
Abstract
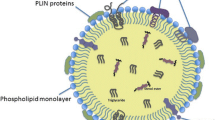
Lipid droplets have a unique structure among organelles consisting of a dense hydrophobic core of neutral lipids surrounded by a single layer of phospholipids decorated with various proteins. Often labeled merely as passive fat storage repositories, they in fact have a remarkably dynamic life cycle. Being formed within the endoplasmic reticulum membrane, lipid droplets rapidly grow, shrink, traverse the cytosol, and engage in contacts with other organelles to exchange proteins and lipids. Their lipid and protein composition changes dynamically in response to cellular states and nutrient availability. Remarkably, their biogenesis is induced when cells experience various forms of nutrient, energy, and redox imbalances, including lipid excess and complete nutrient deprivation. Cancer cells are continuously exposed to nutrient and oxygen fluctuations and have the capacity to switch between alternative nutrient acquisition and metabolic pathways in order to strive even during severe stress. Their supply of lipids is ensured by a series of nutrient uptake and scavenging mechanisms, upregulation of de novo lipid synthesis, repurposing of their structural lipids via enzymatic remodeling, or lipid recycling through autophagy. Importantly, most of these pathways of lipid acquisition converge at lipid droplets, which combine different lipid fluxes and control their usage based on specific cellular needs. It is thus not surprising that lipid droplet breakdown is an elaborately regulated process that occurs via a complex interplay of neutral lipases and autophagic degradation. Cancer cells employ lipid droplets to ensure energy production and redox balance, modulate autophagy, drive membrane synthesis, and control its composition, thereby minimizing stress and fostering tumor progression. As regulators of (poly)unsaturated fatty acid trafficking, lipid droplets are also emerging as modulators of lipid peroxidation and sensitivity to ferroptosis. Clearly, dysregulated lipid droplet turnover may also be detrimental to cancer cells, which should provide potential therapeutic opportunities in the future. In this review, we explore how lipid droplets consolidate lipid acquisition and trafficking pathways in order to match lipid supply with the requirements for cancer cell survival, growth, and metastasis.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
The recently revived interest in cancer metabolism has resulted in the recognition of metabolic reprogramming as one of the major cancer hallmarks (Hanahan and Weinberg 2011). Moving forward from glucose and the classical Warburg effect, recent discoveries have shown that the metabolism of amino acids and lipids is also critical for tumorigenesis (Pavlova and Thompson 2016; Röhrig and Schulze 2016; Ward and Thompson 2012). Additionally, we are now aware that different tumors, and even cells within an individual tumor, display specific metabolic characteristics but also a remarkable metabolic plasticity that enables their adaptation to adverse conditions and drives their malignant potential (Hanahan and Weinberg 2011). However, even genetically distinct cancer types encounter similar stress conditions in the tumor microenvironment and may thus have common metabolic vulnerabilities that present unique therapeutic opportunities (Martinez-Outschoorn et al. 2017). In our quest for new cancer treatments, it is therefore imperative to discover the context-specific responses of cancer cells to nutrient and oxidative fluctuations and thereby expose their metabolic weaknesses.
The roles of lipids in cancer extend well beyond their typically ascribed roles in membrane biogenesis and energy production (Beloribi-Djefaflia et al. 2016; Röhrig and Schulze 2016). In fact, even these seemingly simple roles, as membrane building blocks and energy-rich substrates, are far from being understood at the molecular and functional level. Moreover, we are only beginning to understand the distinct functions of individual species within the enormous variety of lipids and the intricacies of their collective effects in cell metabolism and signaling. The roles of individual lipids are intrinsically tied to the cooperative nature of lipid assemblies, whose function depends on their specific lipid composition and its dynamic changes at particular subcellular locations. Lipid droplets are emerging as novel regulators of many of these processes. These unique and remarkably dynamic organelles respond to nutrient fluctuations and various microenvironmental stress conditions to control the trafficking, storage, and use of lipids for a variety of purposes in the cell (Farese and Walther 2009; Jarc and Petan 2020; Koizume and Miyagi 2016; Krahmer et al. 2013; Olzmann and Carvalho 2019; Petan et al. 2018). They are readily available sources of fatty acids (FAs), sterols, and vitamins that are rapidly released on demand and under specific conditions. These lipids and their metabolites participate in and regulate multiple metabolic and signaling pathways within the cell and in the extracellular space, thereby affecting major cancer hallmarks, including cell growth, proliferation, metabolism, migration, inflammation, and immunity (Attané and Muller 2020; den Brok et al. 2018; Cruz et al. 2020; Currie et al. 2013; Koizume and Miyagi 2016; Petan et al. 2018; Tirinato et al. 2017). Moreover, lipid droplets also participate in the cellular trafficking and quality control of proteins, thereby affecting protein turnover, gene transcription, nuclear function, and various homeostatic and stress responses. Lipid droplets even manage the secretion of proteins that act as danger signals and activate immune cell responses and inflammatory pathways (Veglia et al. 2017; Jarc and Petan 2020). These fat-laden organelles also affect drug efficacy by altering the cellular distribution and activation of lipophilic anti-cancer agents (Dubey et al. 2020; Englinger et al. 2020).
Alterations in lipid droplet metabolism are emerging as important parts of cancer metabolic reprogramming. Their biogenesis and breakdown may either help cancer cells in their constant fight against stress or promote their demise. In this review, we focus on the mechanisms that govern lipid droplet function in response to nutrient and oxygen imbalances. We explore how these highly dynamic organelles consolidate lipid uptake, synthesis, recycling, distribution, and breakdown in order to match these entangled lipid fluxes with the requirements for cancer cell survival, growth, and metastasis.
2 Lipid Droplets Are Dynamic Organelles
2.1 Lipid Droplets Are Versatile Ensembles of Lipids and Proteins
Lipid droplets have a unique structure among organelles with a hydrophobic core consisting of neutral lipids surrounded by a single layer of phospholipids decorated with various proteins (Henne et al. 2018; Olzmann and Carvalho 2019; Walther et al. 2017). Their neutral lipid core stores lipids primarily in their esterified, storage form, e.g., FAs as triacylglycerols (TAGs), cholesterol and other sterols in the form of sterol esters, retinoic acids as retinyl esters, and ceramides esterified into acyl ceramides (Jarc and Petan 2020; Molenaar et al. 2017; Senkal et al. 2017; Thiam and Beller 2017). Lipid droplets from different cells and tissues may display significant differences in the relative proportions of these major lipid species, often reflecting tissue-specific functions and storage requirements. By regulating the storage and release of these various lipids, lipid droplets have a direct impact on their involvement in processes essential for cell survival, growth, and proliferation, including energy production, membrane and organelle biogenesis, cell signaling, and gene transcription.
The lipid droplet proteome in mammalian cells contains approximately 150 proteins and includes proteins involved in lipid metabolism and signaling, redox metabolism, autophagy, gene transcription, ubiquitination, membrane trafficking, and immunity (Bersuker and Olzmann 2017; Bersuker et al. 2018). Many among these lipid droplet-associated proteins have unknown functions, whereas some have known roles in processes as yet unrelated to lipids or lipid droplets. In most cases, the functional importance of their lipid droplet localization is unknown. Furthermore, in some instances, the sequestration of proteins to the lipid droplet surface is a mechanism of control of their involvement in processes occurring at other cellular locations. For example, lipid droplets sequester histones, transcription factors (e.g., NFAT5), and chaperones (e.g., Hsc70 and calreticulin), thereby affecting gene transcription, protein quality control, and immune cell function (Cotte et al. 2018; Gallardo-Montejano et al. 2016; Johnson et al. 2018; Ueno et al. 2013; Veglia et al. 2017; Welte and Gould 2017).
Importantly, the lipid and protein composition of lipid droplets, as well as their size, number, localization, and mobility in the cell, change rapidly in response to cellular states and nutrient availability (Bosch et al. 2020; Herms et al. 2013, 2015; Thiam and Beller 2017). For example, a surge of FAs leads to a rapid activation of TAG synthesis and lipid droplet biogenesis in most cells. This process occurs with a remarkable efficiency within seconds to minutes following FA exposure, whereby the latter may be incorporated into both pre-existing lipid droplets and/or into newly emerging ones (Kassan et al. 2013; Kuerschner et al. 2008). On the contrary, FA and glucose depletion leads to rapid mobilization and redistribution of lipid droplets in the cell, thereby increasing their contacts with the mitochondrial network to couple lipolytic FA release from stored TAGs with mitochondrial FA intake and energy production (Herms et al. 2015; Rambold et al. 2015). However, paradoxically, mitochondria–lipid droplet contacts may also drive TAG synthesis and lipid droplet expansion (Benador et al. 2019). As discussed in this review, the highly dynamic nature of lipid droplet metabolism and its interactions with other organelles endows cells with multiple layers of flexibility, which is often exploited by cancer cells for protection against various stresses.
2.2 Lipid Droplet Biogenesis Occurs at the Crossroads of Membrane and Neutral Lipid Metabolism
The life cycle of the lipid droplet is tightly linked to its mother organelle, the endoplasmic reticulum (ER). TAG synthesis is a prerequisite for de novo lipid droplet formation and occurs between the two leaflets of the ER membrane by sequential addition of FAs to a glycerol backbone, catalyzed by a series of acyltransferase enzymes (Coleman and Mashek 2011). Importantly, the first several steps of the process are common to both phospholipid and TAG synthesis, enabling the cell to rapidly switch between phospholipid and neutral lipid production. This is essential for many aspects of the cellular stress response because it allows, for example, a shift from cell growth and proliferation during nutrient abundance, when the needs for membrane biogenesis are high, to quiescence during starvation, when lipids are syphoned into storage for later use (Bosch et al. 2020; Henne et al. 2018; Natter and Kohlwein 2013). The dephosphorylation of phosphatidic acid into diacylglycerol (DAG) by phosphatidate phosphatases, also called lipins (Zhang and Reue 2017), is the branching-off point between these two pathways and is immediately followed by the last step in TAG biosynthesis: the conversion of DAG into TAG catalyzed by diacylglycerol acyltransferases (DGATs). Cholesteryl ester synthesis also occurs within the ER membrane and is mediated by acyl-coenzyme A:cholesterol acyltransferase (ACAT) enzymes (Chang et al. 2009).
The newly synthesized neutral lipids accumulate in growing lipid “lenses” within the bilayer, eventually giving rise to nascent lipid droplets that bud from the ER membrane and are released into the cytosol (Salo and Ikonen 2019). The budding process is guided by proteins recruited to the nascent droplet, such as the ER membrane protein seipin that is essential for stabilization and growth of the droplet, and requires a particular rearrangement of membrane lipids that drives membrane bending and asymmetrical budding into the cytosol (Chorlay et al. 2019; Henne et al. 2018; Olzmann and Carvalho 2019; Thiam and Beller 2017). Several pathways of phospholipid synthesis and remodeling may contribute to these lipid rearrangements and enable membrane expansion to provide sufficient cover for the growing lipid droplet (Bosch et al. 2020; Penno et al. 2012). Remarkably, some components of the lipid droplet biogenesis machinery required for phospholipid and neutral lipid synthesis are transferred to the nascent lipid droplet and enable its growth independently of the ER (Krahmer et al. 2011; Wilfling et al. 2013). However, lipid droplets may also grow by fusion, and they form transient contacts with the ER, mitochondria, and other organelles, via protein tethers and membrane bridges, thereby allowing bidirectional lipid and protein transfer (Barbosa and Siniossoglou 2017; Bohnert 2020; Schuldiner and Bohnert 2017).
2.3 Lipid Droplet Breakdown Occurs via Lipolysis or Lipophagy
When cells are exposed to nutrient imbalances that lead to a deficit in lipids, lipid droplet breakdown is activated to provide lipids for essential processes (Bosch et al. 2020). At the organismal level, lipid droplet breakdown in adipocytes is hormonally regulated and provides FAs for mitochondrial energy production in non-adipose tissue during fasting and exercise (Haemmerle et al. 2011; Young and Zechner 2013; Zimmermann et al. 2004). However, lipid droplets in most tissues also undergo a dynamic cycle of biogenesis and breakdown in response to hormonal signals and nutrient cues from the environment (Bosch et al. 2020; Jarc and Petan 2019). Intriguingly, upon entry into target cells and tissues, adipose-derived FAs are incorporated into lipid droplets, which become the major platforms that regulate their subsequent use and distribution in the cell (Bosch et al. 2020; Zechner et al. 2012). For example, in the heart, liver, and most other tissues, lipid droplets provide FAs that not only drive mitochondrial energy production, but act as signals that activate transcriptional networks, such as the those mediated by the peroxisome proliferator-activated receptors (PPARs), that are necessary for proper coupling of FA supply with mitochondrial biogenesis, function, and oxidative capacity in the cell (Haemmerle et al. 2011).
Lipid droplet breakdown occurs via two major mechanisms: lipolysis and lipophagy (Currie et al. 2013; Petan et al. 2018; Schulze et al. 2017; Young and Zechner 2013; Zechner et al. 2017). Lipolysis is mediated by cytosolic (neutral) lipases that enable a highly regulated release of FAs from TAGs. Adipose triglyceride lipase (ATGL) is the major TAG lipase in most mammalian cells and catalyzes the first step in TAG lipolysis (Schreiber et al. 2019; Smirnova et al. 2005; Zimmermann et al. 2004), which is followed by the sequential action of hormone-sensitive lipase (HSL) and monoacylglycerol lipase (MAGL) (Grabner et al. 2017). In certain conditions, lipid droplet breakdown also occurs by lipophagy, a form of selective (macro)autophagy that delivers parts of or whole lipid droplets to lysosomes for bulk degradation by hydrolytic enzymes, such as the TAG and cholesteryl ester hydrolase lysosomal acid lipase (LAL) (Schulze et al. 2017; Singh et al. 2009; Zechner et al. 2017).
In principle, while lipolysis generally leads to lipid droplet shrinkage, lipophagy provides a means of complete breakdown of all lipids and proteins within the droplet into basic building blocks, suggesting that each mechanism may serve a distinct purpose in the cell (Ogasawara et al. 2020; Petan et al. 2018; Schulze et al. 2017; Zechner et al. 2017). Lipolysis and lipophagy are regulated by common and complementary signaling pathways, and cells seem to preferentially use one or the other depending on cell type, nutrient status, and current requirements, although concurrent or sequential occurrence is also possible. Indeed, these two mechanisms of lipid droplet breakdown display a considerable crosstalk, whereby the activation of lipolysis may stimulate autophagy/lipophagy, but autophagy may also be activated in a compensatory manner upon inhibition of lipolysis (Goeritzer et al. 2015; Ogasawara et al. 2020; Peng et al. 2016). In addition, chaperone-mediated autophagy may facilitate lipolysis by removing the lipid droplet-coating proteins perilipins 2 and 3 (Kaushik and Cuervo 2015). The main drivers and functions of this intricate interplay of lipid droplet breakdown mechanisms in various cell types and microenvironmental contexts are only beginning to be uncovered (Ogasawara et al. 2020).
3 Lipid Droplets Are at the Core of Cancer Metabolic Reprogramming
3.1 Cancer Cells Use Ingenious Ways of Lipid Acquisition That Converge at the Lipid Droplet
Some of the earliest studies implicating lipids in cancer have shown that aggressive cancers display elevated rates of de novo FA synthesis, revealing that tumors may satisfy their requirements for lipids independently of uptake from the circulation (Menendez and Lupu 2007; Röhrig and Schulze 2016). Ever since, numerous studies have also suggested the involvement of other branches of FA, cholesterol, phospholipid, and neutral lipid metabolism in neoplastic transformation, disease progression, and drug resistance (Carracedo et al. 2013; Currie et al. 2013; Hernández-Corbacho and Obeid 2018; Menendez and Lupu 2007; Petan et al. 2018; Snaebjornsson et al. 2019). Although the first inhibitors of FA synthesis have entered clinical development only recently, some intrinsic drawbacks of targeting this pathway have already been revealed (Röhrig and Schulze 2016).
Namely, cancer cells that have access to lipids from the circulation are resistant to inhibition of FA synthesis, since they may increase lipid uptake to compensate for the lack of endogenous lipogenesis (Martinez-Outschoorn et al. 2017; Röhrig and Schulze 2016; Snaebjornsson et al. 2019). Inhibitors of lipogenesis are also ineffective in cancer cells exposed to hypoxia and nutrient deprivation, because lipogenesis is already blocked under these conditions and cells switch to lipid acquisition from their immediate microenvironment (Ackerman and Simon 2014; Petan et al. 2018). Remarkably, cancer cells engage in opportunistic modes of extracellular nutrient acquisition to satisfy their needs for lipids, amino acids, and carbohydrates by scavenging exosomes, extracellular matrix proteins, and albumin and even engulfing necrotic cell debris and entire living cells (Commisso et al. 2013; Finicle et al. 2018; Jayashankar and Edinger 2020; Kamphorst et al. 2013; Kim et al. 2018; Michalopoulou et al. 2016).
Cancer cells also enter in symbiotic relationships with neighboring cells, including tumor-associated adipocytes, whereby lipid droplet lipolysis in adipocytes provides FAs for energy production in cancer cells (Attané and Muller 2020; Hoy et al. 2017; Nieman et al. 2010; Wang et al. 2017). Furthermore, recent studies have shown that even when extracellular sources of lipids are exhausted, stressed cells may have access to additional endogenous lipid pools. These include lipids that can be recommissioned from their own structural and storage pools via several possible routes, including membrane phospholipid hydrolysis (e.g., by phospholipases A2), autophagic degradation of organelles, and the breakdown of neutral lipids stored within cytosolic lipid droplets (Ackerman et al. 2018; Jarc et al. 2018; Lue et al. 2017; Nguyen et al. 2017; Petan et al. 2018; Pucer et al. 2013; Rambold et al. 2015).
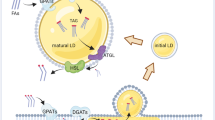
Intriguingly, most if not all of these pathways of lipid acquisition converge at the lipid droplet (Fig. 1). Lipid droplets are perfectly positioned within the metabolic scheme of the cell to control both the acquisition of lipids (from the various internal or external sources mentioned above) and their utilization for various purposes and depending on specific cellular needs. Although lipid droplets are often regarded merely as transient repositories for the trafficking lipids on route to their final destination – and certainly there will be cases when this is true – the syphoning of various lipid fluxes into lipid droplets is in fact required for numerous homeostatic cell functions and, in particular, for the cellular stress response. One of the earliest and most notable examples was reported in cardiomyocytes (Haemmerle et al. 2011). Namely, while extracellular FAs may enter the cell in various ways and bind to different proteins in the cytosol, including nuclear transcription factors, they must first be incorporated into TAGs within lipid droplets and then released by lipolysis in order to bind to and activate PPAR-mediated gene transcription that drives mitochondrial biogenesis and oxidative metabolism in these cells. This seemingly futile cycle of FA esterification and lipolytic release reveals one of the hallmark principles of lipid droplet biology, whereby the organelle acts as a focal point that coordinates lipid flux with metabolic and signaling pathways essential for cell function and resistance to stress (Fig. 1) (Jarc and Petan 2020; Khan et al. 2015; Mottillo et al. 2012; Ong et al. 2011; Zechner et al. 2012).

Lipid droplets integrate lipid uptake and usage pathways in cancer cells. Based on the context and current conditions, cancer cells may use several lipid acquisition pathways, which all converge at the lipid droplet. Lipid droplets act as buffers that consolidate the various lipid fluxes and finely tune their release and distribution in the cell to drive essential processes that control cancer cell fate
Similarly, cancer cells exposed to extracellular FAs form lipid droplets that in turn regulate mitochondrial redox metabolism to increase NADPH production and protect cancer cells from hypoxic damage (Bensaad et al. 2014). Lipid droplets are also formed in breast and ovarian cancer cells exposed to lipids derived from neighboring adipocytes and provide a consistent supply of FAs that drives FA oxidation, sustains metabolic reprogramming, and promotes tumor aggressiveness (Nieman et al. 2010; Wang et al. 2017). Moreover, lipid droplet biogenesis is also activated when exogenous lipids are limiting but endogenous lipids are present in excess, such as following autophagic breakdown of membranous organelles, in order to finely tune their uptake by mitochondria, thereby preventing mitochondrial damage and ensuring efficient energy production (Herms et al. 2015; Nguyen et al. 2017; Rambold et al. 2015). In this review, we discuss these and related studies that describe the various essential roles of lipid droplets in the response of cancer cells to stress and their ability to regulate downstream lipid fluxes depending on cellular requirements.
3.2 Lipid Droplets and Nutrient Scavenging
To ensure their survival and promote growth in a nutrient-poor environment, cancer cells use multiple nutrient scavenging strategies to obtain various macromolecules and break them down to their basic constituents in the lysosome, thereby ensuring the supply of energy substrates and anabolic building blocks (Finicle et al. 2018). Some cancer cell types, in particular those driven by oncogenic mutations in the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways (Commisso et al. 2013; Jayashankar and Edinger 2020; Kamphorst et al. 2013; Kim et al. 2018; Palm et al. 2015), use macropinocytosis, a non-selective endocytotic uptake mechanism of different material, including extracellular fluid, proteins, vesicles, and cellular debris (Finicle et al. 2018; Jayashankar and Edinger 2020; Kim et al. 2018). Macropinocytosis is supported by activation of AMP-activated protein kinase (AMPK) and inhibition of mammalian target of rapamycin (mTOR) pathways; it promotes cancer cell proliferation and confers resistance to therapies that target cancer anabolism.
Remarkably, macropinocytosis enables the extraction of amino acids, nucleotides, and FAs even from dying cell corpses, a process termed necrocytosis (Jayashankar and Edinger 2020; Kim et al. 2018). Necrocytosis has been shown to help amino acid-deprived prostate cancer cells maintain lipid droplet levels, but it remained unknown if extracellular lipids are de facto scavenged from cell debris (Kim et al. 2018). Indeed, some of the other types of acquired nutrients could provide energy and building blocks for essential cellular processes, thereby sparing existing lipid droplets. It was shown recently by tracing experiments that necrotic debris-derived FAs are indeed incorporated into breast cancer cells, thereby reducing their dependence on de novo FA synthesis and rendering them insensitive to inhibitors of FA synthase (Fig. 2a) (Jayashankar and Edinger 2020). These studies hint at the possibility that lipid droplets act as transient buffers for lipids taken up via macropinocytosis. It will be interesting to see in future studies whether FA release from lipid droplets is responsible for the observed reduced dependence on FA synthesis. Given their similar role in cells exposed to FA surges from lysosomal breakdown via autophagy (Nguyen et al. 2017; Rambold et al. 2015), it is possible that lipid droplets serve as central lipid buffering and distribution hubs that carefully balance lipid input with the requirements of these “voracious,” macropinocytic cancer cells.

Lipid droplets, lipid fluxes, and cancer cell fate. (a) Macropinocytosis of extracellular material, including necrotic cell debris and extracellular vesicles (EVs), provides amino acids, nucleotides, and lipids for cancer cell survival and resistance to drugs that target anabolic pathways, including inhibitors of FA synthesis; the macropinocytosis-derived FAs are incorporated into lipid droplets, whose role in mediating the effects of FAs is not yet clear. (b) Lipid droplets are important repositories of unsaturated FAs that are used by cancer cells to maintain proper membrane saturation and prevent endoplasmic reticulum (ER) stress, particularly when demands for lipids are elevated, such as during Ras oncogene- and mTOR signaling-driven rapid cell growth, or when the synthesis of unsaturated lipids is compromised, e.g., due to hypoxia-induced inhibition of stearoyl-CoA desaturase-1 (SCD-1); during hypoxic stress, lipid droplets also drive mitochondrial oxidative metabolism to provide energy and reducing equivalents that reduce oxidative stress. (c) During nutrient replete conditions, when mTOR is active, lipid uptake and de novo FA synthesis drive both membrane synthesis and lipid droplet biogenesis; when lipids become limiting, lipid droplets support membrane synthesis, thereby sustaining cell growth and proliferation. Upon nutrient depletion, cells experience a fall in energy levels, leading to AMPK activation, which in turn blocks de novo lipogenesis and stimulates rapid lipid droplet dispersion to mitochondrial contact sites; AMPK also promotes the lipolytic release and transfer of FAs into mitochondria for oxidation, thereby restoring energy levels and the redox balance through ATP and NADPH production. (d) Distinct populations of mitochondria and lipid droplets may engage in opposing purposes in the same cell: mitochondria, tightly anchored to lipid droplets, provide citrate, ATP, and NADPH to support FA and TAG synthesis driving lipid droplet formation, whereas “free,” cytosolic mitochondria dynamically interact with lipid droplets to take up and oxidize FAs. (e) In the tumor microenvironment, cancer cells take up FAs and EVs released by neighboring adipocytes and store them in lipid droplets, whose breakdown via (1) lipophagy or (2, 3) lipolysis drives mitochondrial energy production, thereby promoting tumor growth and invasion. Under these lipid-rich conditions, AMPK supports lipolysis, lipophagy, and mitochondrial FA oxidation, which may be (3) coupled to or (2) uncoupled from ATP production via uncoupling protein 2 (UCP2); this uncoupling is instigated by the influx of lipid droplet-derived fatty acids and drives a feedback circuit that sustains AMPK activation. (f) In cells exposed to amino acid starvation or to inhibitors targeting the PI3K/Akt/mTOR pathway, mTORC1 is inhibited leading to activation of autophagy, which breaks down membranous organelles to release FAs that trigger lipid droplet biogenesis; rapid lipid droplet biogenesis protects mitochondria from excess FAs; lipid droplets provide an efficient way to gradually deliver FAs via ATGL-mediated lipolysis into fused mitochondria and enable cell survival during starvation; the process is supported by AMPK, which sustains autophagic flux and oxidative metabolism; the hypoxia-inducible lipid droplet-associated protein (HILPDA), an endogenous inhibitor of ATGL, is upregulated in response to autophagy-driven lipid droplet biogenesis, and it may participate in the fine regulation of lipolysis to prevent oxidative stress and lipotoxicity
3.3 Lipid Droplets Maintain Membrane Unsaturation During Stress
Rapidly proliferating cancer cells rely on several oncogenic signal transduction pathways that activate mTOR signaling to maintain high levels of protein and lipid synthesis, which are prerequisites for cell growth and proliferation (Liu and Sabatini 2020). The mTOR pathway is activated in response to amino acid availability and drives cell growth by stimulating numerous anabolic pathways, including protein translation and nucleotide synthesis. It also promotes FA, cholesterol, and glycerolipid synthesis via the sterol regulatory element-binding protein (SREBP) transcription factors (Yecies and Manning 2011). This strong anabolic drive requires a coordination between nutrient availability, metabolic pathways, and the various oncogene-driven mitogenic signals. The survival of cancer cells is thus compromised when biosynthetic pathways, such as lipid and protein production, are not synchronized.
For example, in cancer cells exposed to limited oxygen availability, the conversion of palmitate, the principal product of de novo FA synthesis, into unsaturated FAs is compromised due to inactivation of the oxygen-dependent lipid desaturase stearoyl-coenzyme A desaturase 1 (SCD1) (Fig. 2b) (Kamphorst et al. 2013; Scaglia et al. 2009). Under these conditions, constitutive mTOR activity causes an imbalance between the elevated protein synthesis and the lagging membrane expansion, which ultimately leads to ER stress and cell death (Young et al. 2013). Consequently, these cells become dependent on the uptake of unsaturated FAs from extracellular sources in order to compensate for the diminished desaturase activity and restore the balance between protein and lipid synthesis (Ackerman and Simon 2014; Young et al. 2013). Even in normoxic conditions, elevated Ras oncogene signaling, which imposes a potent growth impetus to cancer cells by activating the MAPK pathway and mTOR complex 1 (mTORC1) signaling, drives the uptake of serum lysophospholipids as sources of unsaturated FAs to reduce the dependence of cancer cells on SCD1 activity (Kamphorst et al. 2013). Intriguingly, upregulated lysophospholipid uptake in cancer cells with Ras oncogenic mutations leads to increased lipid droplet storage (Fig. 2b) (Qiao et al. 2020). The latter is in turn coupled to elevated FA oxidation and improved redox metabolism that promotes tumor aggressiveness in vitro and in vivo, indicating that lipid droplets might mediate the effects of exogenous lysophospholipids in aggressive Ras-driven tumors.
Clearly, the provision of unsaturated FAs is critical for cancer cell survival and growth. There is accumulating evidence that lipid droplets are important sources and regulators of unsaturated FA trafficking. Indeed, recent studies in kidney cancer have found that lipid droplets play an important role in the maintenance of membrane unsaturation levels during hypoxia (Ackerman et al. 2018; Qiu et al. 2015). Constitutive hypoxia-inducible factor (HIF) signaling and abundant lipid storage are hallmarks of clear-cell renal cell carcinoma (ccRCC). It was found that HIF2α-dependent lipid droplet accumulation protects ccRCC cells from ER stress, thereby promoting cell proliferation and xenograft tumor growth (Qiu et al. 2015). Intriguingly, even in cells depleted of HIF2α, overexpression of the lipid droplet-coating protein PLIN2 is sufficient to restore lipid storage and protect from ER stress, which occurs at least in part due to mTOR-driven protein synthesis. Furthermore, it was found recently that lipid droplets formed in nutrient-replete ccRCC cells are rich in serum-derived unsaturated FAs and are gradually broken down when cells are exposed to low serum and oxygen stress (Ackerman et al. 2018). This delayed lipolytic release of unsaturated FAs is dependent on HSL activity and is responsible for replacing saturated acyl chains in cell membranes and prevention of ER stress (Fig. 2b). Concurrently, lipid droplets reduce the dependence on de novo FA synthesis, revealing that targeting lipid droplet biogenesis, e.g., via inhibition of DGATs, may be a more relevant therapeutic target than FA synthesis in ccRCC (Ackerman et al. 2018).
The dependence of cancer cells on the supply of unsaturated FAs from lipid droplets for long-term maintenance of membrane homeostasis and protection against ER stress is very likely not limited to kidney cancer. In rapidly proliferating yeast cells, lipid droplet turnover is essential for providing a balanced supply of saturated and unsaturated FAs for membrane synthesis (Natter and Kohlwein 2013; Petschnigg et al. 2009; Zanghellini et al. 2008), hinting at a conserved, essential function of lipid droplets across the eukaryotic kingdom. Lipid droplets are unique in their ability to consolidate different FA fluxes and regulate their input into phospholipid synthesis and remodeling pathways that are necessary for membrane homeostasis. Collectively, these studies suggest that lipid droplets are important repositories of unsaturated FAs that may be utilized by cancer cells to maintain membrane and organelle function particularly when demands for lipids are elevated, such as during oncogene-driven rapid cell growth, or when the synthesis of unsaturated lipids is compromised, e.g., due to hypoxia.
3.4 Lipid Droplets Match Nutrient Fluctuations with Cell Growth and Survival
Lipid droplet biogenesis and turnover are dynamically altered in response to changes in nutrient and energy status. Recent studies have significantly increased our understanding of the integration of lipid droplet turnover in the general cellular response to nutrient imbalances (Bosch et al. 2020), but new evidence is also emerging regarding their roles in the context of metabolic reprogramming in cancer. Cancer cells often have constitutively activated pathways of nutrient sensing and uptake and display oncogene-driven, growth factor-independent signaling that stimulates cell growth and survival irrespective of nutrient levels. AMPK and mTOR are two major intracellular kinases that reciprocally regulate adaptive cellular responses to nutrient stress and cell growth. They sense metabolite availability, energy and stress levels and integrate these signals with those coming from growth factor and oncogene-driven pathways (González et al. 2020; Liu and Sabatini 2020; Palm and Thompson 2017). AMPK detects glucose and energy levels and responds to starvation by inhibiting anabolic pathways and cell growth and activating catabolic pathways to restore the energy balance. AMPK blocks de novo FA, cholesterol, and TAG synthesis; it activates lipolysis and FA oxidation and engages gene transcription programs responsible for mitochondrial biogenesis and oxidative metabolism (Hardie et al. 2012; Muoio et al. 1999; Wendel et al. 2009; Zechner et al. 2017). The amino acid-sensitive complex mTORC1 is positively regulated by the PI3K/Akt and MAPK pathways to promote cell growth and survival and is inactivated when amino acids are limiting. Because AMPK negatively regulates mTORC1, energy or glucose depletion also inhibits mTORC1 activity; however, amino acid deficiencies do not activate AMPK. Both kinases are often dysregulated in cancer, thereby allowing cancer cells to evade metabolic checkpoints and strive even in nutrient-limiting conditions. Emerging studies are beginning to reveal how lipid droplets respond to nutrient and energy fluctuations and how they are integrated in the sensing and regulatory networks that orchestrate the metabolic rewiring of stressed cancer cells.
3.4.1 Lipid Droplets Are Rapidly Mobilizable Energy Sources During Stress
Many of the hallmark changes in lipid metabolism in cancer cells are shared by rapidly proliferating, fermenting yeast cells (Natter and Kohlwein 2013). Both types of cells depend on lipogenic pathways for cell growth and viability. The synthesis of FAs and their incorporation into complex lipids, most notably phospholipids, drives membrane expansion, which is required for cell growth, cell cycle progression, and cell division. In yeast, TAG lipolysis has been linked with the cell cycle and provides FAs for membrane synthesis (Kurat et al. 2009; Zanghellini et al. 2008). Upon glucose depletion, the Snf1 protein kinase (the yeast orthologue of AMPK) is activated to engage a switch from glucose fermentation to FA oxidation as a primary source of energy. Intriguingly, this is accompanied by a shift from phospholipid to TAG synthesis resulting in elevated lipid droplet biogenesis (Bosch et al. 2020; Henne et al. 2018). This conserved mechanism of preservation of lipids that is activated at the onset of starvation prepares the cell for the possibility of prolonged periods of nutrient deficiency. Indeed, in starving yeast cells, lipid droplets are gradually consumed by microautophagy, a form of lipophagy involving the vacuole, and become essential for long-term survival (Seo et al. 2017).
Proliferating mammalian and cancer cells with access to nutrients mostly rely on glucose fermentation for energy production and use mitochondria as a biosynthetic organelle. Mitochondria provide building blocks and reducing equivalents for anabolic reactions, including FA synthesis, thereby ensuring a consistent supply of FAs for membrane biogenesis (Natter and Kohlwein 2013; Ward and Thompson 2012). In such nutrient- and lipid-rich conditions, mammalian cells also synthesize TAGs and accumulate lipid droplets (Fig. 2c) (Herms et al. 2015). When extracellular lipids become limiting, lipid droplet-derived FAs may be used for phospholipid synthesis and drive cell proliferation (Herms et al. 2015). When both glucose and lipids are scarce, mammalian cells shut off phospholipid synthesis and turn on mitochondrial oxidative metabolism. Lipid droplet-derived FAs are then syphoned into mitochondria for oxidation and energy production. The decrease in energy levels is detected by AMPK, which not only activates FA oxidation and mitochondrial oxidative metabolism but also directly stimulates the rapid redistribution of lipid droplets along the microtubular network, thereby driving their recruitment to mitochondria and optimizing FA delivery (Herms et al. 2015; Zhu et al. 2019). AMPK activation and associated starvation responses, such as autophagy, mTORC1 inhibition, and protein kinase A (PKA) activation, also promote mitochondrial fusion, which is necessary for efficient FA intake and uniform distribution within the network of tubulated mitochondria (Gomes et al. 2011; Rambold and Pearce 2018; Rambold et al. 2015). Lipid droplets thus provide a rapidly mobilizable form of energy substrates for cell survival following a sudden glucose depletion and energy deficiency.
3.4.2 Cancer Cells Depend on the Long-Term Supply of Lipid Droplet-Derived Lipids
Cancer cells may be exposed to relatively long periods of nutrient deficiency due to insufficient vasculature and rapid tumor growth (Wellen and Thompson 2010). Their nutrient and oxygen supply may also be severely compromised following matrix detachment, migration, and invasion into neighboring tissue. Cancer cells having accumulated lipid droplets during nutrient (and oxygen) sufficiency rely on the long-term supply of lipid droplet-derived lipids not only to survive the immediate stress but also to migrate and resume growth at a new location (Clement et al. 2020; Wang et al. 2017). Indeed, lipid droplets, accumulated in nutrient-rich conditions, enable a prolonged protection from starvation by undergoing gradual lipid droplet breakdown (Jarc et al. 2018; Przybytkowski et al. 2007; Pucer et al. 2013). Aggressive breast cancer cells harboring Ras oncogenic mutations increase their lipid droplet storage upon exposure to even minute amounts of monounsaturated or polyunsaturated FAs when grown in nutrient replete conditions. When these cells are switched to lipid- and serum-free starvation medium, but still rich in glucose and amino acids, lipid droplets undergo gradual breakdown over several days in culture resulting in an increased resistance to cell death (Jarc et al. 2018; Przybytkowski et al. 2007; Pucer et al. 2013). In comparison with control cells without initial lipid loading, these cells also activate AMPK, decrease their dependence on de novo lipogenesis, and upregulate FA oxidation (Brglez et al. 2014; Jarc et al. 2018; Pucer et al. 2013). In fact, preloading aggressive breast cancer cells with lipid droplets suppresses the strong surge in lipogenic signaling that occurs at the onset of lipid and serum starvation. The activation of lipogenesis is driven by the major lipid sensor and transcription factor sterol regulatory element-binding protein-1c (SREBP-1c) and its target genes involved in FA and cholesterol synthesis, including FA synthase (FASN), acetyl-coenzyme A carboxylase (ACC), SCD1, and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) (Jarc et al. 2018; Pucer et al. 2013). The biosynthesis of FAs and other lipids consumes large amounts of ATP and reducing power in the form of NADPH (Natter and Kohlwein 2013). Therefore, the breakdown of pre-accumulated lipid droplets at the onset of starvation spares important cellular resources by reducing the need for de novo lipogenesis. The starving cancer cell may thus redirect the saved energy and redox equivalents to other essential processes that protect against starvation.
In addition, the concurrent increase in the levels of FA oxidation enzymes, including carnitine palmitoyltransferase 1A (CPT1A), whose inhibition is lethal to serum-starved breast cancer cells, suggests that the pre-accumulated lipid droplets provide a long-term supply of FAs for mitochondrial oxidation to support cell survival (Pucer et al. 2013). Indeed, a combined depletion of the major TAG lipase ATGL and pharmacological targeting of CPT1A abolished the protective effects of lipid droplets in breast cancer cells (Jarc et al. 2018). Moreover, the observed activation of AMPK and the ability of its activator AICAR to protect breast cancer cells from starvation-induced cell death (Pucer et al. 2013) are in line with the fact that AMPK supports cancer cell survival by stimulating FA oxidation, blocking lipogenesis, and driving both ATP and NADPH production (Buzzai et al. 2005; Carracedo et al. 2013; Jeon et al. 2012; Pike et al. 2011). Such changes in the metabolic landscape involving AMPK, mitochondria, and the lipid droplet may render cancer cells particularly well-equipped to handle prolonged periods of nutrient limitation. Collectively, these studies suggest that lipid droplets support Ras-driven cancer cell survival in lipid-limiting conditions by (1) reducing the need for energy-depleting de novo lipogenesis and (2) driving mitochondrial oxidative metabolism that replenishes cellular energy and redox capacity.
3.4.3 Devouring and Creating Fat: Metabolic Flexibility Driving Tumorigenesis
Recent studies suggest that the interactions between mitochondria and lipid droplets, besides optimizing FA transfer and rates of FA oxidation (Herms et al. 2015; Rambold et al. 2015), in fact enable the formation of complex metabolic and signaling “synapses.” These are endowed with sophisticated feedback mechanisms that finely tune both lipid droplet and mitochondrial metabolism (Benador et al. 2019; Bohnert 2020; Bosch et al. 2020; Freyre et al. 2019; Jarc and Petan 2019). In fact, lipid droplet-mitochondria contacts may also reflect an essential role of mitochondria in the synthesis of TAG and lipid droplet biogenesis. Benador et al. have recently discovered that brown adipose tissue cells contain two segregated and functionally distinct subpopulations of mitochondria (Fig. 2d): peridroplet mitochondria, which are anchored to lipid droplets and are primarily involved in providing ATP and NADPH to support FA and TAG synthesis driving lipid droplet formation, and “free,” cytosolic mitochondria that primarily take up and oxidize FAs (Benador et al. 2018, 2019). Furthermore, in white adipocytes, a tripartite lipid droplet–mitochondria–ER interaction couples FA synthesis from glycolytic precursors processed in the citric acid cycle with their esterification into TAGs within the ER membrane and TAG storage in the growing lipid droplet (Freyre et al. 2019). Thus, overturning the classical biochemical dogma of the exclusively unidirectional mode of FA metabolism, cells may simultaneously engage in antagonistic biochemical processes, such as FA oxidation and synthesis, or lipid droplet expansion and breakdown, using distinct subpopulations of mitochondria and lipid droplets. Emerging studies hint at the possibility that such organelle and metabolic flexibility is also used by cancer cells to trigger and sustain metabolic reprogramming. Indeed, cancer cells grown in various nutrient- and lipid-rich conditions increase FA uptake and activate lipid droplet biogenesis in parallel with catabolic lipid droplet consumption and FA oxidation that drives cancer cell survival, growth, and metastasis (Clement et al. 2020; Lazar et al. 2016; Nieman et al. 2010; Pucer et al. 2013; Wang et al. 2017).
In the tumor microenvironment, cancer cells may “trick” neighboring adipocytes into releasing FAs from their large TAG stores, which are then taken up and used by cancer cells to form lipid droplets (Fig. 2e) (Attané and Muller 2020; Balaban et al. 2017; Clement et al. 2020; Nieman et al. 2010; Wang et al. 2017; Wen et al. 2017). These lipid droplets are broken down via lipolysis or lipophagy, thereby syphoning the adipocyte-derived FAs into mitochondria to be used for energy production and likely other purposes. Remarkably, in melanoma cells exposed to adipocyte-derived extracellular vesicles, mitochondria, lipid droplets, and lysosomes are redistributed and proximally located in cell protrusions to promote cancer cell migration via lipophagic lipid droplet breakdown and FA oxidation (Clement et al. 2020). Intriguingly, although typically sensing nutrient depletion, AMPK is activated in cancer cells co-cultured with adipocytes, most likely to promote and regulate the tight cooperation between lipid droplet consumption and FA oxidation, which may be coupled to or uncoupled from ATP production (Nieman et al. 2010; Wang et al. 2017; Wen et al. 2017; Zechner et al. 2017). Furthermore, upregulated ATGL-mediated lipid droplet lipolysis in breast cancer cells may lead to uncoupling of FA oxidation resulting in a drop in ATP levels and sustained AMPK activation, which promotes further FA uptake and mitochondrial biogenesis (Wang et al. 2017).
Another possibility that may explain the activation of AMPK in such lipid-rich conditions is a decrease in energy levels as a consequence of elevated FA/TAG cycling, whereby the influx of exogenous FAs stimulates a cycle of FA esterification into TAG and lipolysis at the expense of ATP (Prentki and Madiraju 2008; Przybytkowski et al. 2007). Namely, free FAs require ATP-dependent activation into FA-CoA by long-chain acyl-CoA synthetase (ACSL) enzymes before entering TAG synthesis or being transported into mitochondria following lipolysis (Cooper et al. 2015). In line with this, the ACSL inhibitor triacsin C suppresses both FA-induced lipid droplet biogenesis and AMPK activation in breast cancer cells during growth in nutrient-rich conditions (Pucer et al. 2013). Moreover, because inhibition of CPT1A with low concentrations of etomoxir (Raud et al. 2018) also reduces both AMPK activation and lipid droplet accumulation, it may be speculated that the exogenous FA supply stimulates FA oxidation that provides ATP and NADPH to support the anabolic branch of FA/TAG cycling (Pucer et al. 2013). The elevated FA/TAG cycling may lead to ATP deficiency that promotes AMPK activation, which in turn further stimulates mitochondrial FA oxidation. AMPK may be required under these conditions to reduce unnecessary de novo lipogenesis, suppress excessive lipid droplet accumulation, activate lipolysis, and increase the mitochondrial capacity of the cell by stimulating gene expression programs responsible for mitochondrial biogenesis and oxidative metabolism.
Whether different subpopulations of mitochondria and lipid droplets enable these antagonistic processes in individual cancer cells remains to be confirmed. Moreover, the intracellular heterogeneity in mitochondrial and lipid droplet function is likely also influenced and combined with intercellular lipid trafficking and population dynamics, whereby individual cells preferentially specialize their lipid droplet function to serve specific roles, e.g., protect from bulk lipid influx or engage in anabolic vs. catabolic lipid metabolism (Herms et al. 2013; Thiam and Beller 2017).
3.5 When the Going Gets Tough, Lipid Droplets Team Up with Autophagy
When cells are exposed to prolonged nutrient deficiency, and in particular when amino acids become limiting, autophagy is typically strongly activated (Bosch et al. 2020; Galluzzi et al. 2017; Kroemer et al. 2010; Nguyen et al. 2017; Ogasawara et al. 2020; Rambold et al. 2015). Lipid droplets and autophagy engage in a complex relationship, which is currently poorly understood: (1) lipid droplets may be the target of autophagic degradation (Schulze et al. 2017), (2) they may be formed as a consequence of autophagic breakdown of other lipid-containing organelles (Lue et al. 2017; Nguyen et al. 2017; Rambold et al. 2015; VandeKopple et al. 2019), and (3) they may support the formation of autophagosomes by providing lipids (Bekbulat et al. 2019; Dupont et al. 2014; Shpilka et al. 2015) or supporting signaling that stimulates the expression of autophagy genes (Ogasawara et al. 2020; Petan et al. 2018; Zechner et al. 2017). Emerging studies suggest that changes in lipid droplet turnover are a conserved cellular response to high autophagic flux, occurring across the eukaryotic kingdom and playing various beneficial roles in cellular homeostatic and stress responses (Jaishy and Abel 2016; Petan et al. 2018; Wang 2016). The opposite is also true, since lipid overload and exogenous unsaturated FAs stimulate autophagy (Niso-Santano et al. 2015). Indeed, cells preloaded with (unsaturated) FA-induced lipid droplets display higher autophagic flux during starvation (Dupont et al. 2014). In accordance with this entangled relationship, it is not surprising that both lipid droplet turnover and autophagy are often simultaneously or sequentially activated by various kinds of stress.
In mouse embryonic fibroblasts (MEFs) exposed to acute amino acid starvation, mTORC1 is inactivated leading to the activation of autophagy, which in turn triggers lipid droplet biogenesis (Fig. 2f) (Nguyen et al. 2017; Rambold et al. 2015). Lipids derived from membranous organelles are delivered into lysosomes by autophagy and broken down by acid phospholipases and lipases. The FAs released from lysosomes are rapidly esterified by DGAT1 into TAGs and stored in growing lipid droplets. Immediate lipid droplet biogenesis is required to avoid the accumulation of autophagy-derived free FAs that could overwhelm the mitochondrial FA transfer mechanism leading to piling up of toxic acylcarnitines at the mitochondrial “gates.” Furthermore, the newly formed lipid droplets provide an efficient way to gradually deliver FAs into the network of fused mitochondria during the ongoing starvation. Indeed, under these conditions, free FAs are released from lipid droplets primarily by ATGL-mediated lipolysis, but not lipophagy (Rambold et al. 2015). Notably, ATGL may not only provide FAs but also stimulate signaling pathways that both activate mitochondrial oxidative metabolism and regulate autophagy/lipophagy (Zechner et al. 2017). Interestingly, rather than in the initiation of autophagy, AMPK seems to be involved in sustaining autophagic flux and oxidative metabolism during the starvation (Nguyen et al. 2017).
Surely, the fine regulation of lipolysis and its coordination with autophagy will be of critical importance for cell survival in starved cells. Indeed, the hypoxia-inducible lipid droplet-associated protein (HILPDA), an endogenous inhibitor of ATGL (Das et al. 2018), is upregulated in MEFs and in cancer cells during acute starvation (VandeKopple et al. 2019). Interestingly, HILPDA is activated in direct response to autophagy-driven lipid droplet biogenesis, thereby suppressing ATGL-mediated lipolysis. In accordance, ablation of HILPDA reduces lipid droplet accumulation and xenograft tumor growth in vivo, possibly by elevating oxidative stress, lipid peroxidation, and apoptosis due to excessive lipolysis (VandeKopple et al. 2019; Zhang et al. 2017). Although additional confirmation is clearly required, these results suggest that autophagy-driven lipid droplet turnover and the fine-tuning of lipolysis by HILPDA promote tumorigenesis.
While physiological levels of autophagy generally play a tumor suppressor role by preventing cell damage, maintaining cellular fitness, and restoring homeostasis, cancer cells may also subvert the autophagic machinery to enhance their resistance to stress. Lipid droplets and autophagy may play a complementary role in both contexts. For example, nutrient deficiency within cancer cells may be induced indirectly by exposing cells to drugs targeting major nutrient sensing and growth pathways, such as the PI3K/Akt/mTOR pathway (Lue et al. 2017). Intriguingly, although tumor growth is restricted by these drugs, cancer cells may circumvent therapeutic inhibition by activating autophagy. Importantly, this cancer treatment-induced autophagy stimulates lipid droplet biogenesis to sustain mitochondrial energy production and redox homeostasis, thereby reducing cancer cell death (Fig. 2f) (Lue et al. 2017). Intriguingly, the supply of FAs for lipid droplet biogenesis and oxidative metabolism is dependent on an unidentified member of the phospholipase A2 family of enzymes, which release free FAs and lysophospholipids from membrane phospholipids (Lambeau and Gelb 2008; Murakami and Lambeau 2013; Murakami et al. 2011). Several phospholipases A2 have been implicated in lipid droplet metabolism and cancer cell survival (Cabodevilla et al. 2013; Guijas et al. 2014; Jarc et al. 2018; Pucer et al. 2013), but it is not clear how they cooperate with autophagy to stimulate lipid droplet and mitochondrial metabolism (Petan et al. 2018). The mechanisms and relevance of autophagy-driven lipid droplet turnover for tumor growth remain to be established.
Several in vitro studies have shown that lipophagy is typically activated under milder, albeit prolonged, starvation conditions than those activating bulk autophagy (Rambold et al. 2015; Wang 2016). For example, in contrast to amino acid-starved MEFs, autophagy-driven lipid droplet biogenesis does not occur in serum-starved MEFs, most likely because mTORC1 is not inhibited under these conditions, but instead lipophagy contributes to lipid droplet breakdown (Nguyen et al. 2017; Rambold et al. 2015). The activation of AMPK may drive lipophagy under such conditions, because it can bypass mTORC1 and activate lipophagy through direct activation of ULK1 even in nutrient-rich conditions (Kim et al. 2011; Li et al. 2019; Zechner et al. 2017). Moreover, AMPK phosphorylates PLIN2 and primes it for chaperone-mediated autophagy, which is an additional mechanism of AMPK-mediated regulation of both lipophagy and lipolysis (Kaushik and Cuervo 2016). AMPK also indirectly activates the deacetylase sirtuin 1 (SIRT1) and its target transcription factors peroxisome proliferator-activated receptor γ co-activator 1α (PGC1α) and forkhead box protein O (FOXO), which regulate both neutral and acid lipolysis (Zechner et al. 2017).
A role for AMPK-driven lipophagy has been suggested in promoting cancer cell growth in the context of metabolic symbiosis between adipocytes and cancer cells (Wen et al. 2017). Adipocyte-derived FAs were found to stimulate AMPK-dependent lipophagy and mitochondrial energy production, which were required for the survival of neighboring cancer cells during starvation. On the contrary, in prostate cancer cells, the activation of lipophagy may occur in response to SIRT1-mediated acetylation of LAMP1 and lead to proliferative senescence, likely as a consequence of elevated oxidative stress (Panda et al. 2019). Accordingly, excessive lipophagy leads to an overflow of free FAs causing mitochondrial damage, ER stress, and cancer cell death in cervical cancer cells (Mukhopadhyay et al. 2017). Lipophagy has also been associated with reduced ccRCC tumor growth and increased patient survival (Xu et al. 2015). In line with these studies suggesting a tumor suppressor role for lipophagy, recent evidence has shown that LAL suppresses inflammation and metastasis in liver and lung cancer (Du et al. 2015; Zhao et al. 2016). With these mostly preliminary studies, we are only beginning to understand the role of lipophagy in cancer, which seems to play a dual, context-dependent role (Kounakis et al. 2019; Maan et al. 2018; Petan et al. 2018). In accordance with the opposing roles of neutral lipolysis in cancer, the role of lipophagy likely depends on the specific metabolic and oncogenic reprogramming of the cancer type in question and the microenvironmental conditions (Petan et al. 2018).
3.6 Lipid Droplets, Lipid Peroxidation, and Ferroptosis in Cancer
One of the primary functions of lipid droplets in most biological systems and conditions is the protection from various forms of lipotoxicity (Listenberger et al. 2003; Schaffer 2003). Lipid droplets have also recently been implicated in the regulation of the cellular distribution of unsaturated and polyunsaturated FAs (PUFAs) (Ackerman et al. 2018; Bailey et al. 2015; Jarc et al. 2018; Petan et al. 2018), which is essential for the maintenance of proper membrane saturation and redox balance. In fact, lipid droplets seem to act as antioxidant organelles by actively regulating the trafficking of PUFAs in order to prevent oxidative stress and cell death. Lipid droplets also regulate the release of PUFAs for their conversion by cyclooxygenases and lipoxygenases into a whole range of oxygenated mediators of inflammation in immune cells, adipocytes, and in cancer cells (Jarc and Petan 2020). The recent discovery of ferroptosis (Dixon et al. 2012), a type of programmed cell death driven by the oxidation of PUFAs in membrane phospholipids, has pinpointed the importance of lipid peroxidation for cellular well-being and protection from stress. Lipid droplets, being implicated in the regulation of PUFA lipotoxicity and trafficking, are thereby emerging as imminent regulators of ferroptotic sensitivity.
Ferroptosis is a form of programmed cell death that depends on the accumulation of lethal levels of oxidized lipids in cell membranes (Fig. 3) (Dixon and Stockwell 2019). Cells possess at least two major antioxidant mechanisms that act in parallel to protect from ferroptotic cell death: (1) the glutathione peroxidase 4 (GPX4) pathway and (2) the ubiquinol (coenzyme Q10) antioxidant system, which depends on the activity of ferroptosis-suppressor-protein 1 (FSP1; previously called AIFM2) (Bersuker et al. 2019; Doll et al. 2019). Currently, it is not clear whether any final executioner proteins of ferroptosis exist, since the process essentially depends on the propagation of lipid peroxidation chain reactions and the ultimate failure of protective antioxidant mechanisms, progressively leading to irreparable membrane and organelle dysfunction. Importantly, induction of ferroptosis by inhibition of GPX4 and/or FSP1 is effective at killing multiple types of cancers in vitro and in vivo (Badgley et al. 2020; Bersuker et al. 2019; Doll et al. 2019; Hangauer et al. 2017; Tousignant et al. 2020; Viswanathan et al. 2017; Zhang et al. 2019; Zou et al. 2019). Thus, the stimulation of ferroptosis in tumors may offer new opportunities for effective cancer treatment. However, certain types of cancer cells are resistant to known ferroptotic inducers suggesting that additional modulators of ferroptotic sensitivity exist.

Ferroptosis is a consequence of lethal membrane lipid peroxidation. Polyunsaturated fatty acids (PUFAs), mostly residing in membrane phospholipids, are particularly susceptible to oxidation by reactive oxygen species (ROS), non-enzymatic Fe2+-mediated reactions, and lipoxygenase (LOX)-mediated peroxidation. The propagation of lipid peroxidation chain reactions along with a failure of antioxidant mechanisms leads to irreparable cell damage and cell death. Cells possess two complementary mechanisms of protection against ferroptosis. The first depends on cystine import, which is necessary for glutathione (GSH) synthesis, the main redox buffer in the cell, that is in turn required for the activity of glutathione peroxidase 4 (GPX4). GPX4 converts toxic PUFA peroxides into harmless lipid alcohols. The second mechanism depends on the activity of ferroptosis-suppressor-protein 1 (FSP1), which is necessary for the NAD(P)H-dependent regeneration of ubiquinol (coenzyme Q10), the major lipophilic antioxidant in cell membranes. Blocking cystine import by erastin or inhibition of GPX4 activity by RSL-3 results in a failure of the GPX4 antioxidant system, accumulation of lipid peroxides, and ferroptotic cell death
Emerging studies point to a crosstalk between ferroptosis and lipid droplets. Diffuse large B cell lymphoma cancer cells treated with imidazole ketone erastin (IKE), which blocks cystine uptake and promotes ferroptosis by depleting glutathione, display a decrease in the levels of PUFA-containing phospholipids and TAGs, possibly as a consequence of a cell protective mechanism that removes oxidized PUFAs from these lipids (Zhang et al. 2019). The decrease in TAGs could be a consequence of elevated lipolysis, since IKE treatments led to a significant upregulation of ATGL expression, along with enzymes involved in de novo FA synthesis, phospholipid remodeling, and several lipoxygenases. This may indicate that PUFAs are first released from lipid droplets by ATGL and then incorporated in membrane phospholipids, thereby contributing to the lethal membrane lipid peroxidation caused by IKE (Fig. 4). In line with this idea, treatments with the lipophilic antioxidant ferrostatin prevented IKE toxicity and increased TAG accumulation in the cells. This is also in accordance with our studies in breast cancer cells showing that depletion of ATGL suppresses PUFA-induced oxidative stress and rescues cells from PUFA lipotoxicity, whereas lipid droplet biogenesis protects against PUFA lipotoxicity (Jarc et al. 2018). These findings suggest that in some cancer cells, lipid droplet breakdown via lipolysis may promote ferroptotic cell death.

Potential crosstalk between lipid droplets and ferroptosis. Lipid droplets may modulate ferroptosis by regulating polyunsaturated fatty acid (PUFA) trafficking. (a) Lipid droplet formation via DGAT-mediated triglyceride (TAG) synthesis may act as a sink for phospholipid-derived PUFAs, thus preventing their peroxidation; lipid droplet biogenesis may also restrict lipid peroxidation by sequestering already damaged, peroxidized PUFAs (ox-PUFAs) to suppress the propagation of lipid peroxidation. (b) ATGL-mediated TAG lipolysis may provide PUFAs for membrane synthesis, thus stimulating lipid peroxidation and sensitizing cells to ferroptosis. Other lipases and phospholipases may also release ox-PUFAs from TAGs or phospholipids. (c) ATGL may also provide monounsaturated fatty acids (MUFAs) that reduce the abundance of oxidizable PUFAs in membranes, thereby restricting lipid peroxidation
Recent findings provide more support for the idea that lipid droplet breakdown regulates ferroptosis sensitivity. Several types of therapy-resistant cancer cells have been shown to be particularly sensitive to ferroptosis (Tousignant et al. 2020; Viswanathan et al. 2017). Namely, drug-resistant prostate cancer cells undergo an extensive metabolic reprogramming characterized by increased lipid uptake that drives lipid droplet accumulation and phospholipid remodeling. The latter results in elevated membrane PUFA content, thereby increasing lipid peroxidation and dependence on GPX4 activity (Tousignant et al. 2020). Counterintuitively, a depletion of TAGs and CEs was also observed, indicating the possibility that lipid droplet-derived lipids are consumed for phospholipid synthesis and thus mediate ferroptosis sensitivity. The study suggests that some other lipid species, such as acylceramides, concurrently drive the formation of a separate population of lipid droplets (Senkal et al. 2017; Tousignant et al. 2020). Interestingly, lipid droplets have also been suggested to sensitize breast cancer cells to ferroptosis via ATGL-mediated lipolysis in a cell density-dependent manner (Panzilius et al. 2018). Moreover, lipid droplet breakdown via lipophagy has recently been shown to promote GPX4 inhibition-induced ferroptotic cell death in hepatocytes (Bai et al. 2019). Finally, ferroptosis has been identified as a specific vulnerability of clear-cell carcinomas, whereby HILPDA, albeit acting in an ATGL-independent manner, mediates a HIF-2α-dependent enrichment of PUFAs into TAGs and phospholipids (Zou et al. 2019). Collectively, these findings suggest that PUFA-TAGs stored within lipid droplets are drivers of ferroptotic sensitivity, most likely by providing PUFAs for phospholipid membrane synthesis (Fig. 4). Moreover, since TAGs stored within lipid droplets may also be oxidized, it is possible that lipid droplets themselves are sites of lipid peroxidation that promote ferroptosis if peroxidized lipids are not efficiently removed (Ramakrishnan et al. 2014; Veglia et al. 2017). In line with this idea, the Spastin/ABCD1/ESCRT-III lipid droplet-peroxisome tethering complex is necessary for the removal of peroxidized lipids from lipid droplets, which implicates both organelles in protecting cells against lipid peroxidation and possibly ferroptosis (Chang et al. 2019).
On the other hand, depending on the fatty acyl composition of lipid droplets and the predominantly released species, lipid droplet breakdown should also be able to protect from ferroptosis (Fig. 4). Accordingly, lipolysis of monounsaturated FA (MUFA)-enriched TAGs protects aggressive breast cancer cells from PUFA-induced oxidative stress and lipotoxicity, likely by reducing the relative abundance of membrane-resident PUFAs available for peroxidation (Ackerman et al. 2018; Jarc et al. 2018). In addition, the lipolytic release of MUFAs has been recently shown to promote mitochondrial biogenesis and oxidative metabolism via PLIN5-mediated allosteric activation of SIRT1 (Najt et al. 2019), which may additionally explain their beneficial effects on redox metabolism. However, lipid droplet biogenesis was not necessary for the ability of exogenous MUFAs to suppress erastin-induced ferroptosis (Magtanong et al. 2018). Instead, their ASCL3-dependent incorporation into plasma membrane phospholipids and displacement of PUFAs was found to be responsible for the effect in several cancer cell lines. The ability of lipid droplet biogenesis and/or breakdown to modulate ferroptotic sensitivity surely requires further exploration, particularly in the sense that combined targeting of lipid droplet turnover and the anti-ferroptotic redox machinery may prove to be a valid therapeutic strategy.
4 Conclusions and Perspectives
Given their central role as coordinators of lipid metabolism with cell growth and stress resistance, lipid droplets are emerging as potentially vulnerable hotspots in numerous cancers. However, we are only beginning to understand how lipid droplets respond to the various stressful conditions encountered by cancer cells and which are the essential tasks that these organelles perform to support the cellular stress response. We have to find out more about the particular mechanisms involved in order to use this knowledge in cancer treatment. Numerous points in their biogenesis and/or breakdown could potentially be targeted in order to either compromise the ability of lipid droplets to protect cancer cells from stress or to purposefully use lipid droplets to cause cell damage. For example, inhibiting lipid droplet biogenesis in starving cells dependent on autophagy for their survival could increase mitochondrial damage due to the build-up of cytosolic FAs and acylcarnitines (Nguyen et al. 2017). The inhibition of lipid droplet biogenesis in poorly vascularized tumors could abolish their function as long-term lipid reservoirs and compromise the ability of cancer cells to survive prolonged periods of starvation or resume growth upon reoxygenation (Bensaad et al. 2014; Jarc et al. 2018; Pucer et al. 2013). During the final stages of revision of this manuscript, two important papers were published showing that DGAT1-mediated lipid droplet biogenesis is a relevant target for the treatment of melanoma and glioblastoma (Cheng et al. 2020; Wilcock et al. 2020). DGAT1 was even identified as a bona fide oncoprotein that enables enhanced lipid uptake and drives melanoma formation. Its ability to protect cancer cells from oxidative stress and membrane lipid peroxidation, which hints at protection from ferroptosis as well, was found pivotal for melanoma aggressiveness (Wilcock et al. 2020). Compromising the ability of cancer cells to form lipid droplets could also impair their chemoresistance and immune evasion (Cotte et al. 2018). Finally, recent studies have revealed that lipid droplets may also regulate drug efficacy by affecting the selective partitioning of lipophilic drugs in their hydrophobic core and even promote drug activation in situ (Dubey et al. 2020; Englinger et al. 2020).
In other cases, the activation of lipid droplet breakdown could be a beneficial strategy. For example, stimulation of lipolysis or lipophagy is detrimental for cancer cells under certain conditions, since it may increase the levels of oxidative and ER stress, elevate lipid peroxidation and even lead to ferroptotic cell death (Jarc et al. 2018; Mukhopadhyay et al. 2017; Zhang et al. 2019; Zou et al. 2019). However, caution should be exerted, because in many instances lipid droplet breakdown in fact promotes the resistance of cancer cells to stress, as discussed at length in this review. Clearly, the feasibility of targeting lipid droplets should be carefully examined in different tumor types and particular contexts. In summary, lipid droplets are highly dynamic compartments that consolidate lipid uptake, synthesis, recycling, distribution, and breakdown pathways in the cell and are emerging as promising targets either to (1) restrict the supply of essential lipids or to (2) promote the accumulation of damaging lipids in order to compromise cancer cell survival, growth, and metastasis.
References
Ackerman D, Simon MC (2014) Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol 24:472–478. https://doi.org/10.1016/j.tcb.2014.06.001
Ackerman D, Tumanov S, Qiu B et al (2018) Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep 24:2596–2605.e5. https://doi.org/10.1016/j.celrep.2018.08.015
Attané C, Muller C (2020) Drilling for oil: tumor-surrounding adipocytes fueling cancer. Trends Cancer. https://doi.org/10.1016/j.trecan.2020.03.001
Badgley MA, Kremer DM, Maurer HC et al (2020) Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368:85–89. https://doi.org/10.1126/science.aaw9872
Bai Y, Meng L, Han L et al (2019) Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun 508:997–1003. https://doi.org/10.1016/j.bbrc.2018.12.039
Bailey AP, Koster G, Guillermier C et al (2015) Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell 163:340–353. https://doi.org/10.1016/j.cell.2015.09.020
Balaban S, Shearer RF, Lee LS et al (2017) Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab 5:195. https://doi.org/10.1186/s40170-016-0163-7
Barbosa AD, Siniossoglou S (2017) Function of lipid droplet-organelle interactions in lipid homeostasis. Biochim Biophys Acta 1864:1459–1468. https://doi.org/10.1016/j.bbamcr.2017.04.001
Bekbulat F, Schmitt D, Feldmann A et al (2019) RAB18 loss interferes with lipid droplet catabolism and provokes autophagy network adaptations. J Mol Biol 432:1216–1234. https://doi.org/10.1016/j.jmb.2019.12.031
Beloribi-Djefaflia S, Vasseur S, Guillaumond F (2016) Lipid metabolic reprogramming in cancer cells. Oncogenesis 5:e189. https://doi.org/10.1038/oncsis.2015.49
Benador IY, Veliova M, Mahdaviani K et al (2018) Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab 27:869–885.e6. https://doi.org/10.1016/j.cmet.2018.03.003
Benador IY, Veliova M, Liesa M, Shirihai OS (2019) Mitochondria bound to lipid droplets: where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab 29:1–11. https://doi.org/10.1016/j.cmet.2019.02.011
Bensaad K, Favaro E, Lewis CA et al (2014) Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 9:349–365. https://doi.org/10.1016/j.celrep.2014.08.056
Bersuker K, Olzmann JA (2017) Establishing the lipid droplet proteome: mechanisms of lipid droplet protein targeting and degradation. Biochim Biophys Acta 1862:1166–1177. https://doi.org/10.1016/j.bbalip.2017.06.006
Bersuker K, Peterson CWH, To M et al (2018) A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev Cell 44:97–112.e7. https://doi.org/10.1016/j.devcel.2017.11.020
Bersuker K, Hendricks J, Li Z et al (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575:688–692. https://doi.org/10.1038/s41586-019-1705-2
Bohnert M (2020) Tethering fat: tethers in lipid droplet contact sites. Contact 3:251525642090814. https://doi.org/10.1177/2515256420908142
Bosch M, Parton RG, Pol A (2020) Lipid droplets, bioenergetic fluxes, and metabolic flexibility. Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2020.02.010
Brglez V, Lambeau G, Petan T (2014) Secreted phospholipases A2 in cancer: diverse mechanisms of action. Biochimie 107:114–123. https://doi.org/10.1016/j.biochi.2014.09.023
Buzzai M, Bauer DE, Jones RG et al (2005) The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 24:4165–4173. https://doi.org/10.1038/sj.onc.1208622
Cabodevilla AG, Sanchez-Caballero L, Nintou E et al (2013) Cell survival during complete nutrient deprivation depends on lipid droplet-fueled β-oxidation of fatty acids. J Biol Chem 288(27777):27788. https://doi.org/10.1074/jbc.m113.466656
Carracedo A, Cantley LC, Pandolfi PP (2013) Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 13:227–232. https://doi.org/10.1038/nrc3483
Chang T-Y, Li B-L, Chang CCY, Urano Y (2009) Acyl-coenzyme A: cholesterol acyltransferases. Am J Physiol Endocrinol Metab 297:E1–E9. https://doi.org/10.1152/ajpendo.90926.2008
Chang C-L, Weigel AV, Ioannou MS et al (2019) Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through ESCRT-III. J Cell Biol 218:2583–2599. https://doi.org/10.1083/jcb.201902061
Cheng X, Geng F, Pan M et al (2020) Targeting DGAT1 ameliorates Glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab 32:229–242.e8. https://doi.org/10.1016/j.cmet.2020.06.002
Chorlay A, Monticelli L, Ferreira JV et al (2019) Membrane asymmetry imposes directionality on lipid droplet emergence from the ER. Dev Cell 50:25–42.e7. https://doi.org/10.1016/j.devcel.2019.05.003
Clement E, Lazar I, Attané C et al (2020) Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J 39:e102525. https://doi.org/10.15252/embj.2019102525
Coleman RA, Mashek DG (2011) Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem Rev 111:6359–6386. https://doi.org/10.1021/cr100404w
Commisso C, Davidson SM, Soydaner-Azeloglu RG et al (2013) Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497:633–637. https://doi.org/10.1038/nature12138
Cooper DE, Young PA, Klett EL, Coleman RA (2015) Physiological consequences of compartmentalized Acyl-CoA metabolism. J Biol Chem 290:20023–20031. https://doi.org/10.1074/jbc.r115.663260
Cotte AK, Aires V, Fredon M et al (2018) Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat Commun 9:322. https://doi.org/10.1038/s41467-017-02732-5
Cruz ALS, de Barreto EA, Fazolini NPB et al (2020) Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Dis 11:105. https://doi.org/10.1038/s41419-020-2297-3
Currie E, Schulze A, Zechner R et al (2013) Cellular fatty acid metabolism and cancer. Cell Metab 18:153–161. https://doi.org/10.1016/j.cmet.2013.05.017
Das KMP, Wechselberger L, Liziczai M et al (2018) Hypoxia-inducible lipid droplet-associated protein inhibits adipose triglyceride lipase. J Lipid Res 59:531–541. https://doi.org/10.1194/jlr.m082388
den Brok MH, Raaijmakers TK, Collado-Camps E, Adema GJ (2018) Lipid droplets as immune modulators in myeloid cells. Trends Immunol 39:380–392. https://doi.org/10.1016/j.it.2018.01.012
Dixon SJ, Stockwell BR (2019) The hallmarks of Ferroptosis. Annu Rev Cancer Biol 3:35–54. https://doi.org/10.1146/annurev-cancerbio-030518-055844
Dixon SJ, Lemberg KM, Lamprecht MR et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Doll S, Freitas FP, Shah R et al (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575:693–698. https://doi.org/10.1038/s41586-019-1707-0
Du H, Zhao T, Ding X, Yan C (2015) Hepatocyte-specific expression of human lysosome acid lipase corrects liver inflammation and tumor metastasis in LAL−/− mice. Am J Pathol 185:2379–2389. https://doi.org/10.1016/j.ajpath.2015.05.021
Dubey R, Stivala CE, Nguyen HQ et al (2020) Lipid droplets can promote drug accumulation and activation. Nat Chem Biol:1–8. https://doi.org/10.1038/s41589-019-0447-7
Dupont N, Chauhan S, Arko-Mensah J et al (2014) Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol 24:609–620. https://doi.org/10.1016/j.cub.2014.02.008
Englinger B, Laemmerer A, Moser P et al (2020) Lipid droplet-mediated scavenging as novel intrinsic and adaptive resistance factor against the multikinase inhibitor Ponatinib. Int J Cancer. https://doi.org/10.1002/ijc.32924
Farese RV, Walther TC (2009) Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 139(855):860. https://doi.org/10.1016/j.cell.2009.11.005
Finicle BT, Jayashankar V, Edinger AL (2018) Nutrient scavenging in cancer. Nat Rev Cancer 18:619–633. https://doi.org/10.1038/s41568-018-0048-x
Freyre CAC, Rauher PC, Ejsing CS, Klemm RW (2019) MIGA2 links mitochondria, the ER, and lipid droplets and promotes De novo Lipogenesis in adipocytes. Mol Cell 76:811–825.e14. https://doi.org/10.1016/j.molcel.2019.09.011
Gallardo-Montejano VI, Saxena G, Kusminski CM et al (2016) Nuclear Perilipin 5 integrates lipid droplet lipolysis with PGC-1α/SIRT1-dependent transcriptional regulation of mitochondrial function. Nat Commun 7:12723. https://doi.org/10.1038/ncomms12723
Galluzzi L, Baehrecke EH, Ballabio A et al (2017) Molecular definitions of autophagy and related processes. EMBO J 36:1811–1836. https://doi.org/10.15252/embj.201796697
Goeritzer M, Vujic N, Schlager S et al (2015) Active autophagy but not lipophagy in macrophages with defective lipolysis. Biochim Biophys Acta 1851:1304–1316. https://doi.org/10.1016/j.bbalip.2015.06.005
Gomes LC, Benedetto GD, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13:589–598. https://doi.org/10.1038/ncb2220
González A, Hall MN, Lin S-C, Hardie DG (2020) AMPK and TOR: the yin and Yang of cellular nutrient sensing and growth control. Cell Metab 31:472–492. https://doi.org/10.1016/j.cmet.2020.01.015
Grabner GF, Zimmermann R, Schicho R, Taschler U (2017) Monoglyceride lipase as a drug target: at the crossroads of arachidonic acid metabolism and endocannabinoid signaling. Pharmacol Ther 175:35–46. https://doi.org/10.1016/j.pharmthera.2017.02.033
Guijas C, Rodríguez JP, Rubio JM et al (2014) Phospholipase A2 regulation of lipid droplet formation. Biochim Biophys Acta 1841:1661–1671. https://doi.org/10.1016/j.bbalip.2014.10.004
Haemmerle G, Moustafa T, Woelkart G et al (2011) ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med 17:1076–1085. https://doi.org/10.1038/nm.2439
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hangauer MJ, Viswanathan VS, Ryan MJ et al (2017) Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551:247–250. https://doi.org/10.1038/nature24297
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13:251–262. https://doi.org/10.1038/nrm3311
Henne WM, Reese ML, Goodman JM (2018) The assembly of lipid droplets and their roles in challenged cells. EMBO J 37:e98947. https://doi.org/10.15252/embj.201898947
Herms A, Bosch M, Ariotti N et al (2013) Cell-to-cell heterogeneity in lipid droplets suggests a mechanism to reduce lipotoxicity. Curr Biol 23:1489–1496. https://doi.org/10.1016/j.cub.2013.06.032
Herms A, Bosch M, Reddy BJN et al (2015) AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun 6:7176. https://doi.org/10.1038/ncomms8176
Hernández-Corbacho MJ, Obeid LM (2018) A novel role for DGATs in cancer. Adv Biol Regul 72:89–101. https://doi.org/10.1016/j.jbior.2018.12.001
Hoy AJ, Balaban S, Saunders DN (2017) Adipocyte-tumor cell metabolic crosstalk in breast cancer. Trends Mol Med 23:381–392. https://doi.org/10.1016/j.molmed.2017.02.009
Jaishy B, Abel ED (2016) Lipids, lysosomes, and autophagy. J Lipid Res 57:1619–1635. https://doi.org/10.1194/jlr.r067520
Jarc E, Petan T (2019) Lipid droplets and the management of cellular stress. Yale J Biol Med 92:435–452
Jarc E, Petan T (2020) A twist of FATe: lipid droplets and inflammatory lipid mediators. Biochimie 169:69–87. https://doi.org/10.1016/j.biochi.2019.11.016
Jarc E, Kump A, Malavašič P et al (2018) Lipid droplets induced by secreted phospholipase A2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim Biophys Acta 1863:247–265. https://doi.org/10.1016/j.bbalip.2017.12.006
Jayashankar V, Edinger AL (2020) Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat Commun 11:1121. https://doi.org/10.1038/s41467-020-14928-3
Jeon S-M, Chandel NS, Hay N (2012) AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485:661–665. https://doi.org/10.1038/nature11066
Johnson MR, Stephenson RA, Ghaemmaghami S, Welte MA (2018) Developmentally regulated H2Av buffering via dynamic sequestration to lipid droplets in Drosophila embryos. elife 7:e36021. https://doi.org/10.7554/elife.36021
Kamphorst JJ, Cross JR, Fan J et al (2013) Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci 110:8882–8887. https://doi.org/10.1073/pnas.1307237110
Kassan A, Herms A, Herms A et al (2013) Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J Cell Biol 203:985–1001. https://doi.org/10.1083/jcb.201305142
Kaushik S, Cuervo AM (2015) Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol 17:759–770. https://doi.org/10.1038/ncb3166
Kaushik S, Cuervo AM (2016) AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 12:432–438. https://doi.org/10.1080/15548627.2015.1124226
Khan SA, Sathyanarayan A, Mashek MT et al (2015) ATGL-catalyzed lipolysis regulates SIRT1 to control PGC-1α/PPAR-α signaling. Diabetes 64:418–426. https://doi.org/10.2337/db14-0325
Kim J, Kundu M, Viollet B, Guan K-L (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141. https://doi.org/10.1038/ncb2152
Kim SM, Nguyen TT, Ravi A et al (2018) PTEN deficiency and AMPK activation promote nutrient scavenging and anabolism in prostate cancer cells. Cancer Discov 8:17–1215. https://doi.org/10.1158/2159-8290.cd-17-1215
Koizume S, Miyagi Y (2016) Lipid droplets: a key cellular organelle associated with cancer cell survival under normoxia and hypoxia. Int J Mol Sci 17:E1430. https://doi.org/10.3390/ijms17091430
Kounakis K, Chaniotakis M, Markaki M, Tavernarakis N (2019) Emerging roles of lipophagy in health and disease. Front Cell Dev Biol 7:185. https://doi.org/10.3389/fcell.2019.00185
Krahmer N, Guo Y, Wilfling F et al (2011) Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab 14:504–515. https://doi.org/10.1016/j.cmet.2011.07.013
Krahmer N, Farese RV, Walther TC (2013) Balancing the fat: lipid droplets and human disease. EMBO Mol Med 5:905–915. https://doi.org/10.1002/emmm.201100671
Kroemer G, Mariño G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40:280–293. https://doi.org/10.1016/j.molcel.2010.09.023
Kuerschner L, Moessinger C, Thiele C (2008) Imaging of lipid biosynthesis: how a neutral lipid enters lipid droplets. Traffic 9:338–352. https://doi.org/10.1111/j.1600-0854.2007.00689.x
Kurat CF, Wolinski H, Petschnigg J et al (2009) Cdk1/Cdc28-dependent activation of the major triacylglycerol lipase Tgl4 in yeast links lipolysis to cell-cycle progression. Mol Cell 33:53–63. https://doi.org/10.1016/j.molcel.2008.12.019
Lambeau G, Gelb MH (2008) Biochemistry and physiology of mammalian secreted phospholipases A2. Annu Rev Biochem 77:495–520. https://doi.org/10.1146/annurev.biochem.76.062405.154007
Lazar I, Clement E, Dauvillier S et al (2016) Adipocyte Exosomes promote melanoma aggressiveness through fatty acid oxidation: a novel mechanism linking obesity and Cancer. Cancer Res 76:4051–4057. https://doi.org/10.1158/0008-5472.can-16-0651
Li Y, Yang P, Zhao L et al (2019) CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway. J Lipid Res 60:844–855. https://doi.org/10.1194/jlr.m090969
Listenberger LL, Han X, Lewis SE et al (2003) Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci 100:3077–3082. https://doi.org/10.1073/pnas.0630588100
Liu GY, Sabatini DM (2020) mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Bio 21:183–203. https://doi.org/10.1038/s41580-019-0199-y
Lue H-W, Podolak J, Kolahi K et al (2017) Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev 31:2067–2084. https://doi.org/10.1101/gad.305292.117
Maan M, Peters JM, Dutta M, Patterson AD (2018) Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun 504:582–589. https://doi.org/10.1016/j.bbrc.2018.02.097
Magtanong L, Ko P-J, To M et al (2018) Exogenous monounsaturated fatty acids promote a Ferroptosis-resistant cell state. Cell Chem Biol 26:1–23. https://doi.org/10.1016/j.chembiol.2018.11.016
Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG et al (2017) Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14:11–31. https://doi.org/10.1038/nrclinonc.2016.60
Menendez J, Lupu R (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7:763–777. https://doi.org/10.1038/nrc2222
Michalopoulou E, Bulusu V, Kamphorst JJ (2016) Metabolic scavenging by cancer cells: when the going gets tough, the tough keep eating. Br J Cancer 115:635–640. https://doi.org/10.1038/bjc.2016.256
Molenaar MR, Vaandrager AB, Helms JB (2017) Some lipid droplets are more equal than others: different metabolic lipid droplet pools in hepatic stellate cells. Lipid Insights 10:1178635317747281. https://doi.org/10.1177/1178635317747281
Mottillo EP, Bloch AE, Leff T, Granneman JG (2012) Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) α and δ in brown adipocytes to match fatty acid oxidation with supply. J Biol Chem 287:25038–25048. https://doi.org/10.1074/jbc.m112.374041
Mukhopadhyay S, Schlaepfer IR, Bergman BC et al (2017) ATG14 facilitated lipophagy in cancer cells induce ER stress mediated mitoptosis through a ROS dependent pathway. Free Radic Bio Med 104:199–213. https://doi.org/10.1016/j.freeradbiomed.2017.01.007
Muoio DM, Seefeld K, Witters LA, Coleman RA (1999) AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem J 338:783–791. https://doi.org/10.1042/bj3380783
Murakami M, Lambeau G (2013) Emerging roles of secreted phospholipase A(2) enzymes: an update. Biochimie 95:43–50. https://doi.org/10.1016/j.biochi.2012.09.007
Murakami M, Taketomi Y, Miki Y et al (2011) Recent progress in phospholipase A2 research: from cells to animals to humans. Prog Lipid Res 50:152–192. https://doi.org/10.1016/j.plipres.2010.12.001
Najt CP, Khan SA, Heden TD et al (2019) Lipid droplet-derived monounsaturated fatty acids traffic via PLIN5 to allosterically activate SIRT1. Mol Cell 77:810–824. https://doi.org/10.1016/j.molcel.2019.12.003
Natter K, Kohlwein SD (2013) Yeast and cancer cells – common principles in lipid metabolism. Biochim Biophys Acta 1831:314–326. https://doi.org/10.1016/j.bbalip.2012.09.003
Nguyen TB, Louie SM, Daniele JR et al (2017) DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev Cell 42:9–21.e5. https://doi.org/10.1016/j.devcel.2017.06.003
Nieman KM, Kenny HA, Penicka CV et al (2010) Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17:1498–1503. https://doi.org/10.1038/nm.2492
Niso-Santano M, Malik SA, Pietrocola F et al (2015) Unsaturated fatty acids induce non-canonical autophagy. EMBO J 34:1025–1041. https://doi.org/10.15252/embj.201489363
Ogasawara Y, Tsuji T, Fujimoto T (2020) Multifarious roles of lipid droplets in autophagy – target, product, and what else? Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2020.02.013
Olzmann JA, Carvalho P (2019) Dynamics and functions of lipid droplets. Nat Rev Mol Cell Bio 20:137–155. https://doi.org/10.1038/s41580-018-0085-z
Ong KT, Mashek MT, Bu SY et al (2011) Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 53:116–126. https://doi.org/10.1002/hep.24006
Palm W, Thompson CB (2017) Nutrient acquisition strategies of mammalian cells. Nature 546:234–242. https://doi.org/10.1038/nature22379
Palm W, Park Y, Wright K et al (2015) The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162:259–270. https://doi.org/10.1016/j.cell.2015.06.017
Panda PK, Patra S, Naik PP et al (2019) Deacetylation of LAMP1 drives lipophagy-dependent generation of free fatty acids by Abrus agglutinin to promote senescence in prostate cancer. J Cell Physiol 235:2776–2791. https://doi.org/10.1002/jcp.29182
Panzilius E, Holstein F, Bannier-Hélaouët M et al (2018) A cell-density dependent metabolic switch sensitizes breast cancer cells to ferroptosis. Biorxiv 417949. https://doi.org/10.1101/417949
Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23:27–47. https://doi.org/10.1016/j.cmet.2015.12.006
Peng Y, Miao H, Wu S et al (2016) ABHD5 interacts with BECN1 to regulate autophagy and tumorigenesis of colon cancer independent of PNPLA2. Autophagy 12:2167–2182. https://doi.org/10.1080/15548627.2016.1217380
Penno A, Hackenbroich G, Thiele C (2012) Phospholipids and lipid droplets. Biochim Biophys Acta 1831:589–594. https://doi.org/10.1016/j.bbalip.2012.12.001
Petan T, Jarc E, Jusović M (2018) Lipid droplets in cancer: guardians of fat in a stressful world. Molecules 23:1941. https://doi.org/10.3390/molecules23081941
Petschnigg J, Wolinski H, Kolb D et al (2009) Good fat, essential cellular requirements for triacylglycerol synthesis to maintain membrane homeostasis in yeast. J Biol Chem 284:30981–30993. https://doi.org/10.1074/jbc.m109.024752
Pike LS, Smift AL, Croteau NJ et al (2011) Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta 1807:726–734. https://doi.org/10.1016/j.bbabio.2010.10.022
Prentki M, Madiraju S (2008) Glycerolipid metabolism and signaling in health and disease. Endocr Rev 29:647–676. https://doi.org/10.1210/er.2008-0007
Przybytkowski E, Joly E, Nolan CJ et al (2007) Upregulation of cellular triacylglycerol - free fatty acid cycling by oleate is associated with long-term serum-free survival of human breast cancer cells. Biochem Cell Biol 85:301–310. https://doi.org/10.1139/o07-001
Pucer A, Brglez V, Payré C et al (2013) Group X secreted phospholipase A2 induces lipid droplet formation and prolongs breast cancer cell survival. Mol Cancer 12:111. https://doi.org/10.1186/1476-4598-12-111
Qiao S, Koh S-B, Vivekanandan V et al (2020) REDD1 loss reprograms lipid metabolism to drive progression of RAS mutant tumors. Genes Dev. https://doi.org/10.1101/gad.335166.119
Qiu B, Ackerman D, Sanchez DJ et al (2015) HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov 5:652–667. https://doi.org/10.1158/2159-8290.cd-14-1507
Ramakrishnan R, Tyurin VA, Tuyrin VA et al (2014) Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol 192:2920–2931. https://doi.org/10.4049/jimmunol.1302801
Rambold AS, Pearce EL (2018) Mitochondrial dynamics at the Interface of immune cell metabolism and function. Trends Immunol 39:6–18. https://doi.org/10.1016/j.it.2017.08.006
Rambold AS, Cohen S, Lippincott-Schwartz J (2015) Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell 32:678–692. https://doi.org/10.1016/j.devcel.2015.01.029
Raud B, Roy DG, Divakaruni AS et al (2018) Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab 28:504–515.e7. https://doi.org/10.1016/j.cmet.2018.06.002
Röhrig F, Schulze A (2016) The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 16:732–749. https://doi.org/10.1038/nrc.2016.89
Salo VT, Ikonen E (2019) Moving out but keeping in touch: contacts between endoplasmic reticulum and lipid droplets. Curr Opin Cell Biol 57:64–70. https://doi.org/10.1016/j.ceb.2018.11.002
Scaglia N, Chisholm JW, Igal RA (2009) Inhibition of stearoylCoA desaturase-1 inactivates acetyl-CoA carboxylase and impairs proliferation in cancer cells: role of AMPK. PLoS One 4:e6812. https://doi.org/10.1371/journal.pone.0006812
Schaffer JE (2003) Lipotoxicity: when tissues overeat. Current opinion in lipidology 14:281–287. https://doi.org/10.1097/01.mol.0000073508.41685.7f
Schreiber R, Xie H, Schweiger M (2019) Of mice and men: the physiological role of adipose triglyceride lipase (ATGL). Biochim Biophys Acta 1864:880–899. https://doi.org/10.1016/j.bbalip.2018.10.008
Schuldiner M, Bohnert M (2017) A different kind of love – lipid droplet contact sites. Biochim Biophys Acta 1862:1188–1196. https://doi.org/10.1016/j.bbalip.2017.06.005
Schulze RJ, Sathyanarayan A, Mashek DG (2017) Breaking fat: the regulation and mechanisms of lipophagy. Biochim Biophys Acta 1862:1178–1187. https://doi.org/10.1016/j.bbalip.2017.06.008
Senkal CE, Salama MF, Snider AJ et al (2017) Ceramide is metabolized to Acylceramide and stored in lipid droplets. Cell Metab 25:686–697. https://doi.org/10.1016/j.cmet.2017.02.010
Seo AY, Lau P-W, Feliciano D et al (2017) AMPK and vacuole-associated Atg14p orchestrate μ-lipophagy for energy production and long-term survival under glucose starvation. elife 6:1025. https://doi.org/10.7554/elife.21690
Shpilka T, Welter E, Borovsky N et al (2015) Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J 34:2117–2131. https://doi.org/10.15252/embj.201490315
Singh R, Kaushik S, Wang Y et al (2009) Autophagy regulates lipid metabolism. Nature 458:1131–1135. https://doi.org/10.1038/nature07976
Smirnova E, Goldberg EB, Makarova KS et al (2005) ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep 7:106–113. https://doi.org/10.1038/sj.embor.7400559
Snaebjornsson MT, Janaki-Raman S, Schulze A (2019) Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab 31:62–76. https://doi.org/10.1016/j.cmet.2019.11.010
Thiam AR, Beller M (2017) The why, when and how of lipid droplet diversity. J Cell Sci 130:315–324. https://doi.org/10.1242/jcs.192021
Tirinato L, Pagliari F, Limongi T et al (2017) An overview of lipid droplets in cancer and cancer stem cells. Stem Cells Int 2017:1656053. https://doi.org/10.1155/2017/1656053
Tousignant KD, Rockstroh A, Poad BL et al (2020) Therapy-induced lipid uptake and remodeling underpin ferroptosis hypersensitivity in prostate cancer. Cancer Metab 8:11. https://doi.org/10.1186/s40170-020-00217-6
Ueno M, Shen W-J, Patel S et al (2013) Fat-specific protein 27 modulates nuclear factor of activated T cells 5 and the cellular response to stress. J Lipid Res 54:734–743. https://doi.org/10.1194/jlr.m033365
VandeKopple MJ, Wu J, Auer EN et al (2019) HILPDA regulates lipid metabolism, lipid droplet abundance, and response to microenvironmental stress in solid tumors. Mol Cancer Res 17:2089–2101. https://doi.org/10.1158/1541-7786.mcr-18-1343
Veglia F, Tyurin VA, Mohammadyani D et al (2017) Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun 8:2122. https://doi.org/10.1038/s41467-017-02186-9
Viswanathan VS, Ryan MJ, Dhruv HD et al (2017) Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 275:28110. https://doi.org/10.1038/nature23007
Walther TC, Chung J, Farese RV Jr (2017) Lipid droplet biogenesis. Annu Rev Cell Dev Biol 33:491–510. https://doi.org/10.1146/annurev-cellbio-100616-060608
Wang C-W (2016) Lipid droplets, lipophagy, and beyond. Biochim Biophys Acta 1861:793–805. https://doi.org/10.1016/j.bbalip.2015.12.010
Wang YY, Attané C, Milhas D et al (2017) Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2:e87489. https://doi.org/10.1172/jci.insight.87489
Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21:297–308. https://doi.org/10.1016/j.ccr.2012.02.014
Wellen KE, Thompson CB (2010) Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell 40:323–332. https://doi.org/10.1016/j.molcel.2010.10.004
Welte MA, Gould AP (2017) Lipid droplet functions beyond energy storage. Biochim Biophys Acta 1862:1260–1272. https://doi.org/10.1016/j.bbalip.2017.07.006
Wen Y-A, Xing X, Harris JW et al (2017) Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis 8:e2593–e2593. https://doi.org/10.1038/cddis.2017.21
Wendel AA, Lewin TM, Coleman RA (2009) Glycerol-3-phosphate acyltransferases: rate limiting enzymes of triacylglycerol biosynthesis. Biochim Biophys Acta 1791:501–506. https://doi.org/10.1016/j.bbalip.2008.10.010
Wilcock D, Badrock A, Owen R et al (2020) DGAT1 is a lipid metabolism oncoprotein that enables cancer cells to accumulate fatty acid while avoiding lipotoxicity. bioRxiv 2020.06.23.166603. https://doi.org/10.1101/2020.06.23.166603
Wilfling F, Wang H, Haas JT et al (2013) Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell 24:384–399. https://doi.org/10.1016/j.devcel.2013.01.013
Xu G, Jiang Y, Xiao Y et al (2015) Fast clearance of lipid droplets through MAP 1S-activated autophagy suppresses clear cell renal cell carcinomas and promotes patient survival. Oncotarget 7:6255–6265. https://doi.org/10.18632/oncotarget.6669
Yecies JL, Manning BD (2011) mTOR links oncogenic signaling to tumor cell metabolism. J Mol Med 89:221–228. https://doi.org/10.1007/s00109-011-0726-6
Young SG, Zechner R (2013) Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev 27:459–484. https://doi.org/10.1101/gad.209296.112
Young RM, Ackerman D, Quinn ZL et al (2013) Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev 27:1115–1131. https://doi.org/10.1101/gad.198630.112
Zanghellini J, Natter K et al (2008) Quantitative modeling of triacylglycerol homeostasis in yeast--metabolic requirement for lipolysis to promote membrane lipid synthesis and cellular growth. FEBS J 275:5552–5563. https://doi.org/10.1111/j.1742-4658.2008.06681.x
Zechner R, Zimmermann R, Eichmann TO et al (2012) FAT SIGNALS - lipases and lipolysis in lipid metabolism and signaling. Cell Metab 15:279–291. https://doi.org/10.1016/j.cmet.2011.12.018
Zechner R, Madeo F, Kratky D (2017) Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol 18:671–684. https://doi.org/10.1038/nrm.2017.76
Zhang P, Reue K (2017) Lipin proteins and glycerolipid metabolism: roles at the ER membrane and beyond. Biochim Biophys Acta 1859:1583–1595. https://doi.org/10.1016/j.bbamem.2017.04.007
Zhang X, Saarinen AM, Hitosugi T et al (2017) Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. elife 6:739. https://doi.org/10.7554/elife.31132
Zhang Y, Tan H, Daniels JD et al (2019) Imidazole ketone Erastin induces Ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol 26:623–633.e9. https://doi.org/10.1016/j.chembiol.2019.01.008
Zhao T, Ding X, Du H, Yan C (2016) Lung epithelial cell-specific expression of human Lysosomal acid lipase ameliorates lung inflammation and tumor metastasis in Lipa(−/−) mice. Am J Pathol 186:2183–2192. https://doi.org/10.1016/j.ajpath.2016.04.014
Zhu J, Xu M, Liu Y et al (2019) Phosphorylation of PLIN3 by AMPK promotes dispersion of lipid droplets during starvation. Protein Cell 10:382–387. https://doi.org/10.1007/s13238-018-0593-9
Zimmermann R, Strauss JG, Haemmerle G et al (2004) Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306:1383–1386. https://doi.org/10.1126/science.1100747
Zou Y, Palte MJ, Deik AA et al (2019) A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 10:354. https://doi.org/10.1038/s41467-019-09277-9
Acknowledgments
I am profoundly grateful to all fellow scientists for their contributions to the current understanding of the topic, and I sincerely apologize to those whose important work could not be cited in this article. This work was supported by P1-0207 Research Programme grant and the J7-1818 Research Project grant to T.P. from the Slovenian Research Agency.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this chapter

Cite this chapter
Petan, T. (2020). Lipid Droplets in Cancer. In: Pedersen, S.H.F., Barber, D.L. (eds) Organelles in Disease. Reviews of Physiology, Biochemistry and Pharmacology, vol 185. Springer, Cham. https://doi.org/10.1007/112_2020_51
Download citation
DOI: https://doi.org/10.1007/112_2020_51
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-22594-9
Online ISBN: 978-3-031-22595-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)