
Abstract
Quantitative systems pharmacology (QSP) places an emphasis on dynamic systems modeling, incorporating considerations from systems biology modeling and pharmacodynamics. The goal of QSP is often to quantitatively predict the effects of clinical therapeutics, their combinations, and their doses on clinical biomarkers and endpoints. In order to achieve this goal, strategies for incorporating clinical data into model calibration are critical. Virtual population (VPop) approaches facilitate model calibration while faced with challenges encountered in QSP model application, including modeling a breadth of clinical therapies, biomarkers, endpoints, utilizing data of varying structure and source, capturing observed clinical variability, and simulating with models that may require more substantial computational time and resources than often found in pharmacometrics applications. VPops are frequently developed in a process that may involve parameterization of isolated pathway models, integration into a larger QSP model, incorporation of clinical data, calibration, and quantitative validation that the model with the accompanying, calibrated VPop is suitable to address the intended question or help with the intended decision. Here, we introduce previous strategies for developing VPops in the context of a variety of therapeutic and safety areas: metabolic disorders, drug-induced liver injury, autoimmune diseases, and cancer. We introduce methodological considerations, prior work for sensitivity analysis and VPop algorithm design, and potential areas for future advancement. Finally, we give a more detailed application example of a VPop calibration algorithm that illustrates recent progress and many of the methodological considerations. In conclusion, although methodologies have varied, VPop strategies have been successfully applied to give valid clinical insights and predictions with the assistance of carefully defined and designed calibration and validation strategies. While a uniform VPop approach for all potential QSP applications may be challenging given the heterogeneity in use considerations, we anticipate continued innovation will help to drive VPop application for more challenging cases of greater scale while developing new rigorous methodologies and metrics.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Virtual population
- Virtual patient
- In silico trial
- Virtual patient cohort
- Sensitivity analysis
- Objective functions
- Prevalence weight
1 Introduction to the Use of Virtual Populations in Quantitative Systems Pharmacology Models
Quantitative systems pharmacology (QSP) is a discipline that incorporates elements of systems biology and pharmacometrics with an emphasis on dynamic systems modeling [1], often with the goal to quantitatively predict the effects of clinical interventions, their combinations, and their doses on clinical biomarkers and endpoints. In practice, QSP model development typically emphasizes biological regulation demonstrated to play a role in pathophysiology and clinical response, integration with reported clinical biomarkers, and clinical endpoints as prime considerations for the initial model design and refinement. In addition, QSP models are inherently both quantitative and mechanistic in that they aim to model the dynamic behaviors of a system in a fashion that is biophysically constrained by factors such as molecular affinities, proliferation rates, and transport rates. QSP is therefore distinct from some approaches to systems biology that may be more qualitative in deriving insights from data and place an initial emphasis on lateral integration of many pathways [1, 2]. In addition to characterizing the biological or physiological system, QSP models relate the quantity of drug dosed, frequency of dosing, pharmacokinetics, and proximal pharmacodynamics with biological or physiological responses. From this perspective, QSP models can be described as physiologic system pharmacodynamic models that incorporate key considerations from pharmacokinetics, and similar to physiologically based pharmacokinetic models often incorporate a reasonable mechanistic strategy for linking serum or plasma concentrations of a drug to concentrations at the modeled site of action.
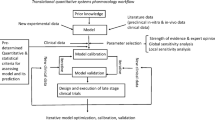
QSP models and their application have been discussed in much detail previously [3,4,5,6], and the goal of this chapter is to both provide background for how Virtual Populations (VPops) form a critical component of QSP research and an introduction to methodology for VPop development. Once established, VPops can be used to generate well-informed predictions on the patient population in new, previously untested, clinical scenarios, such as exploration of new therapeutic interventions. As shown in Fig. 1, VPops can be viewed as forming a critical link between basic pathway information, prior clinical data, quantitative clinical hypotheses, and actionable clinical decisions. Although this chapter will largely focus on clinical development applications, VPops have also served to help evaluate and prioritize new targets and pathways for therapeutic intervention in the setting of drug discovery.

VPops facilitate actionable clinical development insight by integrating pathway knowledge, nonclinical insights, and clinical data into a simulation-based framework that is both in quantitative agreement with these inputs and facilitates modeling new scenarios not included in the source information
2 A Brief Introduction to QSP Model Development
There exists a number of different potential mathematical modeling formalisms for QSP models [7], and QSP models intend to facilitate an understanding of a pathophysiological system. However, not all network-based analyses to identify interaction points for a pharmacological inhibition, although potentially useful, would necessarily be described as QSP modeling approaches. As the name implies, a QSP model intends to capture quantitative outcomes. QSP models mechanistically describe pathophysiological processes, model dynamic quantitative physiologic endpoints, and are used to predict how the dynamics of biomarkers and clinical endpoints change in response to a pharmacological intervention [1]. Systems of ordinary differential equations (ODEs) are commonly used for QSP modeling, and the VPop examples will focus on application with ODE models.
Determining the scale of a QSP model is an important step in planning its development, and scale can vary significantly depending on the scope and application of the model. For many discovery and preclinical applications, small-scale QSP models focusing on very specific biological pathways proximal to the drug target could be sufficient to gain some insight on the target mechanism of action and assist in drug discovery. As will be discussed later, small pathway-level QSP models may be fit to nonclinical data to extract informative model parameters to implement in a more comprehensive QSP model that integrates more of the biological system and feedbacks. The larger model therefore helps with a broader translation of nonclinical knowledge in the context of prior clinical data on targets of interest and related pathways. Small pathway-level models may also find applications in helping to optimize a specific molecule or in late clinical development to simply link a pharmacologic intervention to a biomarker of interest. For clinical applications, middle to large-scale QSP models that describe disease pathophysiology are often well suited to capture biomarker and clinical endpoint diversity representative of real patients, which is often achieved through the development of VPops. Recent progress in computational model development and application packages, advances in computing power, and the availability of richer clinical datasets have made this goal more readily achievable. It is worth noting that larger, more comprehensive models can capture many biomarkers and clinical endpoints of interest, and thus are well suited for identifying new pathways as potential therapeutic targets [8], assisting in biomarker development [9], prioritizing assets for clinical development, and mechanistically characterizing combination therapies and the quantitative contribution of components for combinations. The number of pathways in a model can increase the computational requirements for calibration and application of the model, especially when they span multiple time scales. However, opportunities to calibrate the model against a broader array of data and test salient predictions to validate the model for a particular application can also increase as more pathways and endpoints are incorporated. In the next section, we will provide a brief overview of the development of QSP models and the utility of VPops.
2.1 QSP Model Scope
Scoping is one of the most important factors that shapes the structure and complexity of a QSP model [3, 7]. In practice, an initial QSP model scope can often be achieved by thorough discussions with stakeholders in different scientific groups within pharmaceutical research and development, which may include clinicians, biomarker scientists, clinical pharmacologists, and drug discovery scientists [3]. This also serves to attain alignment on the scope of the model with these important stakeholders from the drug discovery and development matrix teams. Alignment with the matrix teams is crucial, as it helps to set concrete deliverables and timelines for model development and application. An experienced QSP modeler with significant expertise in the disease area is a primary contributor in both scoping and development stages. A thorough understanding of both the mathematics and biology ensures that key mechanistic considerations are incorporated while also resulting in a model with feedbacks structured to give reasonable physiologic behaviors.
Medium to large-scale QSP models that incorporate relevant positive and negative feedbacks based on key target as well as disease cellular and molecular physiology can provide more mechanistic insights while still giving good predictions. This requires a well-informed modeling strategy with suitable information and data to constrain the positive and negative feedbacks. Practically, if reusing and expanding an existing model that is performing well for related applications in a disease area, the expansiveness of the additional pathways to evaluate as well as amount of additional data to inform the model recalibration will have a substantial role in impacting project timelines. Like many multicompartment modeling paradigms in mathematical biology [10], the following elements should be identified to assemble QSP models for application to clinical development and discovery:
-
Compartments. The first step is the identification of biological compartments that usually represent physiological units in the broad sense. For example, a minimal set for an immuno-oncology (I-O) QSP model could include blood compartment, a tumor-draining lymph node, and tumor compartments in order to include key components of the cancer–immunity cycle [11]. Additional physiologic compartments such as a thymus might be added as needed to model specific behaviors or accommodate additional biomarkers.
-
Species. Once the compartment configuration is set up, a list of species tracked in each compartment can be identified based on the model scope. From a mathematical standpoint, a species will generally correspond to a state variable, but during the model development process there may be exceptions to this rule-of-thumb. For an I-O QSP model, these species could include the following.
-
Cancer cells and immune cells, such as CD8 T cells, CD4 Treg cells, antigen presenting cells, and others.
-
Immune checkpoints, possibly including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1), and lymphocyte-activation gene 3 (LAG3).
-
Mediators released by immune cells, including cytokines and chemokines.
-
Other important cell surface receptors, such as targeted cytokine receptors.
-
Therapeutic interventions, such as small molecules or antibodies and other biologics.
-
-
Reactions in each compartment and models for reaction fluxes. Developing a QSP model entails defining accurate mathematical description of the interactions between the species within a compartment. For an I-O QSP model, these interactions could include cell proliferation and apoptosis, cell–cell contact, mediator production, positive and negative feedbacks induced by mediators, cell surface molecule dynamics, and surface molecule induced positive and negative feedbacks, such as those caused by immune checkpoints. The dynamic behaviors of immune checkpoints, including binding, unbinding, and cell membrane diffusion, are of particular interest for linking therapeutic target engagement with pharmacodynamic effects [12].
-
Transport between compartments. Transport processes that bridge different compartments often need to be modeled. We often might not explicitly model these biological compartments spatially, therefore the transport processes should be developed to reflect the underlying biology. This is often accomplished by a series of mathematical approximations, and a relevant example is the tumor penetration of therapeutic molecules [13,14,15]. Still taking an I-O QSP model as an example, important transport processes could include the following.
-
Immune cell exchanges between compartments, such as immune cell extravasation from the blood and immune cell egress from tumor into a tumor-draining lymph node.
-
Molecular transport, such as therapeutic molecules being delivered from blood to tumor and mediator exchanges such as the transport of cytokines.
-
-
Therapeutic interventions. This includes the drug dosing regimens, pharmacokinetic models, target binding, and proximal effects in the model.
There has been much discussion of parameter estimation for systems biology and QSP models [7, 15]. From a practical standpoint, the reliability of applying the model in a predictive context for biomarker and clinical endpoint outcomes is a distinct and often more salient consideration [17, 18]. The systems biology community has been exploring strategies for direct characterization of prediction uncertainty [16,17,18]. We will touch on practical strategies for characterizing uncertainty in QSP model predictions with VPops later in the chapter. It is also worth noting that gathering relevant parameters from fitted pathway models, where a goal is often to characterize the pathway itself as opposed to immediately making a clinical prediction, is a distinct and important consideration. Pathway-level parameters extracted from other sources such as modeling well-controlled in vitro experiments can serve as a basis for parameterizing isolated pathways in a larger QSP model. Additional steps and feedbacks will play a role in translating from the in vitro pathways measured to the clinical disease system. That is, modeled reactions and intercompartment transport can be modeled and parameterized as their own constituents by controlled pathway model development, and careful integration of suitable pathway models into a larger QSP model may improve the process of translating in vitro results.
2.1.1 Pathway Model Development
A pathway model is a small-scale model that focuses on a key biological pathway and its detailed parameterization. During pathway model development, we want to ensure that estimated parameters are identifiable. To facilitate discussion, we consider the process of antibody-dependent cellular cytotoxicity (ADCC). ADCC is a mechanism of cell killing where an effector cell of the immune system binds to the fragment crystallizable (Fc) region of an antibody bound to a specific antigen at the surface of the target cell and releases cytotoxic factors to lyse the target cell. In this example, the effector cell is a natural killer cell (NK), the target cell is a CD4 Treg cell, the antigen is CTLA-4, and the antibody is a member of the class of anti-CTLA-4 antibodies. Here, we desire to get a quantitative estimation of ADCC potencies of different anti-CTLA-4 antibodies. One way to achieve this is through a well-designed in vitro experiment that reports target cell lysis when the assay cocultures different effector to target cell ratios (E:T ratios) as well as different antibody concentrations at physiological conditions (see Fig. 2). Ideally, the effector NK cells and target CD4 Treg cells are derived from human donors so we can better translate the results from this small pathway model to the QSP model.

A scheme of an in vitro ADCC experiment. E: effector cell, here the effector cell is NK cell; T: target cell, here the target cell is CD4 Treg
In order to build a pathway model to describe the in vitro ADCC, we need to develop an ODE system that describes the time course of the cell–cell interaction in an assay well as antibody induced target cell lyses at different E:T ratios and antibody concentrations after a certain time of coculture. Some parameters needed for a pathway model, such as the cell’s radius, may be fixed by data in the literature or direct experimental measure. During the model development and parameter estimation process, we would want to make sure the ODE system describes the in vitro experiments accurately and the model parameters to be estimated are identifiable. For example, if effector–target cell complex formation and dissociation are modeled as a separate step, we could model the lysis rate as:
In this variation of a Hill equation [19], fc_bound_per_cell describes the amount of antibody bound to effector cell which can be modeled by the measured antibody affinities and [ET] is the effector–target cell complex. \( {K}_{\mathrm{kill}}^{\mathrm{max}},\mathrm{fc}\_\mathrm{ec}50 \), and adcc_slope are the parameters that need to be estimated from the in vitro experiment readout at different conditions. Depending on the therapeutic, clinical questions, and available data, one might need additional mechanistic detail for the pathway model. With sufficient data, we may also be able to fit nonlinear mixed-effect statistical models for the pathway model parameters [20]. When employed with an experimental model focused on cells from human subjects, the approach can start to yield estimates of salient variability that could be informative of pathway-level variability in patients when accompanied with a suitable translational strategy.
We give just one example of pathway model development to inform parameterization of a QSP model. In our experience, pathway models could be used more comprehensively through the discovery and clinical stages to support an asset to ensure better quantification of experimental readouts, aid in their interpretation, and inform translational strategies. For example, antibody binding as assessed on cells through flow cytometry and in surface plasmon resonance assays is very different, but the biophysical underpinnings for the differences can be modeled to extract a useful quantitative description that more readily translates to new physiologic scenarios of targeted receptor expression [12, 21]. To extract useful characterizations, the in vitro system often has to be quantitatively well-defined, which can be challenging in coculture designs when many cell types are involved, key quantities are not reported or readily available, or more complicated phenomena are being reported.
2.1.2 In Vitro–In Vivo Translation of Pathway Model Parameterization
Once relevant pathway models are developed and calibrated, the next step is to translate the model pathways and parameters into the QSP platform model. For example, consider if we want to add the ADCC component into the tumor compartment of an I-O QSP model for anti-CTLA-4 antibody. The I-O QSP model should represent the physiological NK cell and CD4 Treg cell interactions in the tumor microenvironment, where in vivo cell movement speeds play important roles in determining cell–cell complex formation. The cell–cell interaction parameters should be updated when translating from the in vitro experiment into the I-O QSP model, and in vivo motility data from techniques such as two-photon microscopy exist to form a rational basis for translation [22]. However, it could be reasonable to use estimated \( {K}_{\mathrm{kill}}^{\mathrm{max}},\mathrm{fc}\_\mathrm{ec}50 \), and adcc_slope in the I-O QSP model. If additional information on inhibition of ADCC processes is available, it can be quantitatively incorporated. It should be noted that other factors in the I-O QSP model are equally important and accounted for when determining in vivo ADCC, including immune cell infiltration into the tumor, antibody delivery into the tumor, cell proliferation, and cell–cell contact effects. The net in vivo ADCC employed in the platform is an integrated outcome that considers all these interactions, and is not solely determined by the in vitro parameterization. It is also worth noting that depending on variability observed in the in vitro parameters and sensitivities observed during model development, one can start to develop reasonable insights into which parameters may ultimately drive some of the biomarkers being calibrated and also formulate reasonable ranges the parameters should vary over during QSP model calibration. It is important to note that this careful translation of the in vitro characteristics is not the sole component of the strategy for clinical prediction, as the population still is ultimately calibrated to clinical variability and response data that can include biomarkers within and effects from this pathway.
Models of responses at the cellular level are additional examples of pathway models that can be integrated into larger QSP models. Such approaches also drive QSP towards a more modular design rationale. Initial advances of modeling cell types with logical network approaches appears promising as one rationale basis for modeling cellular responses in light of multiple inputs [23, 24], and additional steps can be taken to translate logical networks into quantitative ODE models with experimentally tractable and translatable concentration sensitivities [25].
2.2 Integration of Biomarker and Clinical Endpoints
Once a QSP model structure has been integrated and physiologic values or ranges are established for the parameters, the next step is to ensure the model is able to capture and explain observed clinical trial biomarker and response variability. This is important to address before the model is applied to clinical development questions. QSP, in contrast to simpler clinically relevant modeling approaches, therefore often aims to provide a multi-perturbation, multi-output analysis of a disease and one or more therapeutic interventions at a population level. Given that a QSP model usually provides an extensive quantitative framework for characterizing the mechanisms of action of therapies and their physiologic responses in a specified disease area, it can be helpful and sometimes even critical to integrate as many data-supported pharmacodynamic (PD) biomarkers as possible. As this is done, it becomes possible to constrain and calibrate the model with integrated clinical datasets. For example, given an I-O QSP model focusing on CTLA-4 and PD-1 checkpoint inhibitors in a first line melanoma patient population, ideally we would want to integrate essentially any modeled PD markers that were collected from first line melanoma patients in all available clinical trials for modeled therapies. These PD markers could be collected from patients’ blood sample or tumor biopsy. Target tissue data often have an advantage as they may more tightly link the modeled mechanisms and response, whereas data from the blood may differ more from the site of action due to differences in contributions from multiple tissues and different half-lives. Different data sources can play an important role in model development. For our considerations in I-O, the following apply:
-
Immunohistochemistry data from tissue biopsies can inform modeling the composition of tumors [26].
-
Omics data may also be used to inform modeling. In our experience, transcriptomics may correlate well with select, paired markers from immunohistochemistry, but the application context is important for implementing transcriptomics data. Immune signatures may also be useful to indicate the clinical activity of pathways modulated by multiple mediators or with substantial known posttranslational regulation, such as type I interferons and the transforming growth factor beta pathways [27, 28]. Signatures may be helpful for calibrating pharmacodynamic changes in a QSP model if implemented through net effects on the appropriate pathways and cell populations. Immune deconvolution approaches hold promise to estimate cellular compositions from historical clinical transcriptomics data [29, 30]. However, from our experience comparing between different modalities, care should be taken in the interpretation of absolute tissue cell fractions reported from deconvolution.
Expertise in a therapeutic area helps to interpret and apply data in a reasonable fashion. We often would want to integrate patient level data at different time points, such as pretreatment at day 0 vs on-treatment at day X, for multiple therapies. VPops can be used to calibrate a QSP model to clinical data for biomarkers and endpoints that are investigational, accepted as mechanistically important, or established as clinically meaningful for patient outcomes and trials. For the example in I-O, we may also want to include index lesion size and response rate data for calibration. When these data come from original electronic datasets on file, which may more often be readily available in an industry setting, they can provide a multivariate and more detailed resolution of distributions. In contrast, trial results published in journals often present only summary statistics, although they can still be useful to inform VPop calibration. In general, it is important to proactively check and ensure that these integrated biomarker or clinical endpoint data are collected from a well-defined patient population, such as first line or I-O naive melanoma. This is important as it sets the target patient population and clinical data integration criteria. Finally, relationships between the QSP model and the biomarker and clinical endpoint data are integrated from a consistent patient population through the development of virtual patients (VPs), VP cohorts, and VP populations (VPops). Ultimately VPops in conjunction with a QSP model serve to reproduce observed clinical trial outcomes by calibrating to the observed data, and then, if sufficient confidence has been established, often using a fit-for-purpose validation, the QSP model and VPop enable mechanistic extrapolation to new therapeutic regimens or interventions.
3 A Methodological Context for VPop Applications
VPop approaches have been applied to a number of questions relevant for clinical drug development in order to ensure consistency with prior clinical data. Before introducing these studies, we have to define several terms. There has been some divergence in the literature with regards to at what stage a model parameterization should be referred to as a VP [7, 31, 32]. Here, we adopt the standard that a VP is essentially any model parameterization. As we will describe in Subheading 4, if the simulated outputs for a particular model parametrization pass additional checks to verify the VP falls within reasonable clinical observations of ranges and dynamic behaviors, we may further qualify the VP as a plausible VP. A set of plausible VPs is called a VP cohort. We define prevalence weights as a vector of nonnegative weights that sum to one and are equal in length to the number of VPs in a VP cohort. Note that the prevalence weights have an analogy in importance sampling [33,34,35], where one may use sampling from one distribution to characterize another. We generally define a VPop as a set of prevalence weights and an associated VP cohort, although in some of the cases below prevalence weights are not used in the fitting process. Some of the major algorithmic and methodologic considerations and differences are briefly reviewed in order to illustrate different strategies that have been applied for VPops and motivate further discussion of methodology.
Klinke reported on a case study fitting a VPop for type 2 diabetes [36]. In this case study, the model was already well-developed, and Klinke started with a predeveloped set of VPs that were to be calibrated to real patients from the National Health and Nutrition Examination Survey III (NHANES III). Klinke used data from one intervention given in NHANES III for his model calibration, the oral glucose tolerance test. Data from different interventions, hyperinsulemic clamp studies and overnight fasting, were withheld from the calibration, and correlations between the clamp studies and less invasive fasting measures were used to check the VPop calibration. Some unique methodological considerations for Klinke’s approach were the availability of the electronic, multidimensional NHANES III dataset to guide the development of the VPop and the reduction of the dimensionality for fitting with a principal component analysis approach. To develop the VPop, Klinke derived a single variable distribution to fit by adjusting the prevalence weights for his VP cohort. The variable was derived from the radial distance of each real or virtual patient in a four-dimensional principal component space defined by the first four principal components of the original 13 taken from the NHANES III dataset.
A different approach was employed by Howell et al. that incorporated initial assumed parameter distributions and checks of resulting simulations for agreement with data [37]. The most methodological detail is given for their in vivo extrapolation for methapyrilene and to compare acetaminophen and methapyrilene. It is worth noting Howell et al. also made a clear, stepwise distinction when describing their calibration strategy for system and drug-specific parameter calibration. Distributions for each parameter were assumed a priori, VPs were sampled from the assumed distributions, and simulated VPs were compared to data. A fitness score for each VP was generated based on the likelihood of its parameter values from the comparison of simulation outputs to data and the likelihood given the assumed distributions. A genetic algorithm was applied to iteratively generate sets of VPs of improving fitness score. The final set of VPs was selected, with manual filtering to remove VPs with nearly identical parameter values. Variability in drug-specific parameters was incorporated as a second step. Howell et al. did not apply prevalence weights in their VPop calibration strategy. In contrast to many of the other QSP applications discussed here, Howell et al.’s model and VPop approach was used to investigate drug safety. In a related study [38], their group’s approach yielded additional clinically relevant insights, including using the time to reach peak serum alanine aminotransferase as a potential marker for the time to clear the toxic metabolite, N-acetyl-p-benzoquinone imine, from the liver. Their results also suggested improvements for current treatment nomograms, and that the oral N-Acetylcysteine protocol was more efficacious than the intravenous protocol for patients presenting within 24 h of acetaminophen overdose due to the treatment duration.
Later, Schmidt et al. implemented a different algorithm using hypothesis testing strategies to optimize the fits of VPops to data [39]. This approach made use of comparison methods such as t-tests, F-tests, and chi-square tests to weight differently sized clinical datasets in their comparison to VPops. These comparisons were then combined in a composite goodness-of-fit using Fisher’s method. The method was applied to calibrate alternate VPops that matched trial results nearly equally well, but had differences in the assignment of the prevalence of different VPs. The algorithm introduced an effective sample size for the VPop, similar to the effective sample sizes used for weighted sampling strategies [40], with higher effective sample size implying better spreading of the prevalence weights. The algorithm also considered VP parameter values when assigning prevalence weights by grouping parameter ranges into bins and optimizing the binned parameter probabilities. Schmidt et al. applied the method to investigate a type I interferon signature predictive of nonresponse to rituximab in rheumatoid arthritis [41], and proposed a quantitative mechanistic hypothesis for the clinical observation due to anti-inflammatory type I interferon effects that were robust across the alternate VPops. The alternate VPops emphasized different underlying pathophysiologies, exemplified by the mechanistic differences in VPops that tended to respond well to rituximab versus those that did not respond as well. Schmidt et al. also identified candidate baseline synovial biomarkers of response that tended to be selected as predictive of response to rituximab across alternate VPops, including markers of fibroblast-like synoviocyte and B cell activity. Later, Cheng et al. adapted this algorithm for implementation into the QSP Toolbox [32]. In Subheading 6, we will discuss a modification of this algorithm that includes additional types of hypothesis test comparisons, has relaxed the binned parameter prevalence groupings, and implements automated resampling and screening of plausible VPs.
Allen et al. proposed an algorithm that calibrated a VPop based on datasets where a single individual has a number of measured outcomes in a single datasource, such as NHANES III [31]. The strategy from Allen et al. was distinct from Klinke in several aspects. Notably, because the model itself was developed to simulate more quickly and readily facilitated sampling of new VPs, the algorithm was developed to enable additional sampling of the parameter space. Each sampled VP was optimized to better fall within observed physiologic ranges and yield a plausible VP. Allen et al. used estimates of the relative density in the data versus the plausible virtual patients to optimize probabilities to include their VPs in a VPop. That is, the approach from Allen et al. also employed a more direct adjustment for their metric of prevalence, inclusion probabilities, as opposed to optimization of agreement of a PCA-based marginal distribution. The algorithm from Allen et al. optimized the goodness-of-fit as evaluated by an average univariate Kolmogorov-Smirnov test-statistic over all biomarkers. When developing predictions with the virtual population, they sampled VPs according to their inclusion probability.
In a subsequent publication, Rieger et al. compared considerations of the diversity of generated VPs and goodness of fit for the algorithm proposed in Allen et al. with several new proposals [42]. Interestingly, a modified Metropolis-Hastings approach was among the strategies proposed and assessed. One key modification was that their Metropolis-Hastings approach focused on using a well-described target distribution for determining the acceptance probability, that is, distribution of model outputs postsimulation, as opposed to using target parameter distributions as in the canonical version of the algorithm.
Quadratic programming has also been used in VPop approaches. As one example, Kirouac et al. developed a virtual population in a translational application to predict responses to a novel extracellular signal-regulated kinase (ERK) inhibitor, GDC-0994, using data from epidermal growth factor receptor (EGFR), B-Raf, mitogen-activated protein kinase kinase (MEK), and ERK inhibitors [43]. Kirouac et al. implemented their match to clinical data as a constraint in the optimization, with changes in lesion size calibrated across three different targeted oncology therapy combinations. Rather than using the fit to the data as an objective function, which was successfully imposed as a constraint, their minimization objective was the sum of the square of the prevalence weights, essentially maximizing the effective sample size. For the calibration to predict the clinical response of GDC-0994, Kirouac et al. included data from other trials testing combinations of vemurafenib + cetuximab, dabrafenib + trametinib, and dabrafenib + trametinib + panitumumab. Although the clinical trial sample size was small, the approach employed by Kirouac et al. gave good prospective prediction of the observed response rate in a subsequent phase I study of GDC-0994 in colorectal cancer patients.
In addition to calibrating to pharmacodynamic markers and clinical endpoints observed for individual or multiple trials at a population level, calibration to individual responses in individual trials has also been proposed. For example, Milberg et al. fitted a model of response to I-O therapies to lesion response dynamics, and as a first step this included fits to individual patient index lesion dynamics, though not other biomarkers [44]. Once it was established that the model could reproduce the observed lesion trajectories by varying a subset of model parameters, virtual trials were simulated by sampling the subset of parameters within predefined ranges. In another study, Jafarnejad et al. used patient-specific information on tumor mutational burden and antigen affinity while sampling other varied model parameters to guide patient-specific prediction for anti-PD1 therapy responses in a neoadjuvant setting [45].
4 VP Cohorts Are a Precursor to VPop Development
It is often useful to explore the input–output behavior of a QSP model prior to the VPop calibration process. This often includes selecting a subset of the model parameters as well as determining reasonable physiologic ranges in order to generate sufficient variability for the model outputs that are to be matched to clinically observed biomarker data. Part of the parameter selection process is often manual and based on biological insight and expertise developed during model development. Important considerations include:
-
Variation of parameters in key pathways for physiology and quantitative hypothesis testing. In an I-O QSP model, this could be cytokine-mediated versus checkpoint-mediated immune suppression.
-
Characterized variation in pathway models needed to fit literature data or in-house experiments. Taking the ADCC example discussed earlier, different human NK donors might have different maximal ADCC rates. Another example is the expression level of cytokine receptors that may vary from patient to patient.
-
Ability of the model to recreate variability in observed patient characteristics. For an I-O QSP model, that could include immune cell content in the blood, the tumor, or the tumor-draining lymph node.
-
Observed variations between different literature reports.
Appropriate parameter ranges can either be derived from literature values or estimated from pathway model calibrations, as discussed in Subheading 2.1.1. Depending on the size of the QSP model, a global sensitivity analysis (GSA) of desired model outputs with respect to all model parameters, or a related multivariable sensitivity analysis strategy with a parameter subset, can be used to complement manual parameter selection. For example, GSA approaches have been used to identify and rank the most influential parameters for a particular model output given an anticipated range of possible values for the parameter [46]. One may also want to fix noninfluential parameters during the calibration. Varying the “right” set of parameters can be crucial to generate missing phenotypes (e.g., a responder to a specific therapy), which is particularly important if one wants to simultaneously calibrate a QSP model to many biomarkers and endpoints using data from multiple trials and therapies.
By imposing additional restrictions on the model outputs we can avoid behaviors that are known not to be physiologic or take into account published summary information about biomarker ranges, even if individual-level data are not available. As described previously and shown in Fig. 3, a set of plausible VPs is called a VP cohort, and it represents a natural starting point for VPop development.

Virtual patient cohort development. Alternate model parameterizations are sampled and simulated when developing the VP cohort. Multiple interventions (therapies) are simulated for each VP. Biomarker and response data are used to guide reasonable parameter bounds and set acceptance criteria on simulated outcomes. Plausible VPs must pass all acceptance criteria
4.1 VP Cohort Development
In order to illustrate the development process leading to a VP cohort we consider the ODE system of a general QSP model in the form:
Here, Yj is the vector of state variables on intervention j and Θ is a complete set of parameters. In this chapter, we adopt the convention that the term “state variable” refers to mathematical state variables with time derivatives, as defined by the form of eq. (2), and parameters are fixed, time-independent, specified quantities.
In practice, often one will want to simulate J clinical interventions which would most often be implemented as introducing dosing functions, but practically could involve intervention-specific parameter updates or initial values, which we will neglect for now. For notational simplicity, we index the state variable vector over j. Although the functions will mostly be conserved across interventions, we expect changes in considerations like functions to account for dosing that will impact the state variable values. We can also express the model with regards to the M individual state variables:
The N model parameters are denoted as:
Often, the number of parameters is by far larger than the number of state variables, that is, N ≫ M. The right-hand side of the ODE system can also be expressed as:
The right-hand side ODE system includes, for example, the reaction rates within a compartment and the transport rates between compartments.
The set of biomarker data and clinical endpoints being used for calibration may often be mapped to functions of state variables (Y ↦ g(Y)). This occurs, for example, when comparing to cell fractions while using cell numbers as state variables. For each intervention, j = 1, …, J, we can compute the value of the observed biomarkers, b = 1, …, Bj, using the results from the simulations of the ODE system:
The set of biomarker data is often smaller than the number of state variables, that is, Bj ≪ M, and reported biomarkers may also be related to combinations of state variables. The observed biomarker data are:
Each of the Bj observed biomarkers may be reported at a distinct number of Ij, b discrete time instances given by:
Note that we may have multiple observations at each time point in the data, each from different patients. Simulations of alternate model parameterizations, or VPs, will also often be of interest. Note that the number of time points for biomarker b on intervention j, Ij, b, can vary. For example, we might be able to collect many index lesion scans from a cancer patient over the duration of a therapy, but obtain only a baseline and one on-therapy measurement for a biopsy biomarker from the same patient.
To generate variability in the model predictions for biomarker and clinical endpoints we often vary a subset of model parameters:
Each varied parameter is restricted to its physiologically feasible range, that is,
Here, \( {\theta}_{i,L}^a \) and \( {\theta}_{i,U}^a \) are lower and upper bound of the parameter \( {\theta}_i^a \), respectively. We define a parameter axis as a parameter with an associated upper and lower limit that may be varied between virtual patients during model calibration. In some instances, we may wish to combine parameters into groups so they are varied together, but this case is not essential for the present purposes.
The next steps in generating a VP cohort are to simulate different parameter combinations (VPs) and then impose acceptance criteria to ensure plausibility. Often the acceptance criteria can be formulated in the simple form:
That is, we could require the simulated result for biomarker b on intervention j to fall in the same range as the observed data. Often we may want to constrain the simulations to fall within the range of observations matched for the same time point. Constraints imposed on the model outcomes may not have a simple mapping back to the parameter space. The allowed solution space can therefore be nonconvex with regards to parameters, which may pose new considerations in connection with methods such as GSA. Screening functions with boundaries that are time-varying or defined by relationships between the state variables are possible, especially when the dynamic characteristics for plausible physiologic behavior are better established [32].
There are some additional considerations for creating a VP cohort. For example, diversity in the modeled VPs is important, although we will still want to fit to clinical data. Initial iterations of VP cohort development could involve strategies such as Latin Hypercube Sampling or lower discrepancy quasi-random sequences, such as Sobol sequences [47], to more efficiently explore the parameter space. In addition, in some cases we may also incorporate known relationships between parameters based on the data, biology, or known model behaviors. For example, it could be that one model rate must be smaller than another to give realistic asymptotic behaviors or it may be established that the apoptosis rate for one cell type is smaller than another. Therefore, if we want to ensure that samples from \( {\theta}_i^a \) are always smaller than samples from \( {\theta}_j^a \):
Such constraints can be implemented as criteria to prescreen VPs, before running simulations. One could also turn the postsimulation acceptance criteria into an element of a composite objective function if one wants to write an optimization algorithm to create more plausible VPs [31, 32].
4.2 Sensitivity Analysis Techniques, Parallels and Differences with VP Cohorts
Knowledge of model sensitivities developed during pathway and larger model development as well as biological insight and literature research are important guides in developing suitable parameter axes for VP cohort development. It can often be useful to complement these strategies with a systematic approach, that is, based on the strength of the influence a parameter has on the output of interest. Sensitivity analysis (SA) can also help to determine influential model parameters and guide model calibration. Local SA methods often compute the relative change of an output with respect to a small relative deviation of a single input parameter from a nominal value. However, ranges of interest for biological parameters can span orders of magnitude, either due to intrinsic biological variability or uncertainty about the true parameter value, and the impact of changes in combinations of parameters is often of interest. As a result, GSA methods have also been developed, which can be used to quantify changes in the output variable with respect to changes of parameters over their whole range.
In the following we shall focus on three of the GSA methods used in the systems biology literature and their suitability for GSA in the context of VP cohort development in more detail [46, 48, 49]: correlation coefficients (CCs), Morris’s method based on elementary effects, and Sobol indices based on a variance decomposition of the output variable. The Pearson CC is defined as:
Here, \( \mathrm{Cov}\left({\theta}_i^a,y\right) \) denotes the covariance between the input parameter \( {\theta}_i^a \) and an output variable of interest y while \( {\sigma}_{\theta_i^a} \) and σy denote their respective standard deviations. By definition, the Pearson CC varies between −1 (total anticorrelation) and +1 (total correlation), and it measures to what extend \( {\theta}_i^a \) and y are linearly related. If one rank-transforms the data before computing the CC on obtains the Spearman CC. If one additionally discounts linear effects of the remaining parameters \( {\theta}_{j\ne i}^a \) on \( {\theta}_i^a \) and y one obtains the partial rank CC, or PRCC. The PRCC has also been shown to yield a robust global sensitivity measure for nonlinear but monotonic relationships between \( {\theta}_i^a \) and y [48]. If the relationship is nonmonotonic over the sampled parameter range positive and negative effects can average out, yielding a small PRCC and potentially giving an incorrect impression that \( {\theta}_i^a \) has negligible influence on y.
In contrast to PRCCs the applicability of the other two methods, Morris’s elementary effects and Sobol indices, can provide useful sensitivity information in the presence of nonmonotonic input–output relationships. Both methods assume that the axes parameters have been scaled to the unit cube [0, 1]Q via:
Each scaled parameter varies between 0 and 1. This framework is compatible with the bounds often placed on parameter axes. Morris defined an elementary effect of the parameter xi on an output y by the scaled difference:
Here, each of the xj in X = (x1, …, xQ) is randomly chosen from a p-level grid with points:
There is a restriction, xi ≤ 1 − Δ, so that the change in output with respect to xi can be computed with the defined parameter domain. In general, the step size, Δ, as well as the number of grid points per parameter, p, can be different for different parameters. However, the choice Δ = p/[2(p − 1)], with p being an even number, reduces the total number of required model evaluations from 2Qr to (Q + 1)r, where r is the number of independent parameter samples [50]. Efficient sampling strategies have also been proposed in revisions to the method [51]. The mean and the variance of the elementary effect with respect to parameter xi can be estimated from:
While the mean quantifies the average impact of a parameter on the output, the variance contains information about model nonlinearities and parameter interactions. Note that di can change its sign if the relationship between y and xi is nonmonotonic, so that positive and negative contributions to the mean could average out, similar as for PRCCs. However, the Morris method still allows to differentiate between parameters that have negligible impact on the output (|μi| and σi are small) and those that effect the output in a nonmonotonic manner (|μi| is small, but σi is not). Alternatively, the mean can be defined in terms of \( \left|{d}_i^{(k)}\right| \), rather than \( {d}_i^{(k)} \), which then quantifies the average total impact of a parameter on the output independent of its direction.
The elementary effects for a single parameter are defined in a similar manner as local response coefficients. However, by averaging the local sensitivities for this parameter over the whole parameter range (while sampling the other parameters) this method yields global information about the impact of a parameter on an output. Another way to obtain global information about parameter sensitivities is based on a decomposition of the output variance with respect to parameter contributions of increasing order [52]:
The first order effect of parameter xi on the output variance is:
It is computed by first taking the average of Y with respect to all parameters but xi (i.e., by keeping xi fixed). In a second step, the variance of this conditional expectation is computed by averaging over xi. Higher order terms in the expansion of V(Y) can be defined in a similar manner, for example:
Here, Vij quantifies the combined effect of xi and xj on the output variance discounting first order effects of each parameter, that is, Vij quantifies the combined effect of xi and xj due to interactions. By dividing by V(Y), one can define Sobol indices of any order. One computes the first order indices defined by:
Here, by definition several relations hold for the first order indices:
And:
To quantify interactions one can compute higher-order indices, which have an increasing computational cost. Alternatively, one can compute the total effect index associated with parameter xi, given by:
Note that the term in the numerator yields the first order effect of all parameters but xi on the output variance. Hence, V(Y) minus that term should include all terms in the expansion that involve xi, that is, it measures the first order and all higher order effects of xi on the output variance. One can show that:
In addition, parameters with small values of the total effect index can often be neglected when generating variability for the corresponding output. One might expect that variance based methods require r2Q model evaluations, but through an efficient sample design this number can be reduced to (Q + 2)r [53].
Interestingly, when applied to a model for HIV, the three GSA methods introduced here gave similar results with respect to the ranking of parameter importance [46]. This promising result suggests a simple PRCC approach that can yield insight on directionality might also be sufficient for a GSA. However, the monotonic input–output relationships can be a limitation of the PRCC method for many applications, so a combination of methods may give better understanding of the output sensitivities.
Different strategies have been proposed to assess the adequacy of sample size for the GSA methods. One method consists of ranking the parameters and checking for a high correlation of Savage scores, which weight top ranks more heavily [54], between runs of increasing sample size [48, 55]. For PRCC, checking the Symmetrized Blest Measure of Association between runs seems to be a reasonable approach [56]. For the Sobol indices, bootstrapping the simulation results has been proposed to calculate confidence intervals [32, 47], and monitoring the convergence of the sum of the sensitivity indices with increasing model evaluations has also been suggested [57].
There are several additional considerations for simulating VP cohorts that are not explicitly accounted for in the GSA methods as they are originally proposed. All of the GSA methods discussed characterize the effect of multiple parameters (inputs) on a single output for a single modeled intervention. However, when calibrating QSP models to clinical data we normally want to fit the model simultaneously to multiple outputs across multiple interventions, which requires new strategies to combine the single-output parameter sensitivities into a ranking scheme that allows one to choose a suitable set of parameter axes. For example, Campalongo et al. characterize the influence of each parameter on each output in terms of Savage scores, and use the sum of the Savage scores to identify sets of influential and least influential parameters [51].
Another consideration is the set of plausibility constraints for VPs. Although parameter ranges may be well-described by a hypercube, portions of the parameter space may result in simulations run to inform the sensitivities that violate the plausibility constraints. Consequently, the sensitivity metrics may change if we assess only the more relevant plausible regions. Note that the CCs can be computed from a VP cohort since each VP essentially represents a single parameter sample. Computing CCs from plausible VPs in a cohort has the additional advantage that all VPs in the cohort have already passed the acceptance criteria, so that the resulting input–output correlations occur in the physiologically plausible region of parameter space. Since the Sobol indices are often calculated using quasi-Monte Carlo methods, their application should be theoretically robust for sampled points removed from the analysis due to plausibility constraints. One strategy for GSA in VP cohorts could be a combination of PRCCs and Sobol indices because both measures can be directly computed from plausible VP samples. Given the necessities inherent in creating plausible VPs, VP cohorts, and VPops, there remains opportunity for innovation with approaches that facilitate assessment of the sensitivity of multiple simulated biomarkers and endpoints to multiple parameters in an efficient manner while accounting for implausible regions of the parameter space.
5 Considerations for Developing VPop Algorithms
Once the requirements for a VP cohort are established and we begin to generate a cohort of plausible VPs, we still have not yet necessarily ensured the VP simulations also closely match trends in observed data. That is, one goal early in the development of a VPop may be to capture as many distinct plausible model parameterizations as is feasible to better ensure diversity in the modeled pathways [42]. However, the distributions of simulated biomarkers thus sampled may not initially match the target distributions. VPop calibration can be described as the process of generating a VPop that matches the observed data given a QSP model structure and the target data, and may involve a prevalence weighting scheme to accelerate the convergence to the observed data.
There are several important considerations for VPop calibration:
-
It often aims to ensure agreement with data across multiple interventions simultaneously.
-
It often requires a simultaneous fitting of multiple biomarkers and responses for each intervention. In the example presented in Subheading 6, many dozens of biomarker–time point–intervention combinations are fit. One might expect this to grow as a QSP model is reused to support the development of additional therapeutic interventions.
-
It is usually subject to the same considerations for VP plausibility as described previously.
-
It usually requires fairly well-defined patient populations for fitting, for example, I-O naïve and first-line melanoma or first-line non–small cell lung cancer. Defining a population will also help to establish the guidelines for finding, assessing, and integrating clinical data.
Practically, the strategy of VPop calibration is often to collectively fit many salient data, with a goal of optimizing the predictive characteristics of the resulting fit model [16]. Although parameter estimation is often an objective in itself in pharmacometrics modeling, and pharmacometrics parameters have been used in drug labels [58], it is not necessarily a primary objective in many QSP model applications, especially after transiting from pathway models to a QSP model that can simulate the therapeutic responses. A VPop becomes a basis to use a QSP model and outcomes for biomarkers and endpoints it was trained with to predict for new interventions it was not trained with, for example interpolating between dose levels or, if the mechanisms are well-developed, extrapolating outside of dose levels it was calibrated with. In early stages of clinical development, data may be available for VPop calibration for drugs targeted to closely related mechanisms, and the VPop and model then also serve as a mechanism-informed strategy to extrapolate to predict the impact with the new intervention.
It has also been suggested to include intermediate stages of model calibrations with individual reference model parameterizations [7], although this is not always emphasized in QSP workflows [31, 32, 42]. One can define a reference VP as a plausible VP that captures some additional characteristic of interest, such as responsiveness to a therapy. Reference VPs can be mathematically convenient and have been useful in a historical context at initial project stages [59], especially when models are slow to simulate and quantitatively explore. However, with models that may initially capture many of the observed clinical behaviors well, and given advances in strategies to rapidly explore different plausible VPs, we have often found it practical, useful, and rigorous to proceed quickly to the development of VP cohorts and VPops early in a QSP workflow. There are also some cautionary points about the design, interpretation, and robustness of reference VPs. For example, if calibrating a single reference VP to the mean of the observed data, it is worth noting that it may not be feasible that a real single patient would run through the mean of all the multivariate data for the many biomarkers and endpoints from the population, given inherent heterogeneity in patients even within a well-defined clinical population. As one additional example, the relevance of a single mean calibration is challenging to interpret for bimodally distributed biomarkers or endpoints. The process of developing a VP cohort and VPop ensures that the ranges of the observed biomarkers and endpoints can each be captured, and does so simultaneously. As we will show in the example, after calibrating a VPop, one can begin to assess the variability observed in behaviors, including average behaviors, which provides an additional quantitative context for interpreting them. While developing a VPop can take substantial time and resources, the ability to simultaneously capture observed clinical population ranges, population central tendencies, and characteristics of population distributions is an important step to build confidence before making predictions to inform clinical development. It is worth noting that due to the combination of the available data and model behaviors, a retrospective assessment may also reveal some of the plausible VPs in the VPop tend toward population means across multiple biomarkers and endpoints.
As described in Subheading 3, a variety of strategies to fit QSP models have been employed before, including fitting individual patients up to calibrating population behaviors for multiple therapies across different patient sets. There are potential advantages with regards to being able to calibrate model behavior if one can use data from multiple therapies, especially when one wants to combine many mechanisms in a model and they are not generally administered to the same patient. However, in this case, one is not necessarily calibrating the model to an overlapping set of patients across all of the interventions. Care should be taken to ensure the patients being used in the combined fits are of similar background, so information from very different response classes are not being improperly combined.
One salient consideration for developing VPops is the availability of computational resources. The number of simulations needed to develop quantitatively reasonable calibrations can increase as one increases the number of biomarkers and endpoints to fit as well as the number of interventions per VP. An efficient algorithm that is still sufficiently rigorous to give good predictions is a substantial consideration. For example, if one wants to fit 20 biomarkers and responses across multiple interventions simultaneously, one might need to vary a similar number of parameters in the model. Of course, the time to simulate a model may vary substantially depending on the model processes themselves. For example, if a simulation of a VP on an intervention takes 20 s and there are 10 interventions for each VP, then a single VP takes 200 s of compute time. If one wants to simply simulate 1 × 106 VPs and had 1000 compute cores available, the computation would be done in about 55 h. However, VPop algorithms might not only involve an initial sampling but can also include iterative optimization and resampling processes.
5.1 Objective Functions for VPops
An important component to developing a VPop algorithm is the strategy for optimizing the agreement between a VPop and observed clinical data. Therefore, in addition to using objective functions in the step of developing plausible VPs, objective functions can play a role in the VPop development step and prevalence weighting strategies. A closely related consideration is how well the final fitted VPop agrees, and ideally the optimization and assessment strategies are well-aligned. It is possible for the optimization to find a solution that is optimal but does not agree very well with data, for example if two separate constraints on the optimization preclude a good global fit. In such a case, one may also need to assess the source of the problem, for example whether there are underlying issues in the data, the plausibility constraints were not set up in a reasonable fashion, or there is an issue in the model itself.
5.1.1 Objective Functions for Combining Data from Multiple Interventions and Disjoint Patient Groups
Consider a fixed set of simulation results that had already passed plausibility screening, and we want to assess the agreement of those simulation results with observed data. Say we want to assess agreement with a population, pooling together patients that have been given the different therapies that have been simulated. There are a variety of practical quantitative ways to assess the agreement, each with advantages and disadvantages.
As one example, one may use the square error between observed and simulated population values, possibly given a set of associated prevalence weights. Such an error term is often normalized by variability observed in the data to better facilitate comparisons between different types of observations.
Here, we have an objective function applied over J interventions, each with Bj, Tj, b biomarker–time point measures. Joint comparisons, for example involving using information between time points, are not shown here for simplicity. In contrast to the notation used in Eqs. (6)–(8), the individual time points are directly indicated here, but each biomarker–time point may also have distributions of patient data, so the comparison between the set of data for a given time point, dj, b, t, and corresponding weighted model-derived outputs, gj, b, t, is left generally as hj, b rather than directly showing the difference. However, a recognizable least-squares objective function will result given an appropriate selection of hj, b. Here, we have a matrix of the modeled axes coefficients for multiple plausible VPs indicated as \( {\overline{\Theta}}_{\mathrm{axes}} \), and the prevalence weights are indicated by W. E is the square error and sj, b is a normalization factor. One advantage is that such a least squares objective function formulation may be fast to solve even for a large number of biomarkers and interventions, given an appropriate selection for hj,b. Another open consideration for a least-squares approach is how to make a judgement that a fit is considered “acceptable.” One selection for the scaling factor could be:
Here, \( {\sigma}_{j,b}^2 \) is the variance for the biomarker or endpoint b on intervention j. If using this selection for a scaling factor, the minimum in a least square objective function comparing an individual simulated model parameterization with a single target value mathematically coincides with a minimum in a negative log likelihood function for fitting individual time-series data, provided model residuals are independent and normally distributed [17]. However, such a strategy for scaling does not directly take into consideration differences in underlying sample sizes if calibrating multiple datasets. An additional consideration is how to adapt the least-squares approach for fitting population characteristics including observed variability as opposed to individual time series data.
As another example, one could also use frequentist statistical methods to formulate an objective function [39]. For example, p-values are combined and a composite goodness-of-fit is assessed in meta-analysis. A related objective function can be expressed as:
Here, fj, b are functions involving a statistical comparison accounting for the simulation results, such as minus the logarithm of test p-values, and \( S\left({\overline{\Theta}}_{\mathrm{axes}},W\right) \) is a composite objective function. One advantage of such an approach is that, due to the dependence on test statistics against respective data, the normalization for sample size is fixed, and the resulting meta-analysis test statistic may provide a more direct quantitative value for assessment of how good a fit is for each constituent comparison relative to least-squares approaches. With a suitable selection of the fj, b, the p-values for each biomarker–time point also become a diagnostic to help identify any issues in the model or calibration during fitting, and the use of p-values makes it fairly straightforward to compare between them. One disadvantage is that it may be slower to optimize using hypothesis tests in an objective function, given they may necessitate the need for slower, general-purpose optimization methods.
More concretely, if one wants to use Fisher’s method to combine a series of p-values in a meta-analysis of the simulated population versus patient data, the test statistic is:
Here, pj, b, t is a p-value calculated from a statistical comparison to data and \( {S}_F\left({\overline{\Theta}}_{\mathrm{axes}},W\right) \) is a Fisher test statistic. Fisher test statistics have been utilized in meta-analysis approaches [60]. It was noted that the Fisher’s combined method may report overly low p-values when there are mechanistic or other dependencies [61], so for the present case where one wants to minimize the rejection and maximize the agreement with data, Fisher’s method is a conservative approach. Nonetheless, there exist alternative methods for meta-analysis that formally account for nonindependence between the combined tests [61,62,63,64].
Practically, one would like to find VP cohorts and weighting schemes that effectively spread the prevalence weights among many VPs. This helps to ensure a single model parameterization does not drive much of the observed response, and is a prerequisite for resampling strategies and alternate clinical trial simulations. Similar to importance sampling methodologies, one could define the effective sample size as [39, 40, 43]:
Here, N is the number of plausible VPs in the cohort and the weights sum to one.
5.1.2 Objective Functions for Combining Data from a Single Patient Population
If the structure of the data is such that we have comprehensive biomarker and endpoints from a single set of patients, different approaches to calibrating the simulations to data may be more readily used. Situations like this may arise more commonly, for example, in metabolic disease research where a single patient group may be subject to pharmacodynamic studies in addition to the investigational intervention and key indicators of efficacy such as plasma glucose and insulin levels may be subject to frequent sampling. One objective function that was previously proposed for this situation, for example when fitting baseline characteristics of cholesterol across a large database, was to take the average of Kolmogorov–Smirnov (K-S) test statistics calculated independently for each calibrated model output biomarker [31]. In this example, even though the K-S test was calculated for each calibrated biomarker separately, the population fitting step also took into consideration the local multivariate probability density around each VP relative to the observed density across all of the output biomarkers.
5.2 Types of Biomarker and Endpoint Fits
There are many types of information that might be selected for fitting to quantitatively calibrate a VPop, and their choice will depend on the sources of knowledge available for a given clinical intervention, for example whether data are available in-house or must be taken from the literature.
-
Mean and standard deviation are often available in the literature for some endpoints and biomarkers. If the underlying distributions can be verified as lognormal, it is straightforward to calculate and work with the appropriate lognormal summary information.
-
Binned distributions are sometimes natural choices for clinical biomarker and endpoint calibrations. For example, in oncology, clinical response assessments are based in a substantial part on index lesion changes [65], and hence can be grouped as complete responders, partial responders, stable disease or progressive disease.
-
Empirical distributions from source data provide another basis for VPop calibration to patient data. Although individual data points may not be given in many publications, appropriate data may be found from a database or the electronic source clinical trial data.
-
Correlations can also be used in the fitting process.
-
Multivariate distributions can also be fit.
The VPop can be calibrated to many of these characteristics in the data with a related objective function. For example, we previously described a way to calibrate means, variances, binned distributions, and empirical distributions across multiple interventions and disjoint patient groups with a least-squares objective function [66]. As we will describe soon, frequentist statistical methods can be used for these comparisons and more, with methods such as t-tests, F-tests, contingency tables, K-S tests, Fisher’s r-to-z transformation, and multidimensional K-S tests such as the Peacock test and related comparisons [67, 68]. Note that if we have multivariate data, even if not all from the same patients, it is still possible to use these relationships directly in VPop calibration. A calibration of distributions and multivariate relationships is often not possible when using literature data, but can be used with electronic data that might be available within a database or directly from a clinical trial. There are additional options, and an interesting possibility for the multivariate empirical distribution calibration is the earth-mover’s distance [69, 70], although comparisons of one or more pairs of multivariate empirical distributions in many dimensions can be computationally intensive. When combining data across many clinical interventions and trials, it often may not be practical to fit individual patient data due to issues such as the sparsity of data, missing values for many of the biomarkers, and desire to combine observations from disjoint sets of patients. Regardless, multivariate relationships observed in the population from the individual data can often still be calibrated, which we will demonstrate in Subheading 6.
Another consideration is how to best handle time series data, if available, at a population level if not performing fits to individual patient trajectories. Calibrating to distinct marginal distributions at multiple time points might yield reasonable quantitative population behaviors. However, it is worth noting that time series can also be analyzed in terms of autocorrelation characteristics, which perhaps has been more often done for population analyses with stochastic differential equation models [71, 72]. One reasonable proposal to handle this situation could be to directly calibrate, or at least check, that either time series autocorrelation or joint distributions between time point measures for the VPop are similar to the clinical data.
One additional strategy that may be helpful for calibrating QSP models in certain circumstances, especially when one has source data, is imputation. Although one has to be careful when applying data imputation techniques, be transparent about their application, and verify their application is reasonable, they have been useful in a variety of systems biology and medical applications [73,74,75]. There are some situations in QSP where imputation strategies may be useful to help guide model calibration if data are sparse. For example, when many samples from an analyte of interest are below a detection limit but they have clear correlation with other analytes in their observed data [76,77,78]. Another potential application would be to develop quantitative guidance for likely biomarker patterns when there are data for two trials testing the same therapeutic intervention with multivariable individual-level biomarker and endpoint data at multiple time points, but some of the biomarker data are more sparsely sampled in one of the trials.
5.3 Optimization Algorithms
There are two key places where objective functions can play a role in the VPop methods described here. The first is during the cohort development step, when deciding whether a VP is plausible. The second is during the VPop prevalence-weighting step. Following development of suitable objective functions, many optimization strategies have been used in VP and VPop development. For cases where an optimization problem is cast in terms of least squares, fast optimization techniques such as quadratic programming can sometimes be used [43, 66]. Otherwise, more general-purpose optimization techniques such as simulated annealing, particle swarm, or genetic algorithms have been used [31, 32]. As more clinical biomarkers and endpoints are being calibrated, general optimization methods that can scale well across clusters such as parallel asynchronous particle swarm methods may find increasing utilization [79, 80].
5.4 Strategies for Assessing Uncertainty with VPops
As with other modeling approaches, uncertainty is a consideration for VPop approaches. Uncertainty can enter into model development at the steps of defining the model structure, parameterization, and prediction [18]. Although conceptually VP cohorts and VPops can be applied in a controlled manner to address each of these considerations distinctly, we believe the consideration of prediction uncertainty merits the most attention in many practical applications [16, 18], including clinical development. From the model development workflow, considerations for how to structure and parameterize the model are often addressed at the model and cohort development stage, before the VPop is developed. The questions a QSP model is being applied to address during clinical development often depend more on predicted clinical outcomes and biomarkers, as calculated from state variables, rather than structural and parametric insights, which are often explored more during pathway model development.
Prediction uncertainty is a distinct quantitative consideration from population variability. While variability can give rise to uncertainty in a prediction, for example different virtual patients may be recruited onto a virtual trial just as alternate patients can be recruited onto a real trial, there can be uncertainty in a prediction following the calibration not explicitly tied to this source of variability. The mathematical modeling and systems biology communities have proposed a variety of strategies for assessing prediction uncertainty. They have included both propagation methods from the parameters given the model, which often are dependent on convergence in the context of an assumed statistical framework with assumptions for prior parameter distributions, as well as more direct assessments for the predictions of interest using frequentist approaches that, nonetheless, verify different calibrations agree well with data [18, 81,82,83]. One important consideration is that QSP is generally implemented in a fit-for-purpose fashion, and QSP models are usually employed in a specific use context to help with specific decisions. This suggests a targeted approach to assessing uncertainty associated with certain predictions may often be suitable [18]. For example, in the context of QSP modeling, it is possible alternate VPops could fit the data nearly equally well. One general strategy of assessing prediction uncertainty in the context of QSP models is then to characterize alternate VPops that fit the data [39], and assess whether the alternate fits can impact the predicted outcome of interest. If so, additional characterization of the VPops and identification of necessary calibration data to reduce uncertainty in the prediction may be impactful. This analysis of prediction uncertainty is a distinct consideration from variability in trial outcomes due to differences in recruited patients, which can also be addressed with VPop approaches to clinical trial simulation, as we will describe in the example in Subheading 6.
5.5 Overfitting
When developing a QSP model and a VP cohort, selecting parameters to fix and allow to vary during calibration is often guided by knowledge of biology, available data on anticipated parameter-level variability in the population, questions to address, and known sensitivities for modeled biomarkers and endpoints that are supported by data. However, it remains possible one may end up with a VPop that is sensitive to spurious features in a clinical dataset. In the fields of statistical modeling and machine learning, overfitting is described in the context of a bias versus variance trade-off [84]. That is, with few parameters available for fitting and low variance, it could be challenging to calibrate the model across all of the clinical data well, resulting in a biased calibration and prediction. An amount of freedom in model outputs is needed through the parameters that are allowed to vary during the calibration to fit the clinical data across many model outputs. However, if we allow for too much freedom in the fitting, we will still be able to fit the training data very well, but the quality of the fit and importance of the mechanism will be hampered by spurious features and noise in the clinical data. The ability of the model to predict new data may be compromised, corresponding to a high variance situation [84]. In some modeling applications, metrics such as Akaike information criterion and Bayesian information criterion have been used to help balance the model size and number of free parameters versus the fit [20, 84], and other strategies such as Vapnik’s structural risk minimization approach and nested model comparisons are possible [84]. A similar metric has generally not been as extensively applied for VPop approaches as demonstrated by the examples discussed in Subheading 3. Reasons may include the mechanistic rationale and assessment of sensitivities in selection of parameter axes, the time needed to iteratively resample the model to calibrate given different sets of parameter axes, the many model outputs being calibrated, the different scientific applications of the models including capturing varied underlying mechanistic disease pathophysiology, and the different types of objective functions being employed.
One way to avoid overfitting is to employ model averaging, or ensemble, approaches [85]. By balancing the bias versus variance trade-off, ensemble approaches have been shown to improve prediction accuracy in many fields not just for empirical [84], but also for mechanistic models (for example, [86, 87]). We have implemented an ensemble algorithm in the context of VPop calibration, where we bootstrap the data and select random subsets of the VPs in each of many individual calibrations [66]. This is analogous to the random subspace method, which is also called feature bagging, a commonly used ensemble algorithm [84]. Feature bagging reduces variance in fitting by iteratively fitting to bootstrapped data with random subsets of the model’s features, and then averaging the trained predictors into an ensemble model [84, 88]. Using this algorithm, we have observed improvements for prediction of withheld data using cross-validation tests in a scenario where a least-squares objective function was employed [66].
5.6 Additional Considerations for VPop Algorithms
Additional considerations for developing VPops merit special discussion. The first is the calibration of subpopulations. We can define a subpopulation as a subset of VPs with a common characteristic, such as nonresponse to a therapy, and the associated prevalence weights for this subset of VPs. Subpopulations are an important consideration, as they enable one to use additional clinical data for calibration and build confidence in VPop predictive capability. For example, in the case of an I-O therapy, a subpopulation could be defined as the anti-PD-1 progressed population. One might want to calibrate this portion of the VPop response to a second therapy postprogression specifically with clinical data from an anti-PD-1 progressed population, or one might want to develop predictions for how these VPs respond to a second line therapy.
When calibrating a VPop with subpopulations, the full VPop is still calibrated with a full set of clinical data for the clinical population of interest, but the subpopulation is simultaneously optimized such that it is distinctly calibrated to the subpopulation data. One can modify the objective functions described previously, for example:
Here, the prevalence weights are adjusted based on subpopulation assignments. That is, for a specific intervention and biomarker–time point combination, the weights may be rescaled based on whether a VP meets the criteria to be included in the calibration set. An indicator vector assigning a VP as belonging to a subpopulation for each intervention and biomarker–time point combination is useful to relate the subpopulation weights, Wj, b, t, to the weights described previously. That is, we will define sj, b, t as a binary column vector of the same length of the number of VPs with elements equal to one if a virtual patient falls in the subpopulation and zero otherwise. Given the indicator vector, the weights accounting for subpopulation assignment are determined as:
Here, we assume W is a column vector of prevalence weights of length equivalent to the number of VPs. The Hadamard product is indicated in the numerator and the dot product in the denominator. During VPop calibration, there is one set of weights to optimize, since the subpopulation assignments, sj, b, t, are determined based on each VP’s simulated phenotype.
Dropouts in clinical data are another consideration when developing VPops. One special case arises when the dropout criteria are directly related to the efficacy measures. This often happens in oncology trials, where an increase in lesion volumes on a scanner may result in a patient being removed from the trial. The length of time a patient stays in the calibration dataset, as assessed relative to the initiation of the trial, becomes related to the observed efficacy of the therapy. It is indeed possible to adjust VPop calibration algorithms to account for these considerations, and we have demonstrated adjustments to calculation of clinical responses and biomarkers at the VPop level while allowing for index lesion progression to result in exit from a trial [89]. In this approach, when a VP exhibits progressive disease, they are excluded from subsequent calibration time points and later observations in the data are calibrated using the VPs that do not yet exhibit progressive disease. The strategy essentially involves the use of subpopulations to include only VPs that have not exited from the virtual trial. This is one additional advantage to working with source data as opposed to using summary data from the literature during VPop calibration. For example, in oncology, waterfall plots of lesion responses are often reported, which show only the maximal lesion decrease. While one may need to use literature information to inform some aspects of calibration of a systems model, there is much additional dynamic response information that could be useful for model calibration that is not directly represented on many literature readouts such as waterfall plots.
Finally, it is useful to make an additional distinction between calibration and prediction steps. VPop calibration is performed with the clinical data to give a set of model parameterizations and prevalence weights. When simulating a new intervention, these can be applied directly to give a prediction. However, we expect some variability in trial outcomes based on which patients are recruited, and VPop approaches have explored this [31, 89]. One strategy to characterize variability in trial outcomes is to resample from the VPop according to the prevalence weight, thereby allowing the simulation of alternate clinical trials with differing patient recruitment.
5.7 Validation
Model validation is a subsequent step to model calibration, and selection of appropriate calibration and validation data is an important component of QSP modeling. Although a practitioner is free to decide how to apply and validate their QSP model, it is important to increase awareness and agreement on strategies for QSP model validation to improve buy-in and acceptance of quantitative model insights from new stakeholders. QSP model validation has been a topic of perspectives and guidelines from a variety of practitioners with different applications in mind for differing stages and cases of drug development [90,91,92,93]. Regulatory agencies are also interested in QSP modeling approaches, and the U.S. Food and Drug Administration cohosted a scientific exchange on the topic, which was held virtually on July 1, 2020 [94]. In the eight case studies presented by industry stakeholders at the meeting, one striking outcome was the general strategy of checking model behaviors and predictions with data withheld from the model calibration to help build confidence in the model, especially in the context of relevancy for application to a new, specific prediction.
We focus this discussion of validation on application of QSP models in a clinical development setting. In this setting, QSP is often applied in a context-specific manner to support specific clinical decisions. Similar to early stage clinical trials, predictions for some modeled biomarkers or endpoints may be considered key decision outputs, whereas others may not be considered as critical for a decision even if informed with additional molecular and cellular clinical pharmacodynamic data. The additional pathways are included so that the drug’s mechanism of action is modeled in the appropriate physiological context (e.g., feedback loops). They can also facilitate a rapid pivot to apply the model to address questions of stratification markers, alternate combination regimens, and contribution of components.
Establishing confidence in the ability of a model to extrapolate and predict in support for the decisions to be made appears to be one path forward for formal model calibration and subsequent validation while enabling one to use a QSP model as a strategy to more flexibly incorporate a breadth of emerging, relevant clinical response and biomarker data. Although additional considerations of model documentation, design decisions, submodule and pathway calibration, and model behaviors are very important, establishing more direct confidence in the predictive capabilities of the model can be critical to interface with nonmodelers and generate confidence to use the model in a specific application. Important considerations that can help to guide a validation strategy for a QSP model for a specific clinical development decision include:
-
Defining the insight required from the model and predictions to be made will influence a QSP project early, often before additional model development and calibration is begun. This step can also serve to focus the model, control timelines, and avoid overpromising the potential QSP analysis deliverables.
-
Reviewing related clinical information available is important to conduct as soon as the clinical questions are clear, and could also have an impact on priorities for model development. For example, if predictions for a drug for which no clinical data are available are intended to be made, one can begin to look to whether clinical data are available for other approved drugs that impact a similar set of targets or closely related pathways. The QSP model can then effectively become a stepping-stone to address new clinical questions based on available data. This can also guide the prioritization and scope of additional pathways into the model, as one might place more caution on the addition of pathways for which little clinical biomarker data are available. One also must be judicious in the selection of data, ensuring it is from a salient patient population and assessing the quality and any apparent biases or irregularities. Although electronic source data often has a number of advantages, sometimes salient clinical data from targeting related mechanisms may only be available in the literature.
-
Delineating calibration and validation information is an important consideration. One would like to use enough calibration information to constrain the intended predictions of the model, but then one would also like to withhold information to be used as a check of the model’s predictive power. In early clinical development, one might have data from two therapies that target pathways related to the intended target. In such a scenario, one might use the richer dataset with the therapy with more biomarker data to calibrate the model, and then the dataset for the related therapy with less comprehensive biomarker coverage as a validation test.
-
Quantitative criteria for model validation have generally varied in different contexts, likely due in part to differences in workflows and computational strategies. For example, one might need to predict responses more accurately for a phase 3 design than an early go–no go decision, or a decision related to which dose expansion arms to explore. For phase 2 trial design decisions, we have generally sought to ensure the efficacy for related interventions withheld from the calibration fall within the 95% confidence interval for alternate simulated trials of a comparable size.
6 A VPop Example
In this section, we will discuss a VPop application example in immuno-oncology (I-O) using the QSP Toolbox [32]. The example features a number of common challenges for QSP model calibration. It includes incorporating nonclinical information from multiple pathways and clinical information from multiple targeted mechanisms, simultaneous calibration of many model outputs from different interventions, and VP plausibility considerations.
6.1 QSP Toolbox
Software tools are available that can implement many of the considerations for QSP workflows discussed here [32, 96–97]. We have developed a freely available solution in MATLAB® with a permissive license and fully visible code that is frequently updated online (see: https://github.com/BMSQSP/QSPToolbox). Although the implementation we have developed requires familiarity with MATLAB’s scripting language, scripted toolbox strategies have also been successfully adopted in the academic systems modeling community [98,99,100]. In general, although the tool does not have a graphical user interface that would make it more friendly for those without scripting experience, the approach offers a straightforward path to repeatable analyses, generally simplifies the development of new algorithms, and offers flexibility in adapting existing algorithms.
QSP Toolbox includes a set of functions, structure array conventions, and class definitions that computationally implement critical elements of QSP workflows including data integration, model calibration, and assessing the fit to clinically observed variability [32]. It reads QSP models developed in SimBiology®, a VP definition table file, an intervention definition table file, and various available experimental dataset files. By providing the functionality necessary for a more comprehensive QSP workflow, the toolbox helps to run sensitivity analyses, develop a cohort of VPs, implement plausibility considerations, develop VPops calibrated to clinical data, and also provides tools for many useful visualizations and diagnostics.
6.2 I-O QSP Platform
The I-O QSP platform used in the case study presented here is a combination therapeutic response model that captures key CTLA-4 and PD-1 pathways, immune cells including CD8 T cells and regulatory T cells, checkpoints and their ligands, and important clinically measured dynamics including lesion responses and changes in immune content. The platform was developed in SimBiology and, at the time of this writing, included 131 ODEs, 370 reactions, 402 rules and 350 references. Many pathway-model fits were used to help determine the model structure and parameters. Some key components in the model include cell–cell contact, confinement and two-dimensional molecular interactions, immune cell life cycle, recruitment, cytokine-mediated feedback loops, and cancer killing. The model is shown in Fig. 4. The model has been developed in a stepwise fashion, adding new pathways to support clinical development as additional pathways are of interest. In addition to pathway-level fits to help constrain model behaviors, as new pathways are added new clinical data are identified to calibrate the model.

Diagram of the I-O QSP platform. The platform includes cell types and pathways important for immuno-oncology therapies
6.3 The Automated, Algorithmic, Iterative VPop Development Workflow
We have developed an algorithmic, iterative VPop development workflow that enables calibration to a breadth of clinical data sources with a variety of comparison types from multiple interventions. That is, the approach does not require that the data come from one single set of real patients with a single, analytically well-defined multivariate distribution. The algorithm calibrates the VPop to population data of biomarker, pharmacodynamic, and index lesion Response Evaluation Criteria in Solid Tumors (RECIST) scoring, including off-study considerations with progressive disease, for immuno-oncology therapies.
As shown in Fig. 5, the workflow and code has been refined to run on a cluster, not just a single server. VPop development started with a computationally demanding initial sampling, here involving over 1 × 107 simulations, screening for plausible VPs, and then many iterations of new simulations with screening and prevalence-weighting to generate new VPops with increasing numbers of VPs with well-spread prevalence weights that quantitatively matched clinical observations. We have found that the computational demands of iterative simulation and optimization make it much more practical to run on a cluster as the number of interventions and calibrated endpoints increases. Plausibility constraints were developed from multiple observed clinical biomarker and response ranges, implemented as objective function 1 in Fig. 5 to screen for plausible VPs, and an objective function based on multiple statistical comparisons with clinical data was applied as objective function 2 to optimize prevalence weights. We have incorporated the following features to efficiently develop the VPop:
-
The large initial sample and subsequent screening step results in a cohort of plausible VPs that spans many, but not all of the biomarker and response ranges. The algorithm automatically detects and directs resampling in the simulated VP cohort as it iteratively expands. This is done by checking the ranges in the data against the ranges in the cohort and guiding additional sampling to ensure ranges are sufficiently covered.
-
Simultaneous fitting of multiple biomarkers and endpoints is performed with different summary information and more detailed information on distributions. This includes tests for calibration of the mean, standard deviation, marginal distributions, binned distributions, correlations, and joint distributions. Subpopulations can also be defined and calibrated, but were not used in this example except to capture the dropout effects described previously.
-
Averaged ensemble solutions from a least-squares version of the prevalence weight calibration problem are included to initialize the prevalence weight optimization step that then uses multiple statistical comparisons [66].
-
The algorithm utilizes automated clustering across the parameter axes and fitted model outputs to reduce the number of VPs as needed with a correlation-based distance metric [101, 102], which maintains diversity in the plausible cohort and expedites the prevalence weighting steps.

Automated, algorithmic, iterative VPop development. The diagram also shows the names of functions in QSP Toolbox used to perform each step. Setting up a worksheet data structure and running simulations has been described previously. The plausible VP screening steps were automated here (screenWorksheetVPs.m). After screening, a clustering method was used maintain diversity in the plausible VPs while expediting fitting steps (pickClusterVPs.m). Then, the cohort was iteratively expanded (expandVPopEffN.m serves as a wrapper to many functions to carry out the steps, as shown in the diagram). Beginning with a low effective sample size requirement, the algorithm optimizes the prevalence weights directly. Then, it identifies highly weighted VPs and VPs near missing ranges in the data, and directs resampling around these points, screening for plausible VPs, and selection for inclusion in the plausible VP cohort. It then increments the minimum required effective sample size and restarts the prevalence weight optimization. If the algorithm ever has issues finding a good fit after increasing the effective sample size, it will reduce the number of VPs through clustering and the effective sample size requirement, and restart the iterative expansion from a lower effective sample size requirement. The algorithm is generally considered complete when a clinically meaningful effective sample size is achieved with a sufficient composite goodness-of-fit
6.4 VPop Calibration
By applying the automated VPop development workflow to a custom I-O QSP platform, we developed a VPop that captures population variability of biomarkers and lesion size response at various time points simultaneously for anti-PD1 and multiple anti-CTLA-4 therapies. The calibrated VPop captures clinical distributions (1D and 2D), biomarker statistics, and clinical overall response rates assessed from the index lesions simultaneously across multiple interventions at multiple time points (Figs. 6, 7 and 8). The composite goodness-of-fit p-value is 0.92 with an effective sample size of 140. Note higher p-values are a reflection of a better fit and less disagreement with the clinical data. The high effective sample size both ensures the prevalence weights are spread and imposes stronger agreement for the comparisons between data and the VPop. We include comparisons for:
-
Mean
-
Thirty-four comparisons to clinical data are made.
-
The method uses a t-test of prevalence-weighted means to the clinical means.
-
The t-test assumes normality but is robust.
-
-
Variance
-
Thirty-four comparisons to clinical data are made.
-
The method uses an f-test of prevalence-weighted variances to the clinical variances.
-
The F-test assumes normality and is not robust. Simulated population data can also be log-transformed for comparison if specified.
-
-
Distribution
-
Thirty-three comparisons to clinical data are made.
-
The method uses a K-S test for comparing prevalence-weighted distributions to the clinical distributions.
-
The K-S test is a direct comparison of empirical distributions, so specific distribution assumptions such as normality are not required.
-
-
Binned distribution
-
Twenty-five comparisons to clinical data are made, including the best overall response and programmed death-ligand 1 (PD-L1) expression.
-
The method uses the Fisher exact test for small sample sizes and a faster-to-compute chi-square approximation otherwise.
-
-
Two-dimensional distribution
-
Nineteen comparisons to clinical data are made.
-
A variation on Peacock test [67], similar to Fasano and Franceschini’s method [68], for prevalence-weighted 2D distributions versus observed is used.
-
The method uses an empirical distribution comparison and can account for non-Gaussian multivariate relationships in response data.
-
-
Correlation
-
Nineteen comparisons to clinical data are made.
-
A modification of Fisher’s r-to-z transformation to compare two correlation coefficients is used.
-

The VPop simultaneously captures clinical 1D distributions. Each plot represents one clinical biomarker or endpoint at one time on one therapy. The black curve represents the cumulative distribution function (CDF) extracted from internal clinical data, and the red curve represents the cumulative distribution function (CDF) of the calibrated VPop. The VPop matches the marginal distributions observed in the data well, and no assumptions for the distribution need be made for these endpoints given the empirical comparison strategy

The VPop simultaneously captures 2D (joint) clinical distributions. Each plot represents the joint distribution between clinical biomarkers or endpoints. The red dots show the data extracted from internal clinical trials, and blue circles show the VPs, where larger circles indicate greater prevalence weight. The plots demonstrate good agreement between the VPop and the clinical data, and no assumptions of the joint distribution need be made given the empirical comparison strategy

The VPop captures best index lesion response at multiple time points and for multiple interventions. The white bars show the clinical data and the black bars show the VPop response
The use of p-values for the comparisons to clinical data in the algorithm has several useful implications. They provide an objective and nonarbitrary tradeoff for sample sizes when integrating data from multiple sources, they are flexible to allow different tests to incorporate multiple data (whether they are simply summary information from the literature or electronic data from in-house clinical trials), and the individual p-values serve as useful diagnostics during the fitting process. However, when assessing if a fit is good at a given effective sample size iteration and for the final fit, given the number of comparisons being made, a composite metric is often, overall, more valuable than the individual p-values when assessing the acceptability.
Another important aspect of the calibration process is the iterative nature. Assumptions for prior parameter distributions play a more limited role during the iterative fitting process, but here it is required that many simulated model distributions ultimately match observed data. Indeed, one could also use estimated population pharmacokinetic parameter distributions as target distributions to match as well. The approach balances generating diversity in the cohort with the large initial sample, filling of clinical ranges, and use of clustering with ensuring close agreement with observed biomarker and response distributions. The approach also does not require that we develop a single analytical description of the many empirical distributions and data being matched a priori, and is flexible to allow calibration using disjoint source data from patients undergoing different therapies that are not or could not be combined into a single trial. This helps to ensure the model behaves reasonably and can leverage available clinical information from targeting related mechanisms. In the initial sample, the distributions are often not well-filled. However, through the many rounds of resampling and simulation, the prevalence-weighted distributions converged to the observed distributions, and the spread of the prevalence weights increased. In this case, there are also relatively few “drug-specific” parameters aside from PK and rates for target binding and dissociation. For example, the effect of PD-1 ligation to inhibitory ligands on downstream cellular processes is an intrinsic property of the system and implicitly impacts the course of the disease whether the patient is untreated or given an anti-CTLA-4 therapy. The simultaneous nature of the VPop calibration with clinical data available from multiple therapies makes this approach particularly tractable. Although not explored in the example presented, it is also possible to fix the minimum required effective sample size once it is acceptable and continue to run additional sampling, prevalence weighting, as well as clustering and VP cohort reduction steps with a desirable minimal VP cohort size.
6.5 Model Validation
Validation will often be context-specific, depending on the intended application of the model. For this test case, in order to demonstrate one strategy for validation, we decided to withhold and test two pieces of data. We tested model predictions for the response to combination therapy and the response to anti-CTLA-4 therapy post–PD1 progression. Of course, we anticipate variability in clinical outcomes simply due to differences in the patients recruited onto the trial and finite trial sample sizes. We therefore validated the model and calibration by resampling VPs, with replacement, according to prevalence in order to simulate patient recruitment differences in multiple trials. The observed data generally fell within the modeled 95% confidence intervals for index lesion response at multiple time points across multiple therapies (Figs. 9 and 10). In-house clinical trial data were used to calculate the index lesion RECIST classification for the combination therapy, and literature reports were used to calculate the anti-CTLA-4 therapy response post–anti-PD1 progression [103,104,105,106,107].

Prediction of combination therapy response. The boxplots were generated from the VPop, where we simulated 200 virtual trials and each virtual trial includes 200 virtual patients. In the boxplots, the red line indicates the median, the box indicates the interquartile range, and the whiskers indicate the 95th percentile range from alternate simulated trials. The purple circles are the derived rates from source electronic clinical trial data based on index lesion RECIST criteria

Prediction of anti-CTLA-4 response rate after anti-PD1 progression. The boxplot is generated from a VPop where we simulated 100 virtual trials and each virtual trial includes 100 virtual patients. The purple circle is the estimated response rate from literature. The red line indicates the median, the box indicates the interquartile range, and the whiskers indicate the 95th percentile range from alternate simulated trials
7 Conclusions
QSP is a rapidly developing field that integrates elements of mechanistic modeling, dynamic systems analysis, lateral integration of multiple biological pathways, and clinical information on pathophysiology and pharmacology to help guide drug development and clinical application. VPops play a critical role bridging QSP models with available clinical biomarker and efficacy data. They enable the development of new quantitative hypotheses and predictions, including the development of stratification markers for responders, dose selection and efficacy prediction for new therapies, prediction and analysis of the contribution of components, and an assessment of combination regimens.
The science and algorithms for developing VPops are rapidly advancing. Although QSP may be able to learn from systems biology, pharmacometrics, and engineering disciplines, there are unique considerations for VPop development in QSP that may also guide which approach is best in a given context of the model and available data. In some instances, data from a single trial with a set of patients with comprehensive marker and response data collected may be of interest, and an algorithm to handle this case will be sufficient. However, often a QSP modeler may have data from multiple therapies given to different sets of patients with different data collected, and using this additional clinical data can be helpful for model development and calibration. Other considerations for QSP model calibration include VP plausibility, diversity in the VPs, and ensuring many model outputs are in agreement with clinical observations. Efforts to compare VPop strategies are warranted, but care should be taken not to stifle innovation, and it is very likely different algorithms will be optimal in different situations. Given the successful quantitative insights gained from VPop approaches, we anticipate their increasing adoption in clinical development.
References
Sorger P, Allerheiligen SB, Abernethy DR et al (2011) Quantitative and systems pharmacology in the post-genomic era: new approaches to discovering drugs and understanding therapeutic. In: An NIH White Paper by the QSP Workshop Group. p 48
Musante C, Abernethy D, Allerheiligen S et al (2016) GPS for QSP: a summary of the ACoP6 symposium on quantitative systems pharmacology and a stage for near-term efforts in the field: GPS for QSP. CPT Pharmacometrics Syst Pharmacol 5:449–451. https://doi.org/10.1002/psp4.12109
Leil TA, Bertz R (2014) Quantitative systems pharmacology can reduce attrition and improve productivity in pharmaceutical research and development. Front Pharmacol 5:247. https://doi.org/10.3389/fphar.2014.00247
Musante C, Ramanujan S, Schmidt B et al (2017) Quantitative systems pharmacology: a case for disease models. Clin Pharmacol Ther 101:24–27. https://doi.org/10.1002/cpt.528
Ramanujan S, Gadkar K, Kadambi A (2016) Quantitative systems pharmacology: applications and adoption in drug development. In: Mager DE, Kimko HHC (eds) Systems pharmacology and pharmacodynamics. Springer International Publishing, Cham, pp 27–52
Bai JPF, Earp JC, Pillai VC (2019) Translational quantitative systems pharmacology in drug development: from current landscape to good practices. AAPS J 21:72. https://doi.org/10.1208/s12248-019-0339-5
Gadkar K, Kirouac D, Mager D et al (2016) A six-stage workflow for robust application of systems pharmacology. CPT Pharmacometrics Syst Pharmacol 5:235–249. https://doi.org/10.1002/psp4.12071
Rullmann JAC, Struemper H, Defranoux NA et al (2005) Systems biology for battling rheumatoid arthritis: application of the Entelos PhysioLab platform. IEE Proc Syst Biol 152:256. https://doi.org/10.1049/ip-syb:20050053
Meeuwisse CM, van der Linden MP, Rullmann TA et al (2011) Identification of CXCL13 as a marker for rheumatoid arthritis outcome using an in silico model of the rheumatic joint. Arthritis Rheum 63:1265–1273. https://doi.org/10.1002/art.30273
Somersalo E, Cheng Y, Calvetti D (2012) The metabolism of neurons and astrocytes through mathematical models. Ann Biomed Eng 40(11):2328–2344. https://doi.org/10.1007/s10439-012-0643-z
Chen DS, Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39:1–10. https://doi.org/10.1016/j.immuni.2013.07.012
Schmidt BJ, Bee C, Han M et al (2019) Antibodies to modulate surface receptor systems are often bivalent and must compete in a two-dimensional cell contact region. CPT Pharmacometrics Syst Pharmacol 8:873–877. https://doi.org/10.1002/psp4.12468
Schmidt MM, Wittrup KD (2009) A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther 8:2861–2871. https://doi.org/10.1158/1535-7163.MCT-09-0195
Thurber GM, Dane Wittrup K (2012) A mechanistic compartmental model for total antibody uptake in tumors. J Theor Biol 314:57–68. https://doi.org/10.1016/j.jtbi.2012.08.034
Schmidt B, Pan C, Vezina H et al (2016) Nonclinical pharmacology and mechanistic modeling of antibody–drug conjugates in support of human clinical trials. In: Antibody-drug conjugates: fundamentals, drug development, and clinical outcomes to target cancer. Wiley, Hoboken, NJ, pp 207–243
Gutenkunst RN, Waterfall JJ, Casey FP et al (2007) Universally sloppy parameter sensitivities in systems biology models. PLoS Comput Biol 3:1871–1878. https://doi.org/10.1371/journal.pcbi.0030189
Mannakee BK, Ragsdale AP, Transtrum MK et al (2016) Sloppiness and the geometry of parameter space. In: Geris L, Gomez-Cabrero D (eds) Uncertainty in biology. Springer International Publishing, Berlin, pp 271–299
Cedersund G (2016) Prediction uncertainty estimation despite unidentifiability: an overview of recent developments. In: Geris L, Gomez-Cabrero D (eds) Uncertainty in biology. Springer International Publishing, Berlin, pp 449–466
Reeve R, Turner JR (2013) Pharmacodynamic models: parameterizing the hill equation, Michaelis-Menten, the logistic curve, and relationships among these models. J Biopharm Stat 23:648–661. https://doi.org/10.1080/10543406.2012.756496
Pinheiro J, Bates DM (2000) Theory and computational methods for nonlinear mixed-effects models. In: Mixed-effects models in S and S-PLUS. Springer, New York, pp 305–336
Harms BD, Kearns JD, Iadevaia S et al (2014) Understanding the role of cross-arm binding efficiency in the activity of monoclonal and multispecific therapeutic antibodies. Methods 65:95–104
Deguine J, Breart B, Lemaître F et al (2010) Intravital imaging reveals distinct dynamics for natural killer and CD8+ T cells during tumor regression. Immunity 33:632–644. https://doi.org/10.1016/j.immuni.2010.09.016
Rex J, Albrecht U, Ehlting C et al (2016) Model-based characterization of inflammatory gene expression patterns of activated macrophages. PLoS Comput Biol 12:e1005018. https://doi.org/10.1371/journal.pcbi.1005018
Palma A, Jarrah AS, Tieri P et al (2018) Gene regulatory network modeling of macrophage differentiation corroborates the continuum hypothesis of polarization states. Front Physiol 9:1659. https://doi.org/10.3389/fphys.2018.01659
Kirouac DC, Du JY, Lahdenranta J et al (2013) Computational modeling of ERBB2-amplified breast cancer identifies combined ErbB2/3 blockade as superior to the combination of MEK and AKT inhibitors. Sci Signal 6:ra68. https://doi.org/10.1126/scisignal.2004008
Erdag G, Schaefer JT, Smolkin ME et al (2012) Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res 72:1070–1080. https://doi.org/10.1158/0008-5472.CAN-11-3218
Lavoie TB, Kalie E, Crisafulli-Cabatu S et al (2011) Binding and activity of all human alpha interferon subtypes. Cytokine 56:282–289. https://doi.org/10.1016/j.cyto.2011.07.019
Rifkin DB (2005) Latent transforming growth factor-β (TGF-β) binding proteins: orchestrators of TGF-β availability. J Biol Chem 280:7409–7412. https://doi.org/10.1074/jbc.R400029200
Schelker M, Feau S, Du J et al (2017) Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat Commun 8:2032. https://doi.org/10.1038/s41467-017-02289-3
Finotello F, Trajanoski Z (2018) Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol Immunother 67:1031–1040. https://doi.org/10.1007/s00262-018-2150-z
Allen R, Rieger T, Musante C (2016) Efficient generation and selection of virtual populations in quantitative systems pharmacology models: generation and selection of virtual populations. CPT Pharmacometrics Syst Pharmacol 5:140–146. https://doi.org/10.1002/psp4.12063
Cheng Y, Thalhauser CJ, Smithline S et al (2017) QSP toolbox: computational implementation of integrated workflow components for deploying multi-scale mechanistic models. AAPS J 19:1002–1016. https://doi.org/10.1208/s12248-017-0100-x
Tokdar ST, Kass RE (2010) Importance sampling: a review. WIREs Comp Stat 2:54–60. https://doi.org/10.1002/wics.56
Sisson SA, Fan Y, Tanaka MM (2007) Sequential Monte Carlo without likelihoods. Proc Natl Acad Sci U S A 104:1760–1765. https://doi.org/10.1073/pnas.0607208104
Catanach TA, Beck JL (2018) Bayesian updating and uncertainty quantification using sequential tempered MCMC with the rank-one modified Metropolis algorithm. arXiv:180408738 [stat]
Klinke DJ (2008) Integrating epidemiological data into a mechanistic model of type 2 diabetes: validating the prevalence of virtual patients. Ann Biomed Eng 36:321–334. https://doi.org/10.1007/s10439-007-9410-y
Howell BA, Yang Y, Kumar R et al (2012) In vitro to in vivo extrapolation and species response comparisons for drug-induced liver injury (DILI) using DILIsym™: a mechanistic, mathematical model of DILI. J Pharmacokinet Pharmacodyn 39:527–541. https://doi.org/10.1007/s10928-012-9266-0
Woodhead JL, Howell BA, Yang Y et al (2012) An analysis of N -acetylcysteine treatment for acetaminophen overdose using a systems model of drug-induced liver injury. J Pharmacol Exp Ther 342:529–540. https://doi.org/10.1124/jpet.112.192930
Schmidt BJ, Casey FP, Paterson T et al (2013) Alternate virtual populations elucidate the type I interferon signature predictive of the response to rituximab in rheumatoid arthritis. BMC Bioinformatics 14:221. https://doi.org/10.1186/1471-2105-14-221
Robert C, Casella G (2010) Introducing Monte Carlo methods with R. Springer, New York, New York, NY
Thurlings RM, Boumans M, Tekstra J et al (2010) Relationship between the type I interferon signature and the response to rituximab in rheumatoid arthritis patients: type I IFN signature and response to rituximab in RA. Arthritis Rheum 62:3607–3614. https://doi.org/10.1002/art.27702
Rieger TR, Allen RJ, Bystricky L et al (2018) Improving the generation and selection of virtual populations in quantitative systems pharmacology models. Prog Biophys Mol Biol 139:15–22. https://doi.org/10.1016/j.pbiomolbio.2018.06.002
Kirouac DC, Schaefer G, Chan J et al (2017) Clinical responses to ERK inhibition in BRAF V600E-mutant colorectal cancer predicted using a computational model. NPJ Syst Biol Appl 3:14. https://doi.org/10.1038/s41540-017-0016-1
Milberg O, Gong C, Jafarnejad M et al (2019) A QSP model for predicting clinical responses to monotherapy, combination and sequential therapy following CTLA-4, PD-1, and PD-L1 checkpoint blockade. Sci Rep 9:11286. https://doi.org/10.1038/s41598-019-47802-4
Jafarnejad M, Gong C, Gabrielson E et al (2019) A computational model of neoadjuvant PD-1 inhibition in non-small cell lung cancer. AAPS J 21:79. https://doi.org/10.1208/s12248-019-0350-x
Wentworth MT, Smith RC, Banks HT (2016) Parameter selection and verification techniques based on global sensitivity analysis illustrated for an HIV model. SIAM/ASA J Uncertain Quantif 4:266–297. https://doi.org/10.1137/15M1008245
Saltelli A, Ratto M, Andres T et al (2007) Global sensitivity analysis. The primer. Wiley, Chichester, UK
Marino S, Hogue IB, Ray CJ et al (2008) A methodology for performing global uncertainty and sensitivity analysis in systems biology. J Theor Biol 254:178–196. https://doi.org/10.1016/j.jtbi.2008.04.011
Zhang X-Y, Trame MN, Lesko LJ et al (2015) Sobol sensitivity analysis: a tool to guide the development and evaluation of systems pharmacology models. CPT Pharmacometrics Syst Pharmacol 4:69–79
Morris MD (1991) Factorial sampling plans for preliminary computational experiments. Technometrics 33:161–174. https://doi.org/10.1080/00401706.1991.10484804
Campolongo F, Cariboni J, Saltelli A (2007) An effective screening design for sensitivity analysis of large models. Environ Model Softw 22:1509–1518. https://doi.org/10.1016/j.envsoft.2006.10.004
Sobol′ IM (2001) Global sensitivity indices for nonlinear mathematical models and their Monte Carlo estimates. Math Comput Simul 55:271–280. https://doi.org/10.1016/S0378-4754(00)00270-6
Saltelli A, Annoni P, Azzini I et al (2010) Variance based sensitivity analysis of model output. Design and estimator for the total sensitivity index. Comput Phys Commun 181:259–270. https://doi.org/10.1016/j.cpc.2009.09.018
Iman RL, Conover WJ (1987) A measure of top – down correlation. Technometrics 29:351–357. https://doi.org/10.1080/00401706.1987.10488244
Lebedeva G, Sorokin A, Faratian D et al (2012) Model-based global sensitivity analysis as applied to identification of anti-cancer drug targets and biomarkers of drug resistance in the ErbB2/3 network. Eur J Pharm Sci 46:244–258. https://doi.org/10.1016/j.ejps.2011.10.026
Chalom A, Prado PIKL (2012) Parameter space exploration of ecological models arXiv:12106278
Nossent J, Bauwens W (2012) Optimising the convergence of a Sobol’ sensitivity analysis for an environmental model: application of an appropriate estimate for the square of the expectation value and the total variance. In: Proceedings of the iEMSs 2012 International congress on environmental modelling and software, managing resources of a limited planet: pathways and visions under uncertainty. Leipzig, Germany, p 8
Duan JZ (2007) Applications of population pharmacokinetics in current drug labelling. J Clin Pharm Ther 32:57–79. https://doi.org/10.1111/j.1365-2710.2007.00799.x
Powell LM, Lo A, Cole MS, et al (2007) Application of predictive biosimulation to the study of atherosclerosis: development of the cardiovascular PhysioLab® platform and evaluation of CETP inhibitor therapy. Proceedings of the FOSBE 8
Fisher RA (1941) Statistical methods for research workers, 8th edn. Oliver and Boyd, Edinburgh
Poole W, Gibbs DL, Shmulevich I et al (2016) Combining dependent P- values with an empirical adaptation of Brown’s method. Bioinformatics 32:i430–i436. https://doi.org/10.1093/bioinformatics/btw438
Kost JT, McDermott MP (2002) Combining dependent P-values. Stat Probability Lett 60:183–190. https://doi.org/10.1016/S0167-7152(02)00310-3
Brown MB (1975) A method for combining non-independent, one-sided tests of significance. Biometrics 31:987–992. https://doi.org/10.2307/2529826
Wilson DJ (2019) The harmonic mean p-value for combining dependent tests. Proc Natl Acad Sci U S A 116:1195–1200. https://doi.org/10.1073/pnas.1814092116
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247. https://doi.org/10.1016/j.ejca.2008.10.026
Alnaif A, Schmidt B, Thalhauser C, et al (2019) Linear and ensemble approaches for calibrating quantitative systems pharmacology models more quickly and with reduced variability. In: ACoP10. Orlando, FL, ISSN:2688-3953.
Peacock JA (1983) Two-dimensional goodness-of-fit testing in astronomy. Mon Not R Astron Soc 615–627
Fasano G, Franceschini A (1987) A multidimensional version of the Kolmogorov-Smirnov test. Mon Not R Astron Soc 225:155–170
Blum J, del Sol C (2018) An investigation of distribution distance measures. In: PharmaSUG 2018 conference proceedings. Seattle, WA, p AA-06
Ramdas A, Trillos N, Cuturi M (2017) On Wasserstein two-sample testing and related families of nonparametric tests. Entropy 19:47. https://doi.org/10.3390/e19020047
Ellner SP, Seifu Y, Smith RH (2002) Fitting population dynamic models to time-series data by gradient matching. Ecology 83:2256–2270. https://doi.org/10.1890/0012-9658(2002)083[2256:FPDMTT]2.0.CO;2
Brooks ME, McCoy MW, Bolker BM (2013) A method for detecting positive growth autocorrelation without marking individuals. PLoS One 8:e76389. https://doi.org/10.1371/journal.pone.0076389
Clarke ND, Bourque G (2010) Success in the DREAM3 signaling response challenge using simple weighted-average imputation: lessons for community-wide experiments in systems biology. PLoS One 5:e8417. https://doi.org/10.1371/journal.pone.0008417
Guex N, Migliavacca E, Xenarios I (2010) Multiple imputations applied to the DREAM3 phosphoproteomics challenge: a winning strategy. PLoS One 5:e8012. https://doi.org/10.1371/journal.pone.0008012
Barnard J, Meng X-L (1999) Applications of multiple imputation in medical studies: from AIDS to NHANES. Stat Methods Med Res 8:17–36
MICRO-Obes Consortium, Kayser BD, Prifti E et al (2019) Elevated serum ceramides are linked with obesity-associated gut dysbiosis and impaired glucose metabolism. Metabolomics 15:140. https://doi.org/10.1007/s11306-019-1596-0
Douglas GM, Hansen R, Jones CMA et al (2018) Multi-omics differentially classify disease state and treatment outcome in pediatric Crohn’s disease. Microbiome 6:13. https://doi.org/10.1186/s40168-018-0398-3
Palarea-Albaladejo J, Martín-Fernández JA (2015) zCompositions — R package for multivariate imputation of left-censored data under a compositional approach. Chemom Intell Lab Syst 143:85–96. https://doi.org/10.1016/j.chemolab.2015.02.019
Koh B-I, George AD, Haftka RT et al (2006) Parallel asynchronous particle swarm optimization. Int J Numer Methods Eng 67:578–595. https://doi.org/10.1002/nme.1646
Venter G, Sobieszczanski-Sobieski J (2006) A parallel particle swarm optimization algorithm accelerated by asynchronous evaluations. J Aerosp Comput Inf Commun 3:16
Cedersund G (2012) Conclusions via unique predictions obtained despite unidentifiability - new definitions and a general method. FEBS J 279:3513–3527. https://doi.org/10.1111/j.1742-4658.2012.08725.x
Villaverde AF, Raimúndez E, Hasenauer J et al (2019) A comparison of methods for quantifying prediction uncertainty in systems biology. IFAC PapersOnLine 52:45–51. https://doi.org/10.1016/j.ifacol.2019.12.234
Hagen DR, Apgar JF, Witmer DK et al (2011) Reply to Comment on “Sloppy models, parameter uncertainty, and the role of experimental design”. Mol BioSyst 7:2523. https://doi.org/10.1039/c1mb05200d
Hastie T, Tibshirani R, Friedman J (2017) The elements of statistical learning: data mining, inference, and prediction, 2nd edn. Springer, New York
Fletcher D (2018) Model averaging. Springer, Berlin
Zhu Y (2005) Ensemble forecast: a new approach to uncertainty and predictability. Adv Atmos Sci 22:781–788. https://doi.org/10.1007/BF02918678
Parker WS (2013) Ensemble modeling, uncertainty and robust predictions: ensemble modeling. WIREs Clim Change 4:213–223. https://doi.org/10.1002/wcc.220
Breiman L (1996) Bagging predictors. Mach Learn 24:123–140. https://doi.org/10.1007/BF00058655
Cheng Y, Schmidt BJ (2019) An automated iterative virtual population development workflow for calibration of multi-therapy immunooncology quantitative systems pharmacology models (I-O QSP platforms) to population data from the clinical setting. In: ACoP10. Orlando, FL, ISSN:2688-3953.
Friedrich C (2016) A model qualification method for mechanistic physiological QSP models to support model-informed drug development: a model qualification method for QSP models. CPT Pharmacometrics Syst Pharmacol 5:43–53. https://doi.org/10.1002/psp4.12056
Ramanujan S, Chan JR, Friedrich CM et al (2019) A flexible approach for context-dependent assessment of quantitative systems pharmacology models. CPT Pharmacometrics Syst Pharmacol 8:340–343. https://doi.org/10.1002/psp4.12409
Agoram B (2014) Evaluating systems pharmacology models is different from evaluating standard pharmacokinetic–pharmacodynamic models. CPT Pharmacometrics Syst Pharmacol 3:e101. https://doi.org/10.1038/psp.2013.77
American Diabetes Association Consensus Panel (2004) Guidelines for computer modeling of diabetes and its complications. Diabetes Care 27:2262–2265. https://doi.org/10.2337/diacare.27.9.2262
Bai JPF, Schmidt BJ, Gadkar K et al (2021) FDA-industry scientific exchange on assessing quantitative systems pharmacology models in clinical drug development: a meeting report, summary of challenges/gaps, and future perspective. AAPS J 23:10. https://doi.org/10.1208/s12248-021-00585-x
Ermakov S, Schmidt BJ, Musante CJ, Thalhauser CJ (2019) A survey of software tool utilization and capabilities for quantitative systems pharmacology: what we have and what we need. CPT Pharmacometrics Syst Pharmacol 8:62–76. https://doi.org/10.1002/psp4.12373
Hosseini I, Feigelman J, Gajjala A et al (2020) gQSPSim: a SimBiology-based GUI for standardized QSP model development and application. CPT Pharmacometrics Syst Pharmacol 9:165–176. https://doi.org/10.1002/psp4.12494
Ermakov S, Forster P, Pagidala J et al (2014) Virtual systems pharmacology (ViSP) software for simulation from mechanistic systems-level models. Front Pharmacol 5:232. https://doi.org/10.3389/fphar.2014.00232
Agren R, Liu L, Shoaie S et al (2013) The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput Biol 9:e1002980. https://doi.org/10.1371/journal.pcbi.1002980
Heirendt L, Arreckx S, Pfau T et al (2019) Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat Protoc 14:639–702. https://doi.org/10.1038/s41596-018-0098-2
Sové RJ, Jafarnejad M, Zhao C et al (2020) QSP-IO: a quantitative systems pharmacology toolbox for mechanistic multiscale modeling for immuno-oncology applications. Clin Pharmacol Ther 9:484–497. https://doi.org/10.1002/psp4.12546
Kaufman L, Rousseeuw PJ (2005) Finding groups in data: an introduction to cluster analysis. Wiley, Hoboken, NJ
Park H-S, Jun C-H (2009) A simple and fast algorithm for K-medoids clustering. Expert Syst Appl 36:3336–3341. https://doi.org/10.1016/j.eswa.2008.01.039
Jacobsoone-Ulrich A, Jamme P, Alkeraye S et al (2016) Ipilimumab in anti-PD1 refractory metastatic melanoma: a report of eight cases. Melanoma Res 26:153–156. https://doi.org/10.1097/CMR.0000000000000221
Bowyer S, Prithviraj P, Lorigan P et al (2016) Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer 114:1084–1089. https://doi.org/10.1038/bjc.2016.107
Aya F, Gaba L, Victoria I et al (2016) Ipilimumab after progression on anti-PD-1 treatment in advanced melanoma. Future Oncol 12:2683–2688. https://doi.org/10.2217/fon-2016-0037
Zimmer L, Apuri S, Eroglu Z et al (2017) Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer 75:47–55. https://doi.org/10.1016/j.ejca.2017.01.009
Robert C, Long GV, Schachter J et al (2017) Long-term outcomes in patients (pts) with ipilimumab (ipi)-naive advanced melanoma in the phase 3 KEYNOTE-006 study who completed pembrolizumab (pembro) treatment. J Clin Oncol 35:9504–9504. https://doi.org/10.1200/JCO.2017.35.15_suppl.9504
Acknowledgments
We would like to acknowledge QSP colleagues at Bristol Myers Squibb for insightful discussions as we develop and apply QSP models and VPop methods: Fulya Akpinar, Limei Cheng, Pegy Foteinou, Alexander Ratushny, and Huilin Ma.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Cheng, Y., Straube, R., Alnaif, A.E., Huang, L., Leil, T.A., Schmidt, B.J. (2022). Virtual Populations for Quantitative Systems Pharmacology Models. In: Bai, J.P., Hur, J. (eds) Systems Medicine. Methods in Molecular Biology, vol 2486. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2265-0_8
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2265-0_8
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2264-3
Online ISBN: 978-1-0716-2265-0
eBook Packages: Springer Protocols