
Abstract
The computational analysis of enzymes that participate in lipid metabolism has both common and unique challenges when compared to the whole protein universe. Some of the hurdles that interfere with the functional annotation of lipid metabolic enzymes that are common to other pathways include the definition of proper starting datasets, the construction of reliable multiple sequence alignments, the definition of appropriate evolutionary models, and the reconstruction of phylogenetic trees with high statistical support, particularly for large datasets. Most enzymes that take part in lipid metabolism belong to complex superfamilies with many members that are not involved in lipid metabolism. In addition, some enzymes that do not have sequence similarity catalyze similar or even identical reactions. Some of the challenges that, albeit not unique, are more specific to lipid metabolism refer to the high compartmentalization of the routes, the catalysis in hydrophobic environments and, related to this, the function near or in biological membranes.
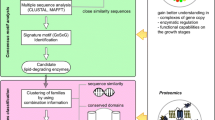
In this work, we provide guidelines intended to assist in the proper functional annotation of lipid metabolic enzymes, based on previous experiences related to the phospholipase D superfamily and the annotation of the triglyceride synthesis pathway in algae. We describe a pipeline that starts with the definition of an initial set of sequences to be used in similarity-based searches and ends in the reconstruction of phylogenies. We also mention the main issues that have to be taken into consideration when using tools to analyze subcellular localization, hydrophobicity patterns, or presence of transmembrane domains in lipid metabolic enzymes.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others


References
Eirin-Lopez JM, Rebordinos L, Rooney AP, Rozas J (2012) The birth-and-death evolution of multigene families revisited. Genome Dyn 7:170–196. doi:10.1159/000337119
Beligni MV, Bagnato C, Prados MB, Bondino H, Laxalt AM, Munnik T, Ten Have A (2015) The diversity of algal phospholipase D homologs revealed by biocomputational analysis. J Phycol 51(5):943–962. doi:10.1111/jpy.12334
Brown JR, Auger KR (2011) Phylogenomics of phosphoinositide lipid kinases: perspectives on the evolution of second messenger signaling and drug discovery. BMC Evol Biol 11:4. doi:10.1186/1471-2148-11-4
Cao H (2011) Structure-function analysis of diacylglycerol acyltransferase sequences from 70 organisms. BMC Res Notes 4:249. doi:10.1186/1756-0500-4-249
Das A, Davis MA, Rudel LL (2008) Identification of putative active site residues of ACAT enzymes. J Lipid Res 49(8):1770–1781. doi:10.1194/jlr.M800131-JLR200
Wilson D, Pethica R, Zhou Y, Talbot C, Vogel C, Madera M, Chothia C, Gough J (2009) SUPERFAMILY–sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res 37(Database issue):D380–D386. doi:10.1093/nar/gkn762
Schomburg I, Chang A, Placzek S, Sohngen C, Rother M, Lang M, Munaretto C, Ulas S, Stelzer M, Grote A, Scheer M, Schomburg D (2013) BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA. Nucleic Acids Res 41(Database issue):D764–D772. doi:10.1093/nar/gks1049
Aoki KF, Kanehisa M (2005) Using the KEGG database resource. Curr Protoc Bioinformatics Chapter 1:Unit 1 12. doi:10.1002/0471250953.bi0112s11
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD (2012) The Pfam protein families database. Nucleic Acids Res 40(Database issue):D290–D301. doi:10.1093/nar/gkr1065
Sigrist CJ, Cerutti L, Hulo N, Gattiker A, Falquet L, Pagni M, Bairoch A, Bucher P (2002) PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform 3(3):265–274
Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics (Oxford, England) 30(9):1236–1240. doi:10.1093/bioinformatics/btu031
Haft DH, Selengut JD, White O (2003) The TIGRFAMs database of protein families. Nucleic Acids Res 31(1):371–373
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28(1):235–242
UniProt Consortium (2015) UniProt: a hub for protein information. Nucleic Acids Res 43(Database issue):D204–D212. doi:10.1093/nar/gku989
Information NCBI (2016) http://www.ncbi.nlm.nih.gov/. Accessed 15 May 2016
Mashima J, Kodama Y, Kosuge T, Fujisawa T, Katayama T, Nagasaki H, Okuda Y, Kaminuma E, Ogasawara O, Okubo K, Nakamura Y, Takagi T (2016) DNA data bank of Japan (DDBJ) progress report. Nucleic Acids Res 44(D1):D51–D57. doi:10.1093/nar/gkv1105
Nordberg H, Cantor M, Dusheyko S, Hua S, Poliakov A, Shabalov I, Smirnova T, Grigoriev IV, Dubchak I (2014) The genome portal of the department of energy joint genome institute: 2014 updates. Nucleic Acids Res 42(Database issue):D26–D31. doi:10.1093/nar/gkt1069
Institute TB (2016) https://www.broadinstitute.org/. Accessed 10 May 2016
Aurrecoechea C, Heiges M, Wang H, Wang Z, Fischer S, Rhodes P, Miller J, Kraemer E, Stoeckert CJ Jr, Roos DS, Kissinger JC (2007) ApiDB: integrated resources for the apicomplexan bioinformatics resource center. Nucleic Acids Res 35(Database issue):D427–D430. doi:10.1093/nar/gkl880
Institute WTS (2016) http://www.sanger.ac.uk/. Accessed 15 May 2016
Institute JCV (2016) http://www.jcvi.org/cms/home/. Accessed 1 May 2016
Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR (2015) HMMER web server: 2015 update. Nucleic Acids Res 43(W1):W30–W38. doi:10.1093/nar/gkv397
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410. doi:10.1016/s0022-2836(05)80360-2
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Zhang Z, Schaffer AA, Miller W, Madden TL, Lipman DJ, Koonin EV, Altschul SF (1998) Protein sequence similarity searches using patterns as seeds. Nucleic Acids Res 26(17):3986–3990
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics (Oxford, England) 28(23):3150–3152. doi:10.1093/bioinformatics/bts565
Notredame C, Higgins DG, Heringa J (2000) T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302(1):205–217. doi:10.1006/jmbi.2000.4042
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30(14):3059–3066
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. doi:10.1093/nar/gkh340
Pei J, Kim BH, Grishin NV (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36(7):2295–2300. doi:10.1093/nar/gkn072
Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39(Web Server issue):W29–W37. doi:10.1093/nar/gkr367
Nicholas KB, Nicholas HB, Deerfield D (1997) GeneDoc: analysis and visualisation of genetic variation. EMBnet News 4:14
Larsson A (2014) AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics (Oxford, England) 30(22):3276–3278. doi:10.1093/bioinformatics/btu531
Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27(2):221–224. doi:10.1093/molbev/msp259
Criscuolo A, Gribaldo S (2010) BMGE (block mapping and gathering with entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol 10:210. doi:10.1186/1471-2148-10-210
Thompson JD, Thierry JC, Poch O (2003) RASCAL: rapid scanning and correction of multiple sequence alignments. Bioinformatics (Oxford, England) 19(9):1155–1161
Muller J, Creevey CJ, Thompson JD, Arendt D, Bork P (2010) AQUA: automated quality improvement for multiple sequence alignments. Bioinformatics (Oxford, England) 26(2):263–265. doi:10.1093/bioinformatics/btp651
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics (Oxford, England) 21(9):2104–2105. doi:10.1093/bioinformatics/bti263
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics (Oxford, England) 30(9):1312–1313. doi:10.1093/bioinformatics/btu033
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59(3):307–321. doi:10.1093/sysbio/syq010
Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ (2016) W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res 44(W1):W232–W235. doi:10.1093/nar/gkw256
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61(3):539–542. doi:10.1093/sysbio/sys029
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29(8):1969–1973. doi:10.1093/molbev/mss075
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10(6):845–858. doi:10.1038/nprot.2015.053
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I-TASSER suite: protein structure and function prediction. Nat Methods 12(1):7–8. doi:10.1038/nmeth.3213
Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2(4):953–971. doi:10.1038/nprot.2007.131
Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K (2007) WoLF PSORT: protein localization predictor. Nucleic Acids Res 35(Web Server issue):W585–W587. doi:10.1093/nar/gkm259
Emanuelsson O, Nielsen H, von Heijne G (1999) ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci 8(5):978–984. doi:10.1110/ps.8.5.978
Claros MG, Vincens P (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem 241(3):779–786
Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S (2004) Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel 17(4):349–356. doi:10.1093/protein/gzh037
Tardif M, Atteia A, Specht M, Cogne G, Rolland N, Brugiere S, Hippler M, Ferro M, Bruley C, Peltier G, Vallon O, Cournac L (2012) PredAlgo: a new subcellular localization prediction tool dedicated to green algae. Mol Biol Evol 29(12):3625–3639. doi:10.1093/molbev/mss178
Gschloessl B, Guermeur Y, Cock JM (2008) HECTAR: a method to predict subcellular targeting in heterokonts. BMC Bioinformatics 9:393. doi:10.1186/1471-2105-9-393
Zuegge J, Ralph S, Schmuker M, McFadden GI, Schneider G (2001) Deciphering apicoplast targeting signals--feature extraction from nuclear-encoded precursors of Plasmodium falciparum apicoplast proteins. Gene 280(1–2):19–26
Gasteiger E, Hoogland C, Gattiker A, Se D, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The proteomics protocols handbook. Humana Press, Totowa, NJ, pp 571–607. doi:10.1385/1-59259-890-0:571
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305(3):567–580. doi:10.1006/jmbi.2000.4315
Tusnady GE, Simon I (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics (Oxford, England) 17(9):849–850
Dobson L, Remenyi I, Tusnady GE (2015) CCTOP: a consensus constrained TOPology prediction web server. Nucleic Acids Res 43(W1):W408–W412. doi:10.1093/nar/gkv451
Kanz C, Aldebert P, Althorpe N, Baker W, Baldwin A, Bates K, Browne P, van den Broek A, Castro M, Cochrane G, Duggan K, Eberhardt R, Faruque N, Gamble J, Diez FG, Harte N, Kulikova T, Lin Q, Lombard V, Lopez R, Mancuso R, McHale M, Nardone F, Silventoinen V, Sobhany S, Stoehr P, Tuli MA, Tzouvara K, Vaughan R, Wu D, Zhu W, Apweiler R (2005) The EMBL nucleotide sequence database. Nucleic Acids Res 33(Database issue):D29–D33. doi:10.1093/nar/gki098
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680
Kemena C, Notredame C (2009) Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics (Oxford, England) 25(19):2455–2465. doi:10.1093/bioinformatics/btp452
Thompson JD, Plewniak F, Ripp R, Thierry JC, Poch O (2001) Towards a reliable objective function for multiple sequence alignments. J Mol Biol 314(4):937–951. doi:10.1006/jmbi.2001.5187
Anisimova M, Gascuel O (2006) Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol 55(4):539–552. doi:10.1080/10635150600755453
Nylander JA, Wilgenbusch JC, Warren DL, Swofford DL (2008) AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics (Oxford, England) 24(4):581–583. doi:10.1093/bioinformatics/btm388
Small I, Peeters N, Legeai F, Lurin C (2004) Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4(6):1581–1590. doi:10.1002/pmic.200300776
Jiroutova K, Horak A, Bowler C, Obornik M (2007) Tryptophan biosynthesis in stramenopiles: eukaryotic winners in the diatom complex chloroplast. J Mol Evol 65(5):496–511. doi:10.1007/s00239-007-9022-z
Argos P, Rao JK, Hargrave PA (1982) Structural prediction of membrane-bound proteins. Eur J Biochem 128(2–3):565–575
Eisenberg D, Schwarz E, Komaromy M, Wall R (1984) Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol 179(1):125–142
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157(1):105–132
Moller S, Croning MD, Apweiler R (2001) Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics (Oxford, England) 17(7):646–653
Lewin TM, Wang P, Coleman RA (1999) Analysis of amino acid motifs diagnostic for the sn-glycerol-3-phosphate acyltransferase reaction. Biochemistry 38(18):5764–5771. doi:10.1021/bi982805d
Wendel AA LT, Coleman RA (2010) NIH public access 1791: pp 380–386
Zharkikh A, Li WH (1992) Statistical properties of bootstrap estimation of phylogenetic variability from nucleotide sequences: II. Four taxa without a molecular clock. J Mol Evol 35(4):356–366
Kall L, Krogh A, Sonnhammer EL (2004) A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338(5):1027–1036. doi:10.1016/j.jmb.2004.03.016
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media LLC
About this protocol
Cite this protocol
Bagnato, C., Have, A.T., Prados, M.B., Beligni, M.V. (2017). Computational Functional Analysis of Lipid Metabolic Enzymes. In: Bhattacharya, S. (eds) Lipidomics. Methods in Molecular Biology, vol 1609. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-6996-8_17
Download citation
DOI: https://doi.org/10.1007/978-1-4939-6996-8_17
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-6995-1
Online ISBN: 978-1-4939-6996-8
eBook Packages: Springer Protocols