
Abstract
In addition to its ability to regenerate any amputated body part, the Hydra freshwater polyp shows the amazing ability to regenerate as a full polyp after a complete dissociation of its tissues. The developmental processes at work in reaggregates undergoing whole-body regeneration can be investigated at the molecular level by RNA interference (RNAi). Here we provide a protocol that combines β-catenin RNAi with reaggregation. This protocol serves as a basis to generate “RNAi-reaggregates,” followed by the extraction of high-quality RNA for the precise quantification of gene expression by real-time PCR. This protocol is efficient, providing both a molecular signature, with the significant downregulation of β-catenin and Wnt3, as well as a robust phenotype, the lack of axis formation, which is observed in all reaggregates.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
- Hydra
- Reaggregation
- Whole-body regeneration
- siRNA electroporation
- Gene knockdown
- β-catenin
- Wnt3
- qPCR
- Patterning
1 Introduction
Hydra is a small freshwater organism that belongs to the phylum Cnidaria (Fig. 1a). The animals exhibit a tube shape with a head at the apical pole and a foot (basal disk) at the basal one. The head region is composed of an apical dome-shaped structure centered around the mouth opening, named hypostome , and at its base, a ring of tentacles (Fig. 1b). When a Hydra is cut into two halves, each half will regenerate within 3–4 days a new complete body, including a fully functional head from the lower half and a foot from the upper half [2]. This is achieved through the rapid formation of an organizer (a group of cells that can induce and pattern adjacent cells) at the regenerating tip as reviewed in [3]. Over the last two decades, it has been demonstrated that Wnt/β-catenin signaling is a component of the head organizer, with the growth factor Wnt3 acting as a head activator [4,5,6,7,8]. In brief, Wnt3 is mainly expressed in the hypostome, is the earliest upregulated Wnt gene during head regeneration, and acts in an auto-regulatory loop to maintain Wnt/β-catenin activity in the apical region [6, 7]. Noteworthy, Wnt3 expression and thus head formation outside the head region are suppressed by the transcription factor Sp5 [8]. At the basal pole, BMP signaling seems to play a key role in basal regeneration [9]. Overall, two regulatory networks organize each pole of a regenerating Hydra.

Phylogenetic position and Hydra anatomy. (a) Hydra is a member of the phylum Cnidaria and the class Hydrozoa. Phylogenetic tree, after Collins et al. [1]. (b) The Hydra head is composed of a hypostome (dome-shaped structure surrounding the mouth opening) with a mouth opening at the apex and a tentacle ring at the basis. The body column separates the head from the basal region with the basal disk also named foot. Scale bar: 500 μm
The extreme regenerative capacity of Hydra culminates in the regeneration from reaggregates. The phenomenon of Hydra cells to self-organize into a new animal has fascinated researchers since its discovery in the 1970s [10, 11]. Once Hydra tissues are dissociated into a single-cell suspension, reaggregates either form spontaneously by keeping the cells at a high density or can be induced by mechanically compacting the cells in capillary tubes or by gentle centrifugation [10]. In the first immediate phase of the reaggregation process, epidermal and gastrodermal cells sort out to re-establish the original cell layers [10]. Over the next days, new polyps emerge from the mass of cells, equipped with tentacles, and a hypostome at the apical pole that become fully functional (i.e., able to feed) around day 6 [10]. At a later stage, a basal disk develops on each polyp, which will eventually detach 1–4 weeks later. The number of polyps that develop from a given reaggregate depends on the initial number of cells that form the reaggregate (classically several polyps emerge from a 70,000-cell reaggregate).
Hydra reaggregates can be seen as the forefather of organoids as they share common features, i.e., their formation relies on self-organization and requires Wnt/β-catenin signaling for symmetry breaking [2]. In Hydra reaggregates, the transition from a ball shape to an elongated shape of the reaggregate is a critical first axis-defining step, characterized by the emergence of Wnt3 expressing clusters that will develop into apical poles [12]. To functionally assess the involvement of genes that act in the patterning of a reaggregate, RNA interference (RNAi) serves as a powerful tool to silence gene expression.
RNAi can be induced in intact Hydra by electroporating small interfering RNAs (siRNAs) [8, 13]. In short, siRNAs are loaded onto an RNA-induced silencing complex (RISC), whereas the “passenger” strand is removed by Argonaute-2 (Ago-2). This leads to an activation of the RISC complex with a single-stranded “guide” RNA molecule that targets mRNAs in a sequence-specific manner. Due to the action of the RNase-H like activity of Ago-2, mRNAs are degraded, which results in gene silencing [14, 15]. We recently demonstrated that gene silencing persists over several days even when RNAi animals are dissociated to a single-cell level, which has opened up new perspectives to study the developmental processes at work in reaggregates [8].
In this chapter, we provide a detailed protocol that combines RNAi-mediated gene silencing with reaggregation (Fig. 2). The effectiveness of this protocol is illustrated by the case of β-catenin, leading to a failure of axis formation. Indeed, the quantification of β-catenin transcripts, in reaggregates exposed to β-catenin siRNAs, by real-time PCR (qPCR) shows a significant downregulation of β-catenin. In addition, these β-catenin RNAi reaggregates show a reduced Wnt3 expression and do not develop axes, which is consistent with a function of Wnt/β-catenin signaling in the formation of the Hydra body axis.

Method overview. See text for details
2 Materials
Prepare all solutions with ultrapure water (Milli-Q system 18.2 MΩ-cm at 25 °C) at room temperature except noted otherwise.
2.1 Electroporation and Reaggregation
-
1.
Hydra medium (HM) stock solution A: 109.54 g CaCl2•6 H2O, 29.2 g NaCl, 3.7 g KCl in 1 L water. Autoclave and store at 4 °C.
-
2.
HM stock solution B: 60.57 g Tris–HCl, pH 7.7 in 1 L water. Autoclave and store at 4 °C.
-
3.
HM stock solution C: 246.5 g MgSO4•7 H2O in 1 L water. Autoclave and store at 4 °C.
-
4.
Hydra medium (HM): 2 mL stock solution A, 2 mL stock solution B, 0.1 mL stock solution C in 1 L water. Store at 18 °C.
-
5.
Electroporation dissociation medium (E-DM): 0.27 g KCl, 1.31 g CaCl2•6 H2O, 0.30 g MgSO4•7 H2O, 1.77 g Na-citrate•2 H2O, 0.66 g Na-pyruvate, 1.08 g glucose, 2.87 g TES in 950 mL water. Stir until all components are dissolved and adjust pH to 6.9 with HCl. Fill up to 1 L and store at 4 °C.
-
6.
121.5 mM rifampicin stock solution: 0.1 g rifampicin in 1 mL DMSO. Store at −20 °C.
-
7.
Restoration medium (RM): 40 mL HM, 10 mL E-DM, 4.9 μL 121.5 mM rifampicin stock solution.
-
8.
HEPES solution : 10 mM HEPES–HCl, pH 7.0. Sterile filter and store at RT.
-
9.
Reaggregation dissociation medium (R-DM): 0.2684 g KCl, 0.6647 g CaCl2, 0.296 g MgSO4•7 H2O, 1.765 g Na-citrate•2 H2O, 0.666 g Na-pyruvate, 0.721 g glucose, 2.865 g TES in 950 mL water. Stir until all components are dissolved and adjust pH to 6.9 with HCl. Add 0.05 g rifampicin, 0.10 g streptomycin, and 0.05 g kanamycin. Make up to 1 L with water and stir for 2 h (see Note 1). Store at 4 °C.
-
10.
70% R-DM: 35 mL R-DM, 15 mL HM.
-
11.
50% R-DM: 25 mL R-DM, 25 mL HM.
2.2 RNA Extraction, Small Interfering RNAs, and Real-Time PCR
-
1.
70% Ethanol (EtOH) solution : 35 mL 100% EtOH, 15 mL nuclease-free water.
-
2.
40 μM β-catenin siRNA-1 stock solution: dissolve the lyophilized oligonucleotide UCA ACC UAA CAG ACA A in nuclease-free water and store at −20 °C.
-
3.
40 μM β-catenin siRNA-2 stock solution: UGA GGA GCU AUA CUU AUG A in nuclease-free water, stored at −20 °C.
-
4.
40 μM β-catenin siRNA-3 stock solution: ACG ACU CUC UGU UGA AUU A in nuclease-free water, stored at −20 °C.
-
5.
40 μM scramble siRNA stock solution: AGGUAGUGUAAUCGCCUUG in nuclease-free water, stored at −20 °C.
-
6.
β-catenin siRNA working solution: 6.7 μL 40 μM β-catenin siRNA-1 stock solution, 6.7 μL 40 μM β-catenin siRNA-2 stock solution, 6.7 μL 40 μM β-catenin siRNA-3 stock solution in 180 μL 10 mM HEPES solution. Prepare fresh before use, keep at 18 °C.
-
7.
Scramble siRNA working solution: 20 μL 40 μM scramble siRNA in 180 μL 10 mM HEPES solution. Prepare fresh before use, keep at 18 °C.
-
8.
10 μM β-catenin forward primer: dissolve the lyophilized oligonucleotide TACGCAATGTTGTTGGTGCT in nuclease-free water and store at −20 °C.
-
9.
10 μM β-catenin reverse primer: GCTTCAATTCGATGGCCTAA in nuclease-free water, stored at −20 °C.
-
10.
10 μM Wnt3 forward primer: GAGTTGACGGTTGCGAACTT in nuclease-free water, stored at −20 °C.
-
11.
10 μM Wnt3 reverse primer: ACATGAAACCTTGCAACACCA in nuclease-free water, stored at −20 °C.
-
12.
10 μM TATA-box binding protein (TBP) forward primer: AAGCGATTTGCAGCAGTTAT in nuclease-free water, stored at −20 °C.
-
13.
10 μM TBP reverse primer: GCTCTTCACTTTTTGCTCCA in nuclease-free water, stored at −20 °C.
2.3 Kits and Equipment
-
1.
Total RNA extraction kit (e.g., E.Z.N.A., Omega Bio-Tek).
-
2.
7.5 M lithium chloride precipitation solution.
-
3.
10,000 U DNase I recombinant.
-
4.
cDNA synthesis kit (e.g., qScript cDNA SuperMix, Quantabio).
-
5.
Master mix for real-time PCR detection (e.g., SYBR® Select Master Mix for CFX).
-
6.
4-mm electroporation cuvettes.
-
7.
1.5-mL nuclease-free microcentrifuge tubes.
-
8.
Pasteur pipettes 150 mm.
-
9.
1-mL single-use syringes.
-
10.
23G × 1 needles (0.6 × 25 mm).
-
11.
Real-time PCR detection system.
-
12.
Electroporation system.
3 Methods
3.1 Hydra Electroporation
For one electroporation experiment, use 20 animals. All steps are performed at 18 °C.
-
1.
Rinse the Hydras three times in Milli-Q water (see Note 2).
-
2.
Incubate animals for 45 min in Milli-Q water (see Note 3).
-
3.
Transfer animals by suction with a Pasteur pipette into a 4-mm electroporation cuvette.
-
4.
Remove all the Milli-Q water with the Pasteur pipette from the cuvette (see Note 4).
-
5.
Add either β-catenin or scramble siRNA working solution into the cuvette (see Note 5).
-
6.
Tap the cuvette three times gently to evenly distribute the animals (see Note 6).
-
7.
Place the cuvette into the shocking chamber of the electroporation system.
-
8.
Wait for 3 min until all animals are relaxed (see Note 7).
-
9.
Set the following electroporation parameters: Voltage: 150 V; Pulse length: 50 ms; Number of pulses: 2; Pulse intervals: 0.1 s (see Note 8).
-
10.
Press the pulse button.
-
11.
Immediately after the electroporation , add 200 μL RM into the cuvette.
-
12.
Transfer all animals with a Pasteur pipette into a six-well plate filled with 5 mL of RM.
-
13.
Keep animals for 24 h in RM.
-
14.
Transfer them to HM.
-
15.
Wait for 24 h.
-
16.
Repeat steps 1–15 a second time (see Note 9).
3.2 Hydra Dissociation and Reaggregation
For one reaggregation experiment, use 80 RNAi animals, which will give four reaggregates (see Note 10). Steps 1–16 are performed on ice at 18 °C. All the remaining steps are performed at 18 °C unless stated otherwise.
-
1.
Transfer 80 electroporated animals into a 15-mL tube (Tube 1) and remove all HM from the tube (see Note 11).
-
2.
Add 3 mL of R-DM.
-
3.
Gently pipette up and down 30 times with a burned Pasteur pipette (see Note 12).
-
4.
Wait 5 min to let tissue pieces sediment.
-
5.
Transfer supernatant into a new 15-mL tube (Tube 2) (see Note 13).
-
6.
Add 3 mL of R-DM into Tube 1.
-
7.
Pipette up and down 30 times.
-
8.
Wait for 5 min to let tissue pieces sediment.
-
9.
Transfer supernatant into Tube 2.
-
10.
Add 3 mL of R-DM into Tube 1 and pipette up and down 50 times (see Note 14).
-
11.
Wait for 5 min to let tissue pieces sediment.
-
12.
Transfer supernatant into Tube 2.
-
13.
Add 1 mL of R-DM into Tube 1.
-
14.
Pipette up and down 50 times.
-
15.
Wait for 5 min to let tissue pieces sediment.
-
16.
Transfer supernatant into Tube 2.
-
17.
Centrifuge at 245 rcf for 45 min at 4 °C (see Note 15).
-
18.
Carefully remove supernatant.
-
19.
Resuspend cell pellet in 2 mL R-DM.
-
20.
Distribute cell suspension into 1.5-mL tubes (500 μL per tube).
-
21.
Centrifuge at 209 rcf for 45 min at 4 °C (see Note 15).
-
22.
Horizontally lay down tubes on a bench.
-
23.
Wait until reaggregates detach from the bottom of the tubes (see Note 16).
-
24.
Transfer reaggregates into a six-well plate filled with 75% R-DM.
-
25.
Keep them for 16 h in 70% R-DM (see Note 17).
-
26.
Transfer reaggregates into a six-well plate filled with 50% R-DM.
-
27.
Keep them for 4 h in 50% R-DM (see Note 18).
-
28.
Transfer reaggregates into HM.
- 29.
3.3 RNA Extraction and Real-Time PCR (qPCR)
-
1.
Transfer a reaggregate into a 1.5-mL tube.
-
2.
Add 350 μL of the RNA extract kit’s lysis buffer.
-
3.
Pass the lysate 15 times through a 23G × 1 needle fixed to a single-use syringe.
-
4.
Perform RNA extraction following the manufacturer’s instructions.
-
5.
Elute RNA in 30 μL nuclease-free water.
-
6.
Add 4 μL of 10× DNase buffer, 2 μL of DNase, and 4 μL of nuclease-free water to the eluted RNA.
-
7.
Incubate at 37 °C for 15 min (see Note 21).
-
8.
Add 27 μL 7.5 M lithium chloride precipitation solution.
-
9.
Incubate for 2 h at −80 °C (see Note 22).
-
10.
Centrifuge full speed for 30 min at 4 °C.
-
11.
Carefully remove supernatant.
-
12.
Add 500 μL ice-cold 70% EtOH.
-
13.
Gently wash the pellet by pipetting up and down (see Note 23).
-
14.
Centrifuge full speed for 30 min at 4 °C.
-
15.
Carefully remove 70% EtOH.
-
16.
Air-dry RNA for 5 min (see Note 24).
-
17.
Resuspend RNA in 20 μL nuclease-free water.
-
18.
Store at −80 °C or short term on ice for cDNA synthesis (see Note 25).
-
19.
Transcribe 500 ng of purified RNA into cDNA in a total volume of 20 μL following the kit manufacturer’s instructions.
-
20.
Adjust the cDNA concentration to 3.2 ng/μL by mixing 9 μL of cDNA template with 61 μL of nuclease-free water (see Note 26).
-
21.
Add 16 ng of cDNA template (5 μL of the 3.2 ng/μL cDNA solution) into as many wells of a 96-well plate as qPCR to be done.
-
22.
Per well, add 10 μL of master mix, 0.6 μL of 10 μM qPCR forward primer (300 nM final), 0.6 μL of 10 μM qPCR reverse primer (300 nM final), and 3.8 μL nuclease-free water (see Note 27).
-
23.
Perform qPCR in the 96-well plate following the manufacturer’s instructions (see Note 28).
4 Notes
-
1.
Rifampicin is very hard to dissolve. Stir R-DM for at least 2 h after adding rifampicin.
-
2.
Cool down Milli-Q water to 18 °C prior to usage.
-
3.
Water is hypotonic, which leads to a swelling of the cells. Do not exceed an incubation time of 1 h 15 min as this leads to excessive tissue loss.
-
4.
Remaining HM will dilute the siRNAs. If necessary, use a micropipette to remove the HM.
-
5.
Do not keep siRNA dilutions on ice. The animals will not relax when a cold solution is added onto them. Always keep siRNA dilutions at 18 °C until the electroporations are performed.
-
6.
The electroporation efficiency will be reduced when the animals clump and are not evenly distributed in the cuvette.
-
7.
The surface of the relaxed animals is greater than that of the contracted ones, and thus more siRNAs can be taken up by relaxed Hydra. Never perform an electroporation on contracted animals, as this will reduce the electroporation efficiency. We never use any chemical for relaxing the animals prior to electroporation . Hydras spontaneously relax in in the siRNA dilution after 3 min.
-
8.
The electroporation conditions are optimized to obtain a 100% survival rate for Hydra magnipapillata. Depending on the species used, the conditions might need to be adjusted.
-
9.
The more often animals are electroporated the smaller they get as tissue loss occurs. We do not recommend performing more than two electroporations as the animals become too small to perform a reaggregation experiment.
-
10.
To obtain four reaggregates from wild-type Hydra magnipapillata, 60 animals are sufficient. However, over the electroporations, the animals slightly shrink as tissue loss occurs and thus the number of animals should be increased to 80. In comparison, 120 electroporated Hydra vulgaris of the Basel strain are necessary to obtain four reaggregates [8].
-
11.
24 h after the second electroporation , RNAi animals can also be drug treated for 18 h, i.e., with Alsterpaullone to enhance certain phenotypes, which is then directly followed by reaggregation [8].
-
12.
The sharp end of a Pasteur pipette can easily destroy the Hydra cells. Always slightly burn the Pasteur pipette so that the end is smooth. Excessive pipetting up-and-down can also cause damage to cells. Pipetting up-and-down 30 times is sufficient to dissociate the Hydra tissue.
-
13.
Tissue pieces that are a sign of incomplete dissociation should never be transferred into tube 2. If necessary, increase the sedimentation time to obtain a better separation of single cells (supernatant) from tissue pieces.
-
14.
At that stage, we recommend to start pipetting up-and-down 50 times to achieve a complete dissociation of any remaining tissue pieces.
-
15.
The centrifugation time can be adjusted depending on the centrifuge used. In our hands, a centrifugation of less than 45 min leads to partial cell collection.
-
16.
Do not use a rotating wheel to detach the reaggregates as this leads to excessive cell loss.
-
17.
A critical step in ensuring a 100% reaggregate survival is a long incubation in 70% R-DM. After 16 h in R-DM, the surface of the reaggregates should be smooth (see 1 d time point of Fig. 3), which indicates a successful separation of the two cell layers, epidermis and gastrodermis.
-
18.
The incubation time can be reduced to 1 h if the cell layers are already separated. If not, extend the incubation to 4 h, which will further promote the separation of the cell layers and thus the smoothening of the reaggregates.
-
19.
To prevent any infection, the reaggregates should always be kept in clean HM.
-
20.
We have successfully combined RNAi and reaggregation. Note that axis formation is not visible after silencing β-catenin (Fig. 3).
-
21.
DNase treatment is performed to remove any remaining genomic DNA traces that might falsify the qPCR result. Note that SYBR interacts with any double-stranded DNA.
-
22.
Lithium chloride offers the advantage over other precipitation methods that it does not precipitate contaminants such as DNA, carbohydrates, and proteins [16].
-
23.
Be careful not to lose the RNA pellet. The RNA has a white color and is hard to see.
-
24.
Do not over-dry RNA as this will cause its degradation.
-
25.
The RNA yield of four reaggregates is approximately 700 ng.
-
26.
We recommend using low retention tips in all steps to reduce the adhesion of DNA to the tip surface.
-
27.
We have used qPCR to validate the silencing of β-catenin and to test the expression of Wnt3 . We demonstrate a successful silencing of β-catenin and a downregulation of Wnt3 gene expression (Fig. 4). Note that the expression of Wnt3 in β-catenin RNAi reaggregates can also be tested by in situ hybridization as described in [8].
-
28.
The analysis was carried out as described in [17].

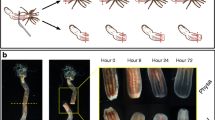
Regeneration of Hydra body axes from reaggregated cells after silencing β-catenin. The reaggregation experiment was performed with animals exposed twice (RNAi1, RNAi2) to scramble (top) and β-catenin (bottom) siRNAs. Shown are four representative reaggregates (agg 1–4). Reaggregates were imaged at indicated time points. The red arrow indicates the time point of animal dissociation, taken as t0 for reaggregation. Note that axes are clearly visible in control RNAi reaggregates on day 4 (white arrowheads), while β-catenin RNAi reaggregates fail to develop axes while forming a few tentacles (white arrows). Scale bars: 200 μm

Real-time PCR of RNAi reaggregates. RNAi was performed as depicted in the scheme. The red arrow indicates the time point of animal dissociation, taken as t0 for reaggregation. RNA was extracted on day 4 of the reaggregation process, followed by real-time PCR (qPCR) to measure the expression of β-catenin (blue symbols) and Wnt3 (purple symbols) in control and β-catenin RNAi reaggregates. Each data point represents one biologically independent experiment. Statistical p-values: * ≤ 0.05, ** ≤ 0.01 (unpaired t-test)
References
Collins AG, Schuchert P, Marques AC, Jankowski T, Medina M, Schierwater B (2006) Medusozoan phylogeny and character evolution clarified by new large and small subunit rDNA data and an assessment of the utility of phylogenetic mixture models. Syst Biol 55(1):97–115. https://doi.org/10.1080/10635150500433615
Vogg MC, Galliot B, Tsiairis CD (2019) Model systems for regeneration: Hydra. Development 146 (21). https://doi.org/10.1242/dev.177212
Vogg MC, Wenger Y, Galliot B (2016) How somatic adult tissues develop organizer activity. Curr Top Dev Biol 116:391–414. https://doi.org/10.1016/bs.ctdb.2015.11.002
Hobmayer B, Rentzsch F, Kuhn K, Happel CM, von Laue CC, Snyder P, Rothbacher U, Holstein TW (2000) WNT signalling molecules act in axis formation in the diploblastic metazoan Hydra. Nature 407(6801):186–189. https://doi.org/10.1038/35025063
Chera S, Ghila L, Dobretz K, Wenger Y, Bauer C, Buzgariu W, Martinou JC, Galliot B (2009) Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell 17(2):279–289. https://doi.org/10.1016/j.devcel.2009.07.014
Lengfeld T, Watanabe H, Simakov O, Lindgens D, Gee L, Law L, Schmidt HA, Ozbek S, Bode H, Holstein TW (2009) Multiple Wnts are involved in Hydra organizer formation and regeneration. Dev Biol 330(1):186–199. https://doi.org/10.1016/j.ydbio.2009.02.004
Nakamura Y, Tsiairis CD, Ozbek S, Holstein TW (2011) Autoregulatory and repressive inputs localize Hydra Wnt3 to the head organizer. Proc Natl Acad Sci U S A 108(22):9137–9142. https://doi.org/10.1073/pnas.1018109108
Vogg MC, Beccari L, Iglesias Olle L, Rampon C, Vriz S, Perruchoud C, Wenger Y, Galliot B (2019) An evolutionarily-conserved Wnt3/beta-catenin/Sp5 feedback loop restricts head organizer activity in Hydra. Nat Commun 10(1):312. https://doi.org/10.1038/s41467-018-08242-2
Wenger Y, Buzgariu W, Perruchoud C, Loichot G, Galliot B (2019) Generic and context-dependent gene modulations during Hydra whole body regeneration. bioRxiv:587147. https://doi.org/10.1101/587147
Gierer A, Berking S, Bode H, David CN, Flick K, Hansmann G, Schaller H, Trenkner E (1972) Regeneration of hydra from reaggregated cells. Nat New Biol 239(91):98–101
Noda K (1971) Reconstruction of dissociated cells of hydra. Zool Mag 80:27–31
Technau U, Cramer von Laue C, Rentzsch F, Luft S, Hobmayer B, Bode HR, Holstein TW (2000) Parameters of self-organization in Hydra aggregates. Proc Natl Acad Sci U S A 97(22):12127–12131. https://doi.org/10.1073/pnas.97.22.12127
Watanabe H, Schmidt HA, Kuhn A, Hoger SK, Kocagoz Y, Laumann-Lipp N, Ozbek S, Holstein TW (2014) Nodal signalling determines biradial asymmetry in Hydra. Nature 515(7525):112–115. https://doi.org/10.1038/nature13666
Kim D, Rossi J (2008) RNAi mechanisms and applications. BioTechniques 44(5):613–616. https://doi.org/10.2144/000112792
Pratt AJ, MacRae IJ (2009) The RNA-induced silencing complex: a versatile gene-silencing machine. J Biol Chem 284(27):17897–17901. https://doi.org/10.1074/jbc.R900012200
Barlow JJ, Mathias AP, Williamson R, Gammack DB (1963) A simple method for the quantitative isolation of undegraded high molecular weight ribonucleic acid. Biochem Biophys Res Commun 13:61–66. https://doi.org/10.1016/0006-291x(63)90163-3
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45
Acknowledgments
The authors thank Charisios Tsiairis for helpful comments and discussions. The research in the Galliot laboratory is supported by the Swiss National Science Foundation (SNF 310030_189122), the Canton of Geneva, and the Claraz donation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Vogg, M.C., Galliot, B. (2022). Combining RNAi-Mediated β-Catenin Inhibition and Reaggregation to Study Hydra Whole-Body Regeneration. In: Blanchoud, S., Galliot, B. (eds) Whole-Body Regeneration. Methods in Molecular Biology, vol 2450. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2172-1_34
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2172-1_34
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2171-4
Online ISBN: 978-1-0716-2172-1
eBook Packages: Springer Protocols