M, Salem M, Boehringer D, Leulliot N, Aebersold R, Panse VG, Ban N (2016) Insertion of the biogenesis factor Rei1 probes the ribosomal tunnel during 60S maturation. Cell 164:91–102
Ma C, Wu S, Li N, Chen Y, Yan K, Li Z, Zheng L, Lei J, Woolford JL Jr, Gao N (2017) Structural snapshot of cytoplasmic pre-60S ribosomal particles bound by Nmd3, Lsg1, Tif6 and Reh1. Nat Struct Mol Biol 24:214–220
Oeffinger M, Wei KE, Rogers R, DeGrasse JA, Chait BT, Aitchison JD, Rout MP (2007) Comprehensive analysis of diverse ribonucleoprotein complexes. Nat Methods 4:951–956
Pillet B, Mitterer V, Kressler D, Pertschy B (2017) Hold on to your friends: dedicated chaperones of ribosomal proteins: dedicated chaperones mediate the safe transfer of ribosomal proteins to their site of pre-ribosome incorporation. BioEssays 39:1–12
Eisinger DP, Dick FA, Denke E, Trumpower BL (1997) SQT1, which encodes an essential WD domain protein of Saccharomyces cerevisiae, suppresses dominant-negative mutations of the ribosomal protein gene QSR1. Mol Cell Biol 17:5146–5155
Iouk TL, Aitchison JD, Maguire S, Wozniak RW (2001) Rrb1p, a yeast nuclear WD-repeat protein involved in the regulation of ribosome biosynthesis. Mol Cell Biol 21:1260–1271
Schaper S, Fromont-Racine M, Linder P, de la Cruz J, Namane A, Yaniv M (2001) A yeast homolog of chromatin assembly factor 1 is involved in early ribosome assembly. Curr Biol 11:1885–1890
West M, Hedges JB, Chen A, Johnson AW (2005) Defining the order in which Nmd3p and Rpl10p load onto nascent 60S ribosomal subunits. Mol Cell Biol 25:3802–3813
Koch B, Mitterer V, Niederhauser J, Stanborough T, Murat G, Rechberger G, Bergler H, Kressler D, Pertschy B (2012) Yar1 protects the ribosomal protein Rps3 from aggregation. J Biol Chem 287:21806–21815
Kressler D, Bange G, Ogawa Y, Stjepanovic G, Bradatsch B, Pratte D, Amlacher S, Strauss D, Yoneda Y, Katahira J, Sinning I, Hurt E (2012) Synchronizing nuclear import of ribosomal proteins with ribosome assembly. Science 338:666–671
Stelter P, Huber FM, Kunze R, Flemming D, Hoelz A, Hurt E (2015) Coordinated ribosomal L4 protein assembly into the pre-ribosome is regulated by its eukaryote-specific extension. Mol Cell 58:854–862
Pillet B, Garcia-Gomez JJ, Pausch P, Falquet L, Bange G, de la Cruz J, Kressler D (2015) The dedicated chaperone Acl4 escorts ribosomal protein Rpl4 to its nuclear pre-60S assembly site. PLoS Genet 11:e1005565
Pausch P, Singh U, Ahmed YL, Pillet B, Murat G, Altegoer F, Stier G, Thoms M, Hurt E, Sinning I, Bange G, Kressler D (2015) Co-translational capturing of nascent ribosomal proteins by their dedicated chaperones. Nat Commun 6:7494
Rossler I, Embacher J, Pillet B, Murat G, Liesinger L, Hafner J, Unterluggauer JJ, Birner-Gruenberger R, Kressler D, Pertschy B (2019) Tsr4 and Nap1, two novel members of the ribosomal protein chaperOME. Nucleic Acids Res 47(13):6984–7002. https://doi.org/10.1093/nar/gkz317
Black JJ, Musalgaonkar S, Johnson AW (2019) Tsr4 is a cytoplasmic chaperone for the ribosomal protein Rps2 in Saccharomyces cerevisiae. Mol Cell Biol 39:e00094-19. https://doi.org/10.1128/MCB.00094-19
Schutz S, Fischer U, Altvater M, Nerurkar P, Pena C, Gerber M, Chang Y, Caesar S, Schubert OT, Schlenstedt G, Panse VG (2014) A RanGTP-independent mechanism allows ribosomal protein nuclear import for ribosome assembly. elife 3:e03473
Faza MB, Chang Y, Occhipinti L, Kemmler S, Panse VG (2012) Role of Mex67-Mtr2 in the nuclear export of 40S pre-ribosomes. PLoS Genet 8:e1002915
Fischer U, Schauble N, Schutz S, Altvater M, Chang Y, Faza MB, Panse VG (2015) A non-canonical mechanism for Crm1-export cargo complex assembly. elife 4:e05745
Bax R, Raue HA, Vos JC (2006) Slx9p facilitates efficient ITS1 processing of pre-rRNA in Saccharomyces cerevisiae. RNA 12:2005–2013
Li Z, Lee I, Moradi E, Hung NJ, Johnson AW, Marcotte EM (2009) Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol 7:e1000213
Yano R, Oakes ML, Tabb MM, Nomura M (1994) Yeast Srp1p has homology to armadillo/plakoglobin/beta-catenin and participates in apparently multiple nuclear functions including the maintenance of the nucleolar structure. Proc Natl Acad Sci U S A 91:6880–6884
Koepp DM, Wong DH, Corbett AH, Silver PA (1996) Dynamic localization of the nuclear import receptor and its interactions with transport factors. J Cell Biol 133:1163–1176
Aitchison JD, Blobel G, Rout MP (1996) Kap104p: a karyopherin involved in the nuclear transport of messenger RNA binding proteins. Science 274:624–627
Sydorskyy Y, Dilworth DJ, Yi EC, Goodlett DR, Wozniak RW, Aitchison JD (2003) Intersection of the Kap123p-mediated nuclear import and ribosome export pathways. Mol Cell Biol 23:2042–2054
Senger B, Simos G, Bischoff FR, Podtelejnikov A, Mann M, Hurt E (1998) Mtr10p functions as a nuclear import receptor for the mRNA-binding protein Npl3p. EMBO J 17:2196–2207
Greiner M, Caesar S, Schlenstedt G (2004) The histones H2A/H2B and H3/H4 are imported into the yeast nucleus by different mechanisms. Eur J Cell Biol 83:511–520
Seedorf M, Silver PA (1997) Importin/karyopherin protein family members required for mRNA export from the nucleus. Proc Natl Acad Sci U S A 94:8590–8595
Schlenstedt G, Smirnova E, Deane R, Solsbacher J, Kutay U, Görlich D, Ponstingl H, Bischoff FR (1997) Yrb4p, a yeast ran-GTP-binding protein involved in import of ribosomal protein L25 into the nucleus. EMBO J 16:6237–6249
Hung NJ, Lo KY, Patel SS, Helmke K, Johnson AW (2008) Arx1 is a nuclear export receptor for the 60S ribosomal subunit in yeast. Mol Biol Cell 19:735–744
Strasser K, Bassler J, Hurt E (2000) Binding of the Mex67p/Mtr2p heterodimer to FXFG, GLFG, and FG repeat nucleoporins is essential for nuclear mRNA export. J Cell Biol 150:695–706
Yao W, Roser D, Kohler A, Bradatsch B, Bassler J, Hurt E (2007) Nuclear export of ribosomal 60S subunits by the general mRNA export receptor Mex67-Mtr2. Mol Cell 26:51–62
Altvater M, Chang Y, Melnik A, Occhipinti L, Schütz S, Rothenbusch U, Picotti P, Panse VG (2012) Targeted proteomics reveals compositional dynamics of 60S pre-ribosomes after nuclear export. Mol Syst Biol 8:628
Hurt E, Hannus S, Schmelzl B, Lau D, Tollervey D, Simos G (1999) A novel in vivo assay reveals inhibition of ribosomal nuclear export in Ran-cycle and nucleoporin mutants. J Cell Biol 144:389–401
Milkereit P, Strauss D, Bassler J, Gadal O, Kühn H, Schütz S, Gas N, Lechner J, Hurt E, Tschochner H (2003) A Noc complex specifically involved in the formation and nuclear export of ribosomal 40 S subunits. J Biol Chem 278:4072–4081
Stage-Zimmermann T, Schmidt U, Silver PA (2000) Factors affecting nuclear export of the 60S ribosomal subunit in vivo. Mol Biol Cell 11:3777–3789
Seiser RM, Sundberg AE, Wollam BJ, Zobel-Thropp P, Baldwin K, Spector MD, Lycan DE (2006) Ltv1 is required for efficient nuclear export of the ribosomal small subunit in Saccharomyces cerevisiae. Genetics 174:679–691
Zemp I, Wild T, O'Donohue MF, Wandrey F, Widmann B, Gleizes PE, Kutay U (2009) Distinct cytoplasmic maturation steps of 40S ribosomal subunit precursors require hRio2. J Cell Biol 185:1167–1180
Wild T, Horvath P, Wyler E, Widmann B, Badertscher L, Zemp I, Kozak K, Csucs G, Lund E, Kutay U (2010) A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol 8:e1000522
Oeffinger M, Dlakic M, Tollervey D (2004) A pre-ribosome-associated HEAT-repeat protein is required for export of both ribosomal subunits. Genes Dev 18:196–209
Bradatsch B, Katahira J, Kowalinski E, Bange G, Yao W, Sekimoto T, Baumgartel V, Boese G, Bassler J, Wild K, Peters R, Yoneda Y, Sinning I, Hurt E (2007) Arx1 functions as an unorthodox nuclear export receptor for the 60S preribosomal subunit. Mol Cell 27:767–779
Yao Y, Demoinet E, Saveanu C, Lenormand P, Jacquier A, Fromont-Racine M (2010) Ecm1 is a new pre-ribosomal factor involved in pre-60S particle export. RNA 16:1007–1017
Bassler J, Klein I, Schmidt C, Kallas M, Thomson E, Wagner MA, Bradatsch B, Rechberger G, Strohmaier H, Hurt E, Bergler H (2012) The conserved Bud20 zinc finger protein is a new component of the ribosomal 60S subunit export machinery. Mol Cell Biol 32:4898–4912
Kunzler M, Hurt EC (1998) Cse1p functions as the nuclear export receptor for importin alpha in yeast. FEBS Lett 433:185–190
Murphy R, Watkins JL, Wente SR (1996) GLE2, a Saccharomyces cerevisiae homologue of the Schizosaccharomyces pombe export factor RAE1, is required for nuclear pore complex structure and function. Mol Biol Cell 7:1921–1937
Santos-Rosa H, Moreno H, Simos G, Segref A, Fahrenkrog B, Pante N, Hurt E (1998) Nuclear mRNA export requires complex formation between Mex67p and Mtr2p at the nuclear pores. Mol Cell Biol 18:6826–6838
Fries T, Betz C, Sohn K, Caesar S, Schlenstedt G, Bailer SM (2007) A novel conserved nuclear localization signal is recognized by a group of yeast importins. J Biol Chem 282:19292–19301
Maurer P, Redd M, Solsbacher J, Bischoff FR, Greiner M, Podtelejnikov AV, Mann M, Stade K, Weis K, Schlenstedt G (2001) The nuclear export receptor Xpo1p forms distinct complexes with NES transport substrates and the yeast Ran binding protein 1 (Yrb1p). Mol Biol Cell 12:539–549
Solsbacher J, Maurer P, Bischoff FR, Schlenstedt G (1998) Cse1p is involved in export of yeast importin alpha from the nucleus. Mol Cell Biol 18:6805–6815
Acknowledgments
V.G.P. is supported by grants from the Swiss National Science Foundation, the NCCR RNA & Disease, the Novartis Foundation and the Olga Mayenfisch Stiftung. M.O-O. is a recipient of a doctoral fellowship from the Boehringer Ingelheim Fonds.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
This chapter is dedicated to the memory of our former graduate student and friend Dr. Cohue Peña.
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Oborská-Oplová, M., Fischer, U., Altvater, M., Panse, V.G. (2022). Eukaryotic Ribosome assembly and Nucleocytoplasmic Transport. In: Entian, KD. (eds) Ribosome Biogenesis. Methods in Molecular Biology, vol 2533. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2501-9_7
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2501-9_7
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2500-2
Online ISBN: 978-1-0716-2501-9
eBook Packages: Springer Protocols