
Abstract
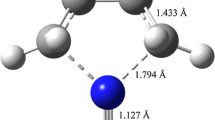
Molecular dissociation of chlorine peroxide (ClOOCl), which consists of two elementary dissociation channels (of Cl–O and O–O), is investigated using molecular dynamics simulations on a neural network-fitted potential energy surface constructed by MP2 calculations with the 6-311G(d,p) basis set. When relaxed scans of the surface are executed, we observe that Cl–O dissociation is extremely reactive with a low barrier height of 0.1928 eV (18.602 kJ/mol), while O–O bond scission is less reactive (0.7164 eV or 69.122 kJ/mol). By utilizing the “novelty sampling” method, 35,006 data points in the ClOOCl configuration hyperspace are collected, and a 40-neuron feed-forward neural network is employed to fit approximately 90% of the data to produce an analytic potential energy function. The mean absolute error and root mean squared error of this fit are reported as 0.0078 eV (0.753 kJ/mol) and 0.0137 eV (1.322 kJ/mol), respectively. Finally, quasi-classical molecular dynamics is executed at various levels of internal energy (from 0.8 to 1.3 eV) to examine the bond ruptures. The two first-order rate coefficients are computed statistically, and the results range from 5.20 to 22.67 ps−1 for Cl–O dissociation and 3.72–8.35 ps−1 for O–O dissociation. Rice-Ramsperger-Kassel theory is utilized to classically correlate internal energies to rate coefficients in both cases, and the plots exhibit very good linearity, thus can be employed to predict rate coefficients at other internal energy levels with good reliability.







Similar content being viewed by others


References
Molina LT, Molina MJ (1987) Production of chlorine oxide (Cl2O2) from the self-reaction of the chlorine oxide (ClO) radical. J Phys Chem 91(2):433–436
Cheng B-M, Lee Y-P (1989) Production and trapping of gaseous dimeric ClO: the infrared spectrum of chlorine peroxide (ClOOCl) in solid argon. J Chem Phys 90(11):5930–5935
Cox RA, Hayman GD (1988) The stability and photochemistry of dimers of the ClO radical and implications for Antarctic ozone depletion. Nature 332(6167):796–800
Barrett JW, Solomon PM, de Zafra RL, Jaramillo M, Emmons L, Parrish A (1988) Formation of the Antarctic ozone hole by the CIO dimer mechanism. Nature 336(6198):455–458
Huder KJ, DeMore WB (1995) Absorption cross sections of the ClO dimer. J Phys Chem 99(12):3905–3908
Demore WB, Golden DM, Hampson RF, Howard CJ, Kolb CE, Kurylo MJ, Molina MJ, Ravishankara AR, Sander SP (1997) Chemical kinetics and photochemical data for use in stratospheric modeling. JPL Publication, Vol 94-26. Jet Propulsion Laboratory, California Institute of Technology, Pasadena, CA
DeMore WB, Tschuikow-Roux E (1990) Ultraviolet spectrum and chemical reactivity of the chlorine monoxide dimer. J Phys Chem 94(15):5856–5860
Stimpfle RM, Wilmouth DM, Salawitch RJ, Anderson JG (2004) First measurements of ClOOCl in the stratosphere: the coupling of ClOOCl and ClO in the Arctic polar vortex. J Geophys Res 109(D3):D03301
Avallone LM, Toohey DW (2001) Tests of halogen photochemistry using in situ measurements of ClO and BrO in the lower polar stratosphere. J Geophys Res 106(D10):10411–10421
Plenge J, Flesch R, Kühl S, Vogel B, Müller R, Stroh F, Rühl E (2004) Ultraviolet photolysis of the ClO Dimer. J Phys Chem A 108(22):4859–4863
Sumińska-Ebersoldt O, Lehmann R, Wegner T, Grooß JU, Hösen E, Weigel R, Volk CM, Borrmann S, Rex M, Stroh F, von Hobe M (2011) ClOOCl photolysis at high solar zenith angles: analysis of the RECONCILE self-match flight. Atmos Chem Phys Discuss 11(7):18901–18926
Huang W-T, Chen AF, Chen IC, Tsai C-H, Lin JJ-M (2011) Photodissociation dynamics of ClOOCl at 248.4 and 308.4 nm. Phys Chem Chem Phys 13(18):8195–8203
Jacobs J, Kronberg M, Mueller HSP, Willner H (1994) An experimental study on the photochemistry and vibrational spectroscopy of three isomers of Cl2O2 isolated in cryogenic matrixes. J Am Chem Soc 116(3):1106–1114
Han Y-K, Kim KH, Lee YS, Baeck KK (1998) Energies and structures of isomers of Cl2O2 calculated by density functional methods. J Mol Struct THEOCHEM 431(1–2):185–189
Ochterski JW, Petersson GA, Montgomery JA (1996) A complete basis set model chemistry. V. Extensions to six or more heavy atoms. J Chem Phys 104(7):2598–2619
Petersson GA, Tensfeldt TG, Montgomery JA (1991) A complete basis set model chemistry. III. The complete basis set-quadratic configuration interaction family of methods. J Chem Phys 94(9):6091–6101
Curtiss LA, Raghavachari K, Trucks GW, Pople JA (1991) Gaussian-2 theory for molecular energies of first- and second-row compounds. J Chem Phys 94(11):7221–7230
Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA (1999) A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J Chem Phys 110(6):2822–2827
Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA (2000) A complete basis set model chemistry. VII. Use of the minimum population localization method. J Chem Phys 112(15):6532–6542
Baboul AG, Curtiss LA, Redfern PC, Raghavachari K (1999) Gaussian-3 theory using density functional geometries and zero-point energies. J Chem Phys 110(16):7650–7657
Jalbout AF (2002) The isomerization of ClOOCl: high level ab initio and density functional theory analysis. J Mol Struct THEOCHEM 594(1–2):1–7
Kaledin AL, Morokuma K (2000) An ab initio direct-trajectory study of the photodissociation of ClOOCl. J Chem Phys 113(14):5750–5762
Hegarty D, Robb MA (1979) Application of unitary group methods to configuration interaction calculations. Mol Phys 38(6):1795–1812
Eade RHA, Robb MA (1981) Direct minimization in mc scf theory. The quasi-newton method. Chem Phys Lett 83(2):362–368
Schlegel HB, Robb MA (1982) MC SCF gradient optimization of the H2CO–>H2+CO transition structure. Chem Phys Lett 93(1):43–46
Bernardi F, Bottoni A, McDouall JJW, Robb MA, Schlegel HB (1984) MCSCF gradient calculation of transition structures in organic reactions. Faraday Symp Chem Soc 19:137–147
Frisch M, Ragazos IN, Robb MA, Bernhard Schlegel H (1992) An evaluation of three direct MC-SCF procedures. Chem Phys Lett 189(6):524–528
Yamamoto N, Vreven T, Robb MA, Frisch MJ, Bernhard Schlegel H (1996) A direct derivative MC-SCF procedure. Chem Phys Lett 250(3–4):373–378
Toniolo A, Granucci G, Inglese S, Persico M (2001) Theoretical study of the photodissociation dynamics of ClOOCl. Phys Chem Chem Phys 3(19):4266–4279
Dewar MJS, Thiel W (1977) Ground states of molecules. 38. The MNDO method. Approximations and parameters. J Am Chem Soc 99(15):4899–4907
Oncák M, Sistík L, Slavícek P (2010) Can theory quantitatively model stratospheric photolysis? Ab initio estimate of absolute absorption cross sections of ClOOCl. J Chem Phys 133(17):174303
Finley J, Malmqvist P-Å, Roos BO, Serrano-Andrés L (1998) The multi-state CASPT2 method. Chem Phys Lett 288(2–4):299–306
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03. Revision C.02 edn., Wallingford, CT
Birk M, Friedl RR, Cohen EA, Pickett HM, Sander SP (1989) The rotational spectrum and structure of chlorine peroxide. J Chem Phys 91(11):6588–6597
Becke AD (1993) Density functional thermochemistry. III. The role of exact exchange. J Chem Phys 98(7):5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37(2):785
Vosko SH, Wilk L, Nusair M (1980) Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can J Phys 58(8):1200–1211
Devlin FJ, Finley JW, Stephens PJ, Frisch MJ (1995) Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields: a comparison of local, nonlocal, and hybrid density functionals. J Phys Chem 99(46):16883–16902
Zhao Y, Truhlar DG (2006) A density functional that accounts for medium-range correlation energies in organic chemistry. Org Lett 8(25):5753–5755
Frisch MJ, Head-Gordon M, Pople JA (1990) A direct MP2 gradient method. Chem Phys Lett 166(3):275–280
Frisch MJ, Head-Gordon M, Pople JA (1990) Semi-direct algorithms for the MP2 energy and gradient. Chem Phys Lett 166(3):281–289
Head-Gordon M, Pople JA, Frisch MJ (1988) MP2 energy evaluation by direct methods. Chem Phys Lett 153(6):503–506
Head-Gordon M, Head-Gordon T (1994) Analytic MP2 frequencies without fifth-order storage. Theory and application to bifurcated hydrogen bonds in the water hexamer. Chem Phys Lett 220(1–2):122–128
Sæbø S, Almlöf J (1989) Avoiding the integral storage bottleneck in LCAO calculations of electron correlation. Chem Phys Lett 154(1):83–89
McLean AD, Chandler GS (1980) Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J Chem Phys 72(10):5639–5648
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys 72(1):650–654
Davidson ER (1996) Comment on “Comment on Dunning’s correlation-consistent basis sets”. Chem Phys Lett 260(3–4):514–518
Tomasello P, Ehara M, Nakatsuji H (2003) Theoretical investigation on the valence ionization spectra of Cl2O, ClOOCl, and F2O by correlation-based configuration interaction methods. J Chem Phys 118(13):5811–5820
Krishnan R, Pople JA (1978) Approximate fourth-order perturbation theory of the electron correlation energy. Int J Quantum Chem 14(1):91–100
Purvis GD, Bartlett RJ (1982) A full coupled-cluster singles and doubles model: the inclusion of disconnected triples. J Chem Phys 76(4):1910–1918
Scuseria GE, Janssen CL, Schaefer HF (1988) An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J Chem Phys 89(12):7382–7387
Scuseria GE, Schaefer HF (1989) Is coupled cluster singles and doubles (CCSD) more computationally intensive than quadratic configuration interaction (QCISD)? J Chem Phys 90(7):3700–3703
Hagan MTD, Beale HB (1996) Neural network design. Colorado Bookstore, Boulder, CO
Handley CM, Popelier PLA (2010) Potential energy surfaces fitted by artificial neural networks. J Phys Chem A 114(10):3371–3383
Malshe M, Raff LM, Rockley MG, Hagan M, Agrawal PM, Komanduri R (2007) Theoretical investigation of the dissociation dynamics of vibrationally excited vinyl bromide on an ab initio potential-energy surface obtained using modified novelty sampling and feedforward neural networks. II. Numerical application of the method. J Chem Phys 127(13):134105
Agrawal PM, Raff LM, Hagan MT, Komanduri R (2006) Molecular dynamics investigations of the dissociation of SiO2 on an ab initio potential energy surface obtained using neural network methods. J Chem Phys 124(13):134306
Le HM, Raff LM (2008) Cis→trans, trans→cis isomerizations and N–O bond dissociation of nitrous acid (HONO) on an ab initio potential surface obtained by novelty sampling and feed-forward neural network fitting. J Chem Phys 128(19):194310
Le HM, Huynh S, Raff LM (2009) Molecular dissociation of hydrogen peroxide (HOOH) on a neural network ab initio potential surface with a new configuration sampling method involving gradient fitting. J Chem Phys 131(1):014107
Le HM, Raff LM (2009) Molecular dynamics investigation of the bimolecular reaction BeH + H2 → BeH2 + H on an ab initio potential-energy surface obtained using neural network methods with both potential and gradient accuracy determination. J Phys Chem A 114(1):45–53
Le HM, Dinh TS, Le HV (2011) Molecular dynamics investigations of ozone on an ab initio potential energy surface with the utilization of pattern-recognition neural network for accurate determination of product formation. J Phys Chem A 115(40):10862–10870
Pukrittayakamee A, Malshe M, Hagan M, Raff LM, Narulkar R, Bukkapatnum S, Komanduri R (2009) Simultaneous fitting of a potential-energy surface and its corresponding force fields using feedforward neural networks. J Chem Phys 130(13):134101
Malshe M, Raff LM, Hagan M, Bukkapatnam S, Komanduri R (2010) Input vector optimization of feed-forward neural networks for fitting ab initio potential-energy databases. J Chem Phys 132(20):204103
Handley CM, Popelier PLA (2009) Dynamically polarizable water potential based on multipole moments trained by machine learning. J Chem Theory Comput 5(6):1474–1489
Caruana R, Lawrence S, Giles CL (2000) Overfitting in neural nets: backpropagation, conjugate gradient, and early stopping. In: Proceedings of neural information processing systems, pp 402–408
Malshe M, Narulkar R, Raff LM, Hagan M, Bukkapatnam S, Agrawal PM, Komanduri R (2009) Development of generalized potential-energy surfaces using many-body expansions, neural networks, and moiety energy approximations. J Chem Phys 130(18):184102
Agrawal PM, Malshe M, Narulkar R, Raff LM, Hagan M, Bukkapatnum S, Komanduri R (2009) A self-starting method for obtaining analytic potential-energy surfaces from ab initio electronic structure calculations. J Phys Chem A 113(5):869–877
MathWorks (2011) MATLAB. Natick, MA
Raff LM (1988) Projection methods for obtaining intramolecular energy transfer rates from classical trajectory results: application to 1,2-difluoroethane. J Chem Phys 89(9):5680–5691
Acknowledgments
We thank Prof. Lionel M. Raff from the Chemistry Department, Oklahoma State University for his helpful advice in this research. The authors also thank the Faculty of Materials Science, College of Science, Vietnam National University (VNU) in Ho Chi Minh City for their computing supports. The research grant for this work is funded by VNU.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Le, A.T.H., Vu, N.H., Dinh, T.S. et al. Molecular dynamics investigations of chlorine peroxide dissociation on a neural network ab initio potential energy surface. Theor Chem Acc 131, 1158 (2012). https://doi.org/10.1007/s00214-012-1158-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-012-1158-2