
Abstract
The ability to regenerate lost body parts is irregularly distributed among animals, with substantial differences in regenerative potential between and within metazoan phyla. It is widely believed that regenerative animal clades inherited some aspects of their capacity to regenerate from their common ancestors but have also evolved new mechanisms that are not shared with other regenerative animals. Therefore, to gain a broad understanding of animal regenerative mechanisms and evolution, a broad sampling approach is necessary. Unfortunately, only few regenerative animals have been established as laboratory models with protocols for functional gene studies. Here, we describe the methods to establish transgenic individuals of the marine cnidarian Hydractinia. We also provide methods for transient gene expression manipulation without modifying the genome of the animals.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
The phylum Cnidaria comprises some 11,000 species of rather diverse animals [1] that share a unique cell type—the cnidocyte, also known as nematocyte or stinging cell. These cells, which belong to the neural lineage, are used for prey capture, defense, and adhesion [2, 3]. Phylogenetically, members of the Cnidaria are divided between two main clades, Anthozoa (e.g., corals, sea anemones, and sea pens) and Medusozoa (e.g., hydrozoans, scyphozoans, and cubozoans), plus a group of parasitic cnidarians, the Myxozoa, that are a sister taxon to Medusozoa [4]. Anthozoans are being characterized by the lack of a medusa stage, which has been lost and gained multiple times in the Medusozoa clade. As a result, many modern medusozoans lack the medusa stage in their life cycle.
Cnidarians possess a remarkable regenerative ability, being able to regrow whole bodies from small tissue fragments, and in some cases also from reaggregated cell suspensions [5,6,7]. However, the mechanisms used for regeneration are different not only between species but also within different body parts of single species [8, 9]. Hence, to gain full understanding of the mechanisms driving regeneration in such a diverse group, it is necessary to study regeneration in as many species and contexts as possible. The main challenge in doing so at the molecular level is the enormous effort required to develop transgenic technologies for each studied species. Contrary to common sense, it appears that substantially different protocols are required to obtain transgenic animals even within one phylum [10,11,12,13]. Here, we present the current protocols used to manipulate gene expression in the colonial hydrozoan Hydractinia .
The genus Hydractinia is represented in the literature primarily by two North Atlantic sibling species, H. echinata and H. symbiolongicarpus. Despite the similarity between the two, they do not readily hybridize [14] and differ in genome size (774 and 514 Mb, respectively) as well as in some aspects of post-metamorphosis growth form. Both the species can be grown in artificial seawater tanks, but selected laboratory strains are only available in H. symbiolongicarpus [15, 16].
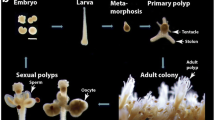
Hydractinia is one of only four cnidarian genera for which transgenesis technologies have been well established (Fig. 1) [10, 11, 17, 18]; the other three being Hydra, Nematostella, and Clytia. First steps to generate transgenic Aiptasia have been reported recently [19]. Transient gene knockdown protocols using short hairpin RNA, RNAi, and morpholino oligonucleotides have been established in Hydractinia as well [20,20,22]. Transient ectopic/overexpression experiments can be achieved with mRNA injection into embryos [15].

A selection of transgenic reporter animals. (a) Tfap2::GFP. This male sexual polyp expresses GFP in early germ cells. (b) Piwi1::GFP. This animal expresses GFP in i-cells and germ cells. The image shows a feeding polyp with no germ cells. (c) A double transgenic female obtained by crossing a Tfap2::GFP animal with a Piwi1::mScarlet partner. (d) A CRISPR-Cas9 knockin animal in which GFP has been inserted in-frame in to the Ef1a endogenous coding sequence. (e) A Rfamide::GFP reporter animal. A subset of neurons express GFP. (f) A G0, Actin1:: GFP mosaic reporter animal. This transgene is epithelial specific. All images were taken from live animals
Hydractinia has been primarily used to study stem/germ cells [15, 20], regeneration [19], neurogenesis [23], and allorecognition [24]. However, recent developments by the Hydractinia research community call for expansion of the usage of this tractable animal model to other disciplines as well.
2 Materials
2.1 Animal Maintenance, Spawning, and Metamorphosis
-
1.
Artificial or filtered natural seawater (FSW): 28–32 ppm, 0.22 μm-filtered, pH 8.15–8.35.
-
2.
CsCl stock solution: 580 mM CsCl in deionized H2O (diH2O).
-
3.
Embryonic water: 20% (v/v) CsCl stock solution in FSW.
-
4.
Self-made oyster puree.
-
5.
Brine shrimp artemia cyst.
-
6.
Brine shrimp hatcher.
-
7.
Culture seawater tank system (Fig. 2).

Hydractinia culture. (a) Overview of a commercial tank system used in our laboratory. (b) A close-up on the slides-in-a-staining-rack system. (c) A close up on a colony growing on a microscope slide
2.2 Embryo Microinjection
-
1.
Injection plate: 35-mm petri dish with 180–200 μm plankton net attached.
-
2.
Injection capillaries with filament: made by flaming micropipette puller.
-
3.
Fluorescence tracer dye (e.g., Dextran AF555 200 ng/μL [ex/em: 555/565 nm], Lucifer Yellow CH 200 ng/μL [ex/em: 428/536 nm]).
-
4.
Injection stock buffer: 2 M KCl in diH2O.
-
5.
Three axis coarse/fine joystick micro-manipulator.
-
6.
Fluorescence stereo microscope.
-
7.
Microinjector.
-
8.
Vannas scissors, curved, 80 mm (e.g., Ted Pella; 1341).
-
9.
Flaming micropipette puller (e.g., Sutter Instrument Co; P-97).
2.3 shRNA
Prepare all solutions using ultrapure nuclease-free water (prepared by purifying deionized water, to attain a resistivity of 18 MΩ/cm at 25 °C) and analytical grade reagents. Diligently follow all waste disposal regulations when disposing waste materials.
-
1.
T7-based in vitro transcription kit (e.g., NEB Arca/NTP T7 mRNA synthesis with poly A tailing).
-
2.
Silica membrane column with 2-mL collection tube.
-
3.
Wash-r1: 1 M Gu-HCl, 10 mM Tris–HCl, pH 7.0.
-
4.
Wash-r2: 10 mM Tris–HCl, pH 7.5, 60 mM potassium acetate in 80% ethanol.
-
5.
3 U/μL DNaseI.
-
6.
DNA digestion buffer: 40 mM Tris–HCl, pH 8.0, 10 mM NaCl, 6 mM MgCl2, 10 mM CaCl2.
-
7.
DNA extraction kit (e.g., Qiagen, Cat. #69504).
-
8.
High-Fidelity DNA Polymerase.
-
9.
Taq polymerase.
-
10.
A/T cloning vector (e.g., pGEM® T-easy Vector System, Promega).
-
11.
Restriction endonuclease.
-
12.
Gel extraction kit.
2.4 CRISPR-Cas9 Editing and Genotyping
CRISPR RNAs (crRNA) for gene targeting experiments are synthesized commercially (e.g., Integrated DNA Technologies, IDT). crRNAs need to be hybridized to tracer RNA (tracr RNA) prior to use and can be stored at −20 °C after hybridization. Cas9 enzyme aliquots are stored at −80 °C and are mixed with crRNAs:tracrRNA prior to use.
-
1.
crRNA targeting sequences (two for performing deletions, see Note 1).
-
2.
tracrRNA and hybridization buffer.
-
3.
Cas9 enzyme.
-
4.
100 g/mL ampicillin in LB broth.
-
5.
LB agar plates: 1.5% (w/v) agar in LB broth. Heat to dissolve, pour hot in petri dishes, store at 4 °C.
-
6.
Plasmid miniprep kit (e.g., GenElute™ Plasmid Miniprep kit, Sigma-Aldrich).
3 Methods
3.1 Animal Maintenance, Spawning and Metamorphosis
-
1.
Stable clones of wild-type/transgenic animals are grown on glass microscope slides at 20–22 °C in artificial seawater (Fig. 2) under a 14:10 light:dark regime (see Note 2).
-
2.
Feed four times a week with freshly hatched Artemia and once a week with oysters (see Note 3).
-
3.
Transfer the Hydractinia colonies to be spawned in water bowls filled with FSW outside of the tanks to facilitate embryo collection . The animals spawn about 90 min after being exposed to light.
-
4.
Collect embryos into a water bowl filled with FSW using a Pasteur pipette (see Note 4).
-
5.
Wait for 3 days for the embryos to reach planula larval stage.
-
6.
Transfer the larvae into a water bowl filled with 110 mM CsCl in FSW.
-
7.
Wait for 3 h.
-
8.
Line a container with the desired substratum (e.g., glass slides) for the larvae to settle onto.
-
9.
Fill the settling container with FSW.
-
10.
Transfer the contracted larvae using a Pasteur pipette into the settling container.
-
11.
Wait for 30 min for the larvae to settle.
-
12.
Metamorphosis is completed within 24 h.
3.2 Capillaries’ Preparation and Injection
-
1.
Fill injection pump with pressurized air and adjust it to 60 psi.
-
2.
Fill the pulled needle with injection solution (see Note 5) using a microloader pipette tip by inserting pipette tip from the back end as deep as possible into the tip of the needle and slowly release the injection solution.
-
3.
Insert filled needle into the holding capillary, which is in turn secured in the capillary holder connected to the injector.
-
4.
Cut/break the edge of the capillary, turn injection pump on, balance and adjust injection pressure/time (see Note 6).
-
5.
Transfer embryos using a glass Pasteur pipette into to the injection plate filled with 4 °C pre-chilled ASW.
-
6.
Place injection plate containing embryos on the stage of the dissection microscope.
-
7.
Once the embryos are in the field of view, make sure they have settled into the pockets of the plankton net (see Note 7).
-
8.
Lower the capillary holder until the tip of the needle becomes visible (through the binoculars) and submerged in ASW.
-
9.
Cut the end of the needle with fine scissors until droplet is visible (see Note 8).
-
10.
Inject embryos by placing the needle in the middle of the embryo. By moving the capillary holder up and down, you will be able to see the needle penetrating the embryo (see Note 9).
-
11.
Move to the next embryo by moving the injection plate—not the capillary stage.
-
12.
Transfer the injected embryos to a petri dish filled with ASW and place in a 16–18 °C incubator.
3.3 Strategy for Reporter Line Cloning
Reporter vector constructs are generated by inserting upstream regulatory sequence (URS) and downstream regulatory sequence (DRS) upstream and downstream of the fluorescent protein coding sequence (CDS), respectively (see Note 10). Cloning of the fragments can be achieved by restriction enzyme-based approach or by Gibson assembly. By using the former method, you can easily remove and add different URS and DRS fragments. All the vectors generated in our lab have the same restriction recognition sites (see Note 11; Fig. 3A).
-
1.
Generate reporter vector by standard molecular cloning.
-
2.
Transfect chemically/electro-competent XL1 Blue E. coli bacteria according to standard protocols.
-
3.
Grow the bacteria and extract plasmids using a commercially available mini-prep kit or use a self-made one (see Note 12).
-
4.
Inject embryos—see Subheading 3.2.
-
5.
Transfer injected embryos from injection plate to large glass petri dishes and washed carefully with ASW.
-
6.
Transfer to an 18 °C incubator.
-
7.
Check for fluorescence the next day using a fluorescence stereomicroscope.
-
8.
Transgenic larvae are separated from the non-injected ones and kept in the 18 °C incubator until metamorphosis induction.
-
9.
Follow steps 6–12 in Subheading 3.1. to induce metamorphosis.
-
10.
Select transgenic animals based on the expected gene expression pattern from previous mRNA in situ hybridization or protein immunofluorescence.
-
11.
Once transgenic animals reach sexual maturity, cross them with wild-type animals to produce G1 non-chimeric transgenic animals (see Note 13).

Constructs structure. (a) Reporter construct. (b) Ectopic expression construct. (c) mRNA synthesis template. (d) shRNA synthesis template. RS restriction site, P2A self-cleavage P2A peptide coding sequence 5′GGTTCAGGTGCTACAAATTTTTCATTATTAAAACAAGCTGGTGATGTTGAAGAAAATCCAGGTCCA3′, LP: linker peptide coding sequence 5′TGGCCAGGAGGCTCCGGCTCC3′, UTR untranslated region 5′TGCAGCCCCGGTAGAAAAA3′. URS: upstream regulatory sequence; DRS: downstream regulatory sequence
3.4 Strategy for Ectopic Expression Construct Cloning
Vectors for transgenic reporter lines can be used to ectopically express or overexpress your gene of interest. URS and DRS regions of the reporter line do not change and a cassette containing UTR + CDS + P2A peptide is inserted upstream of the fluorescence protein (Fig. 3B).
-
1.
Design primers to clone a cassette containing UTR + the coding sequence of your gene of interest + a P2A peptide.
-
2.
Follow the same procedure as in Subheading 3.3 in order to generate the vector.
-
3.
Inject embryos as in Subheading 3.2.
-
4.
Evaluate and maintain transgenic animals as described in Subheading 3.3.
3.5 Designing mRNA Constructs
-
1.
Design template construct (TC) for mRNA synthesis (see Note 14).
-
2.
Use target gene protein sequence from other animals as query to search the homologous transcripts from Hydractinia symbiolongicarpus.
-
3.
Perform TBLASTN search at https://blast.ncbi.nlm.nih.gov/ against transcriptome shotgun assembly (TSA) database (do not use the default nr database ) of Hydractinia symbiolongicarpus. Limit the search by TSA project and type in “GAWH: TSA: Hydractinia symbiolongicarpus, transcriptome shotgun assembly” into the box.
-
4.
From the TBLASTN results page, retrieve the top hit Hydractinia transcript sequence. This is the coding sequence of gene of interest (CDS-GOI).
-
5.
Retrieve the coding sequence of a fluorescence of protein (CDS-FP, e.g., eGFP or mScarlet) from a public database or use the available fluorescence protein sequence that you have already in an existing plasmid in your lab.
-
6.
Codon optimize FP sequences for Hydractinia symbiolongicarpus using http://genomes.urv.es/OPTIMIZER/. Codon usage table can be found at http://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=13093.
-
7.
Arrange these two CDS according to the scheme shown in Fig. 3C (see Note 15).
3.6 Synthesizing the mRNA Template Construct
There are three ways to synthesize the actual fragment: [1] design a plasmid with the construct as insert; [2] PCR several fragments and assembled it with Gibson Assembly process; [3] commercial synthesis. Synthesizing through a commercial service is the easier option, but the other two are more cost effective. We explain the two first options below. Both use plasmids that contain T7 promotor region flanking the insertion site (we use pGEM-T-easy vector plasmid).
3.6.1 Generating Plasmid for Template Construct
-
1.
Design a primer pair to amplify CDS-GOI.
-
2.
Add RS-1 and UTR sequences to the 5′ end of the forward primer and RS-2 to the 3′ end of the reverse primer for CDS-GOI.
-
3.
Amplify CDS-GOI using high-fidelity DNA polymerase using Hydractinia cDNA library as template and the pair of primers design above.
-
4.
Extract the amplicon from agarose gel and add an A-tail to the fragment by Taq polymerase.
-
5.
Perform TA cloning of the A-tailed amplicon inserted into pGEM-T-easy vector.
-
6.
Confirm and assess the plasmid by Sanger sequencing ensure to choose the correct in frame insertion of CDS-GOI to the 3′ region of the T7 promotor.
-
7.
Keep the correct plasmid at −20 °C for step 17.
-
8.
Design a primer pair to amplify CDS-FP.
-
9.
Add RS-2 and P2A/LP sequence to the 5′end of the forward primer and add RS-3 (see Note 16).
-
10.
Amplify CDS-FP using high-fidelity DNA polymerase, plasmid with FP and pair of primers design as in steps 8 and 9.
-
11.
Run the amplicons on an agarose gel.
-
12.
Extract the fragment using standard protocols.
-
13.
Add A-tail to it by Taq polymerase.
-
14.
Perform TA cloning of the A-tailed amplicon inserted into A/T vector.
-
15.
Confirm and assess the plasmid by Sanger sequencing ensure to choose the correct in frame insertion of CDS-FP.
-
16.
Keep the correct plasmid at −20 °C for step 17.
-
17.
Digest plasmid from steps 6 to 7 (pGOI) and from steps 15 to 16 (pFP) with restriction endonuclease corresponding to RS-2 and RS-3. Ligate the small FP fragment from pFP digestion into the backbone fragment from pGOI digestion.
-
18.
Confirm and assess the plasmid by Sanger sequencing and choose the plasmid with correct in-frame ligated template construct as described in Fig. 3C at the 3′ region of the T7 promotor sequence (pTC).
-
19.
Digest and linearize pTC with restriction endonuclease corresponding with RS-3.
-
20.
Extract the linearize pTC with gel extraction system and use this purified fragment as DNA template for in vitro transcription.
3.6.2 Synthesizing Gibson Assembled dsDNA Fragment for Template Construct
-
1.
Design a primer pair to amplify CDS-GOI.
-
2.
Add RS-1 (for Gibson assembly replace the restriction site with 20 nt sequence from the 5′ region of insertion site of your vector plasmid) and UTR sequence to the 5′end of the forward primer for CDS-GOI and add RS-2 and P2A/LP sequence to the 3′end of the reverse primer for CDS-GOI.
-
3.
Amplify CDS-GOI using high-fidelity DNA polymerase, Hydractinia cDNA library as template and pair of primers design as in steps 1 and 2.
-
4.
Keep the PCR mixture at 4 °C until electrophoresis .
-
5.
Design a primer pair to amplify CDS-FP.
-
6.
Add RS-2 and P2A/LP sequence (see Note 17) to the 5′end of the forward primer and add RS-3 and 3′ region of insertion site from the plasmid (see Note 18) to the 3′ end of the reverse primer for CDS-FP.
-
7.
Amplify CDS-FP using high fidelity DNA polymerase, plasmid with fp and pair of primers design as in steps 4 and 5.
-
8.
Keep the PCR mixture at 4 °C until electrophoresis.
-
9.
Electrophorase the PCR mixture from steps 3 and 6 in agarose gel.
-
10.
Extract the amplicons from agarose gel.
-
11.
Use both fragments and a backbone of plasmid for performing Gibson assembly following the manufacturer’s instruction.
-
12.
Transform competent E. coli, grow the colonies, and extract the plasmid.
-
13.
Confirm and assess the plasmid by Sanger sequencing and ensure to choose the plasmid with correct in frame ligated template construct as described in Fig. 3C at the 3′ region of the T7 promotor sequence (pTC).
-
14.
Digest and linearize pTC with restriction endonuclease corresponding with RS-3.
-
15.
Extract the linearize pTC with gel extraction system and use this purified fragment as DNA template for in vitro transcription.
3.7 T7 In Vitro Transcription mRNA Synthesis, Microinjection , and Evaluation
-
1.
Perform T7 in vitro transcription system according to the manufacturer’s instruction to synthesize mRNA.
-
2.
Purify the mRNA with an RNA column system or LiCl.
-
3.
Run the yielded mRNA before and after poly-A-tailing in a formalin-agarose gel and assess the size of the mRNA before injection .
-
4.
Inject the mRNA at 0.8–1.2 μg/μL according to Subheading 3.2.
-
5.
Assess and evaluate the expression of the mRNA by checking the fluorescent protein. Phenotype can best be assessed between 5 and 96 h post injection .
3.8 Short-Hairpin RNA Interference
-
1.
Use target gene protein sequence from other animals as query to search the orthologous transcripts from Hydractinia symbiolongicarpus.
-
2.
Perform TBLASTN search at https://blast.ncbi.nlm.nih.gov/ against transcriptome shotgun assembly (TSA) database (do not use the default nr database ) of Hydractinia symbiolongicarpus. Limit the search set by TSA project and type in “GAWH: TSA: Hydractinia symbiolongicarpus, transcriptome shotgun assembly” to the filling box.
-
3.
From the tblastn results page, retrieve the top hit Hydractinia ’s transcript sequence (only the sequence and not in fasta format).
-
4.
Use the top hit sequence to find siRNA motif at http://www.invivogen.com/sirnawizard/design.php. Choose the desired motif size (21 nt is the default). Leave blanks the option of mRNA database and miRNA SEED database .
-
5.
Choose several sequences from the motif outputs with higher GC content, 5′ GG, and 3′ AT-rich stretches (see Note 19).
-
6.
Perform BLASTN on several motif output sequences against the Hydractinia symbiolongicarpus TSA with loose parameter (e.g., expected value 1) to confirm specificity (see Note 20).
-
7.
Create the shRNA design by converting the selected 21-nt motif (passenger sequence) into RNA by replacing any T with U.
-
8.
Add the reverse-complemented sequence (guide sequence) to the 3′ end and separate them by loop sequences [5′-AUUUACU-3′].
-
9.
Add “UU” to the 3′ of the sequence to create overhang.
-
10.
Select sequences that are not predicted to form secondary structures using http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi. Add one or two mismatches in the middle of the passenger sequence (but not to the guide sequence).
-
11.
Revert the RNA sequence back to DNA by replacing U to T and add T7 promoter [5′-TAATACGACTCACTATA-3′] to the 5′ (Fig. 3).
-
12.
This sequence is the forward oligonucleotides. Create the reverse oligonucleotides sequence by reverse-complementing the forward oligonucleotides sequence. Synthesize the forward and reverse oligonucleotides using commercial services.
-
13.
Dissolve forward and reverse oligonucleotides separately in ultrapure nuclease free water to a final concentration of 100 μM.
-
14.
Dilute these 100 μM oligonucleotides 1:10 in nuclease-free H2O while combining forward and reserved oligos.
-
15.
Denature the combined oligonucleotides at 98 °C for 5 min.
-
16.
Cool it down at room temperature for 10 min; this is the template for T7-based in vitro transcription (IVT) of shRNA.
-
17.
Follow the manufacturer’s protocol for IVT reaction; however, triple the total IVT volume to get enough yield.
-
18.
The IVT reaction mix will turn viscous after the incubation. Thus, dilute the IVT suspension with nuclease-free H2O to made up 100 μL solutions.
-
19.
Add 1 volume of ethanol absolute into the solutions and mix them well.
-
20.
Transfer the whole solution into an RNA column in 2-mL collection tube and bind the RNA into the column-membrane by centrifugation at 8000 rcf for 2 min then discard the flow-through.
-
21.
Add 400 μL of Wash-r1 buffer into the column and centrifuge the column at 11,000 rcf for 1 min, then discard the flow-through.
-
22.
Add 80 μL DNaseI solution into the center of the column and incubate it at room temperature for 1–3 h.
-
23.
Wash the column with 400 μL of Wash-r1 buffer.
-
24.
Further wash the column with 750 μL of Wash-r2 buffer.
-
25.
Dry the column by one more centrifugation at maximum speed for 2 min.
-
26.
Elute the RNA from the column with 40 μL ultrapure nuclease-free water by ≥11,000 rcf centrifugation for 1 min.
-
27.
Assess the eluted RNA solution by miRNA assay of Qubit fluorometer, Nanodrop RNA mode, and RNA-gel electrophoresis using low molecular weight RNA marker (see Note 21).
-
28.
Prepare the mixture of tracer dye in 1 M KCl.
-
29.
Prepare shRNA for microinjection solution with the recipes described at Table 1.
-
30.
Perform microinjection following the standard procedure in Subheading 3.2 (see Note 22).
-
31.
Incubate the injected embryos in a 16 °C incubator until required for phenotype assessment.
-
32.
Confirmation of shRNA-mediated knockdown can be performed by several methods including Western blot, given the antibody for the specific gene is available; utilizing the similar antibody, immunofluorescence can also be performed; if an antibody is not available, RT-qPCR is possible; performing mRNA ectopic expression of a fluorescence protein tagged with the shRNA target sequence, co-injected with specific shRNA, and using shRNA mock as control.
-
33.
Once the shRNA-mediated knockdown in confirmed, phenotype evaluation can be performed as soon as 3–4 h after injection until 10–15 days post injection [15].
-
34.
shRNA action will still be effective after metamorphosis; hence, it can be used to delay reporting the expression of a transgene if its embryonic expression is lethal [15].
3.9 CRISPR/Cas9 Design, Microinjection , and Genotyping
-
1.
Design crRNAs using Geneious or other software.
-
2.
BLAST crRNAs against the Hydractinia genome to confirm their specificity and exclude those with multiple matches in the genome.
-
3.
Hybridize the crRNA with tracrRNA before use to create a viable short guide RNA (sgRNA) that can work with Cas9 for editing. Alternatively, obtain sgRNAs from a commercial supplier.
-
4.
Incubate crRNA:tracrRNA with Cas9 enzyme for 15 min on ice prior to use.
-
5.
Dilute Cas9 to 1 μg/μL and crRNA:tracrRNA to 500 ng/μL.
-
6.
Use the protocol for microinjection described in Subheading 3.2.
-
7.
Keep only injected animals, identified by dextran-AlexaFluor 555 co-injection.
-
8.
Raise and metamorphose all injected embryos.
-
9.
Obtain DNA samples from each individual approximately 10–14 days after metamorphosis by removing a single polyp from the colony and extracting DNA using an extraction kit. Remove excess seawater prior to beginning the extraction protocol .
-
10.
Designed primers for PCR amplification to anneal over 100 bp away from the crRNA cut sites. In rare cases, regions as large as 1000 bp immediately around a single cut site can be deleted by resection. In these cases, multiple primer sets may be necessary for understanding the extent of a deletion.
-
11.
Use a two-step PCR approach for initial amplification of DNA fragments. (denaturation = 98 °C, 3 min; denaturation = 98 °C, 30 s; annealing/extension = 68 °C, 1 min 15 s for 1–2 kb fragments).
-
12.
Use high-fidelity DNA polymerase for the first round of PCR, followed by a short A-tailing step using Taq polymerase.
-
13.
Ligate fragments into the A/T cloning vector.
-
14.
Transform into DH5-alpha E. coli bacteria.
-
15.
Plate on LB-agar plates.
-
16.
Grow overnight at 37 °C.
-
17.
Culture individual clones overnight in LB-broth with ampicillin.
-
18.
Extract plasmid DNA using the plasmid miniprep kit.
-
19.
Sequence plasmid DNA using pGEM primers for T7 or SP6 sequences.
-
20.
Due to the mosaic nature of these experiments, genotyped animals should be crossed to generate non-mosaic knockout individuals for proper characterization of phenotypes.
4 Notes
-
1.
Two crRNA can be selected to delete a specific segment in a gene to increase the probability of loss-of-function mutations.
-
2.
Hydractinia can grow at nearly any light:dark cycle; choose the one most convenient to your work cycle. Several hours of darkness followed by light trigger spawning.
-
3.
Both fresh or frozen oyster puree can be used.
-
4.
Once the embryos are collected, they can be immediately stored in 4 °C for up to 4 h to halt their development , providing a wider timeframe for injections. At room temperature, they will start dividing after 30–60 min.
-
5.
For the generation of transgenic reporter animals, 3–4 μg/μL of plasmid is mixed with 400 mM KCl in 1:5 ratio for better distribution of the plasmid, resulting in lower mosaicism.
-
6.
We use injection pressure around 40–50 psi, but this may depend on the system used and should be determined empirically.
-
7.
If the embryos are not secured in the net’s pockets, injection will be difficult as they will keep rolling around in the plate.
-
8.
Cut the needle bit by bit until a small droplet is visible. Be careful not to cut the needle too high because the solution will then come all out and be lost. When you press the petal, you should be able to see an increase in the droplet.
-
9.
One way to discriminate between injected and uninjected embryos is size: injected ones are bigger. One can also use a fluorescence tracer. Two optional tracer dyes were successfully used in Hydractinia : Dextran-AF555 (Ex/Em: 555/565) and Lucifer yellow CH (Ex/Em: 428/536). Pay attention to the emission spectrum and avoid overlapping with the fluorescent protein used in the co-injected cassettes.
-
10.
5′ URS and 3′ DRS regions are usually identified based on the coding sequence of the gene of interest and forward and reverse primers are designed empirically. Based on the ORFs and by aligning H. echinata and H. symbiolongicarpus genomes, the conserved regions are usually selected for cloning.
-
11.
5′ URS is flanked upstream by BamHI and downstream by NotI. Fluorescence protein CDS is flanked upstream by NotI and downstream by SacI, and 3′ DRS is flanked upstream by SacI and downstream by PacI.
-
12.
Miniprep commercially available kit is used for small-scale cultures in order to sequence the plasmids. Once the presence of the construct is confirmed, large-scale cultures are prepared in order to achieve high yield extracted plasmids.
-
13.
Based on the observations from various transgenic reporter animals raised in our lab, second-generation animals (G1) tend to exhibit lower—or no mosaicism. Mosaicism in G1 probably reflect epigenetic factors.
-
14.
mRNA synthesis can also be done using a PCR fragment as template. However, we recommended to construct a plasmid instead, as it will be more sustainable for long-term experimental plan. This design and plasmid construction is based on the TA-cloning system by Promega (pGEM-T-easy vector).
-
15.
One can choose between P2A or LP to separate CDS-GOI and CDS-FP. LP is a small glycine rich peptide that will allow necessary distance between FP and GOI for correct folding, thus required only if the two proteins are meant to be fused in vivo. P2A is chosen if the two proteins need to be separated in vivo. UTR is a short, AT rich sequence that is necessary for the translational machinery in Hydractinia to effectively initiate translation in vivo. The CDS-GOI can be arranged in CDS-1 or CDS-2 position.
-
16.
Restriction site 3 has to be the same as the restriction site available at the 3′ region of the plasmid insertion site and added to the 3′ end of the reverse primer for CDS-FP.
-
17.
RS-2 and P2A/LP sequences act as the overlapping region for Gibson assembly.
-
18.
Add some 20 nucleotides from the 3′ region of the insertion site of the backbone plasmid to create the overlapping region for the Gibson assembly to be successful.
-
19.
GG dinucleotides at the 5′ is required to optimize the T7 IVT reaction. GGG would be better but GG or GC also work. At 3′ region, AT richness is necessary to enable the cell distinguishing passenger from guide sequence that will be use by the endogenous Argonaut.
-
20.
BLASTN against the Hydractinia TSA database is necessary to prevent off-target effect, i.e., ensure that there is no other targets except the one intended.
-
21.
For the first synthesis of shRNA, confirmation of the shRNA on a gel is recommended. Subsequent syntheses of shRNA from the same oligonucleotide will only require miRNA Qubit measurement.
-
22.
The concentration of injected shRNA depends on the abundance of the target mRNA. For highly expressed gene such as Ef1α, it requires a concentration as high as 900 ng/μL. We have not tested higher concentrations.
References
Zapata F, Goetz FE, Smith SA, Howison M, Siebert S, Church SH, Sanders SM, Ames CL, McFadden CS, France SC, Daly M, Collins AG, Haddock SHD, Dunn CW, Cartwright P (2015) Phylogenomic analyses support traditional relationships within Cnidaria. PLoS One 10(10):e0139068. https://doi.org/10.1371/journal.pone.0139068
Technau U, Steele RE (2011) Evolutionary crossroads in developmental biology: Cnidaria. Development 138:1447–1458. https://doi.org/10.1242/dev.048959
Bosch TC, Klimovich A, Domazet-Loso T, Grunder S, Holstein TW, Jekely G, Miller DJ, Murillo-Rincon AP, Rentzsch F, Richards GS, Schroder K, Technau U, Yuste R (2017) Back to the basics: cnidarians start to fire. Trends Neurosci 40(2):92–105. https://doi.org/10.1016/j.tins.2016.11.005
Chang ES, Neuhof M, Rubinstein ND, Diamant A, Philippe H, Huchon D, Cartwright P (2015) Genomic insights into the evolutionary origin of Myxozoa within Cnidaria. Proc Natl Acad Sci U S A 112(48):14912–14917. https://doi.org/10.1073/pnas.1511468112
Holstein TW, Hobmayer E, Technau U (2003) Cnidarians: an evolutionarily conserved model system for regeneration? Dev Dyn 226:257. https://doi.org/10.1002/dvdy.10227
DuBuc TQ, Traylor-Knowles N, Martindale MQ (2014) Initiating a regenerative response; cellular and molecular features of wound healing in the cnidarian Nematostella vectensis. BMC Biol 12(1):24. https://doi.org/10.1186/1741-7007-12-24
Vogg MC, Galliot B, Tsiairis CD (2019) Model systems for regeneration: Hydra. Development 146(21):dev177212. https://doi.org/10.1242/dev.177212
Chera S, Ghila L, Dobretz K, Wenger Y, Bauer C, Buzgariu W, Martinou J-C, Galliot B (2009) Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell 17(2):279–289
Bradshaw B, Thompson K, Frank U (2015) Distinct mechanisms underlie oral vs aboral regeneration in the cnidarian Hydractinia echinata. eLife 4:e05506. https://doi.org/10.7554/eLife.05506
Renfer E, Amon-Hassenzahl A, Steinmetz PRH, Technau U (2009) A muscle-specific transgenic reporter line of the sea anemone, Nematostella vectensis. Proc Natl Acad Sci 107(1):104–108. https://doi.org/10.1073/pnas.0909148107
Künzel T, Heiermann R, Frank U, Müller WA, Tilmann W, Bause M, Nonn A, Helling M, Schwarz RS, Plickert G (2010) Migration and differentiation potential of stem cells in the cnidarian Hydractinia analysed in GFP-transgenic animals and chimeras. Dev Biol 348:120–129
Klimovich A, Wittlieb J, Bosch TCG (2019) Transgenesis in Hydra to characterize gene function and visualize cell behavior. Nat Protoc 14(7):2069–2090. https://doi.org/10.1038/s41596-019-0173-3
Rentzsch F, Renfer E, Technau U (2020) Generating transgenic reporter lines for studying nervous system development in the Cnidarian Nematostella vectensis. Methods Mol Biol 2047:45–57. https://doi.org/10.1007/978-1-4939-9732-9_3
Buss LW, Yund PO (1989) A sibling species group of Hydractinia in the north-eastern United States. J Mar Biol Assoc UK 69(4):857–874. https://doi.org/10.1017/S0025315400032215
DuBuc TQ, Schnitzler CE, Chrysostomou E, McMahon ET, Febrimarsa, Gahan JM, Buggie T, Gornik SG, Hanley S, Barreira SN, Gonzalez P, Baxevanis AD, Frank U (2020) Transcription factor AP2 controls cnidarian germ cell induction. Science 367(6479):757–762. https://doi.org/10.1126/science.aay6782
Frank U, Nicotra ML, Schnitzler CE (2020) The colonial cnidarian Hydractinia. EvoDevo 11(1):7. https://doi.org/10.1186/s13227-020-00151-0
Wittlieb J, Khalturin K, Lohmann JU, Anton-Erxleben F, Bosch TCG (2006) From the cover: transgenic Hydra allow in vivo tracking of individual stem cells during morphogenesis. PNAS 103(16):6208–6211. https://doi.org/10.1073/pnas.0510163103
Quiroga Artigas G, Lapébie P, Leclère L, Takeda N, Deguchi R, Jékely G, Momose T, Houliston E (2018) A gonad-expressed opsin mediates light-induced spawning in the jellyfish Clytia. eLife 7:e29555. https://doi.org/10.7554/eLife.29555
Jones VAS, Bucher M, Hambleton EA, Guse A (2018) Microinjection to deliver protein, mRNA, and DNA into zygotes of the cnidarian endosymbiosis model Aiptasia sp. Sci Rep 8(1):16437. https://doi.org/10.1038/s41598-018-34773-1
Millane RC, Kanska J, Duffy DJ, Seoighe C, Cunningham S, Plickert G, Frank U (2011) Induced stem cell neoplasia in a cnidarian by ectopic expression of a POU domain transcription factor. Development 138(12):2429–2439. https://doi.org/10.1242/dev.064931
Flici H, Schnitker N, Millane RC, Govinden G, Houlihan A, Boomkamp SD, Shen S, Baxevanis AD, Frank U (2017) An evolutionarily conserved SoxB-Hdac2 crosstalk regulates neurogenesis in a cnidarian. Cell Rep 18:1395–1409. https://doi.org/10.1016/j.celrep.2017.01.019
Karadge UB, Gosto M, Nicotra ML (2015) Allorecognition proteins in an invertebrate exhibit homophilic interactions. Curr Biol 25:2845–2850. https://doi.org/10.1016/j.cub.2015.09.030
Steven MS, Zhiwei M, Julia MH, Brooke MR, Gregory AG, Alan MW, Hakima F, Uri F, Christine ES, Andreas DB, Matthew LN (2018) CRISPR/Cas9-mediated gene knockin in the hydroid Hydractinia symbiolongicarpus. BMC Genomics 19(1) https://doi.org/10.1186/s12864-018-5032-z
Kanska J, Frank U. Novel roles for Nanos in neural cell fate determination revealed by studies in a cnidarian. J Cell Sci https://doi.org/10.1242/jcs.127233
Acknowledgments
Research in the Frank lab is supported by the SFI-HRB-Wellcome Research Partnership (grant No. 210722/Z/18/Z) and by National Science Foundation (grant No. 1827635).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Chrysostomou, E., Febrimarsa, DuBuc, T., Frank, U. (2022). Gene Manipulation in Hydractinia . In: Blanchoud, S., Galliot, B. (eds) Whole-Body Regeneration. Methods in Molecular Biology, vol 2450. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2172-1_22
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2172-1_22
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2171-4
Online ISBN: 978-1-0716-2172-1
eBook Packages: Springer Protocols