
Abstract
Although shotgun sequences of the genomic DNA of common wheat and its ancestors are available, gene discovery in common wheat is primarily based on proof sequencing of expressed full-length (FL) cDNAs. Use of expressed sequence tag (EST) databases including FLcDNA has been recognized as an important method for gene annotation in common wheat. In the large repetitive genome of common wheat, a transcriptome approach is complementary to whole genome sequencing. We have initiated a wheat EST project in Japan and constructed cDNA libraries from various tissues and strains of wheat, including biotic and abiotic stress treatments. We have also generated a high quality full-length cDNA resource for common wheat, an essential element necessary for the ongoing curation and annotation of the wheat genome. After several rounds of screening of CAP-trapped cDNA libraries, 21,408 FLcDNAs have been fully sequenced. The origins of these FLcDNAs were estimated through examination of the RNAseq data of three ancestral diploids, namely, Triticum urartu, Aegilops speltoides, and Aegilops tauschii. In addition, 51 cDNA libraries were constructed with an accumulation of 0.9 million ESTs. The ESTs, including the FLcDNA data, were assembled into contigs with stringent bioinformatic tool parameters. In total, 41,003 gene clusters were classified, in which 27,943 (68.1 %) had homology with other cereal genes. The digital monitoring system was utilized to identify characteristic gene expression patterns among various tissues and stress treatments in common wheat. These transcriptome data comprise a substantial reference for wheat genome sequencing.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
Large-Scale Collection of Genes Expressed in Common Wheat
Wheat is characteristically polyploidic in nature and harbors large complex genomes. Therefore, accumulation of expressed sequence tags (ESTs) for wheat is particularly important for enabling functional genomics and molecular breeding studies. We obtained large collections of ESTs from various tissues in the wheat life cycle and from tissues subjected to stresses. Since full-length cDNAs are indispensable for certifying the ESTs collected and for annotating genes present in the genome, we performed a systematic survey of and sequencing of full-length cDNA clones. The strategy for the collection of ESTs in common wheat is shown in Fig. 10.1. First, total RNAs were extracted from the ten tissues over the course of the wheat life cycle. Subsequently, RNAs were extracted from biotic and abiotic-stressed tissues. The cDNA libraries were constructed from these RNAs by using standard methods. Colonies were randomly picked and sequenced from both ends. At present, 894,756 unbiased ESTs are available. More than 1.2 million wheat ESTs are registered in the NCBI EST database, 70 % of which were contributed from Japan. For full-length cDNAs, the cDNA library was constructed with the CAP-trapper method (Kawaura et al. 2009). After one-path sequencing of cDNA clones, independent clones were selected three times, and the inserts of the cDNA clones were verified. Finally, 22,519 sequence-verified full-length cDNAs were obtained.

MUGEST: Wheat EST project in Japan
Functional Analysis of ESTs in Common Wheat
About one million ESTs comprising 125.3 Mb nucleotides were collected from the 51 cDNA libraries constructed from various tissues (including biotic and abiotic tissues) and organs under a range of conditions (Manickavelu et al. 2012). ESTs were assembled with stringent parameters so as to produce 37,138 contigs and 215,199 singlets, 10.6 % of which had no homology with those in the public databases. Using these EST data, we developed the correspondence analysis (CA) method (Yano et al. 2006), as shown in Fig. 10.2. The CA method enables identification and comparisons of significant gene expression with the specific library. This method was applied for comparison of gene expression analysis of susceptive and resistant near-isogenic lines in common wheat infected by Puccinia triticina (Manickavelu et al. 2010). In Fig. 10.2, four libraries developed for powdery mildew and leaf rust (two susceptible and two resistant) have been compared. Using this method, common and specific genes related to treatments or the library were easily selected.

Correspondence analysis for characterization of gene expression profiles in four libraries. Four cDNA libraries were developed for powdery mildew and leaf rust (two susceptible and two resistant). Each corner refers to each library; the significant genes related to specific libraries are positioned near, and in the same color as, the corresponding library
Based on the gene ontology, characteristic proteins were classified according to molecular functions, cellular localization, and biological processes. Furthermore, the unigenes were classified into susceptible and resistant classes based on the EST members assembled from the respective libraries. Several genes showing specific expression in the resistant and susceptible lines could be selected. The molecular pathogenicity of leaf rust after infection in wheat was evaluated, and the EST data were supplied for future studies.
Full-Length cDNA Collection in Common Wheat
For construction of full-length cDNAs, total RNAs were extracted from the 17 tissues studied (Kawaura et al. 2009). Tese RNAs were mixed together to construct a cDNA library with the CAP-trapper method. We used three rounds of cDNA selection. At first, 19,968 clones were supplied for one-path sequencing from both ends. These sequences were clustered and non-redundant groups were selected. In this step, 6,162 sequence-verified full-length cDNAs were obtained after using the primer-walked Sanger sequencing method. Subsequently, data were obtained for an additional 10,645 cDNAs. After the second round, the already sequenced clones were subtracted from the library. An additional 5,712 redundant cDNAs were sequenced with the 454 FLx + instrument. Finally, 22,519 sequence-verified full-length cDNAs are now available. These FLcDNAs showed a wide size distribution with a range of 64–8,983 bp, and the mean size of these FLcDNAs was 1,848 bp larger than those of barley (1,711 bp; Matsumoto et al. 2011) and rice (1,746 bp; Kikuchi et al. 2003).
We performed RNAseq analysis of diploid ancestors to assign these FLcDNAs to three genomes, namely A, B, and D. Total RNAs were extracted from seedlings, roots and spikes of Triticum urartu Tum., Aegilops speltoides Tausch, and Aegilops tauschii Coss. These RNAs were utilized for RNAseq by using the Illumina Hiseq 2000 system. For each RNA sample, more than 50 Gb were read. These RNA data were grouped to generate contig clusters for each species. In this case, we used data for 16,807 FLcDNAs. These FLcDNA data were compared against contigs from each species. Top hits were selected based on homology and SNP patterns amongst genes from the three genomes. Thus, 4,759 cDNAs were assigned to the A genome, 2,849 to B, and 5,343 to the D genome. In total, 77 % of genes could be assigned. The remaining 23 % had no counterpart in the diploid RNAseq data pool. The B genome had a lesser number of expressed genes than the other genomes. This finding suggests that the lesser sequence homology of genes between the B genome of common wheat and the S genome of Ae. speltoides might disturb, to some extent, the ability to assign groups.
Identification of Standard Transcripts in Common Wheat
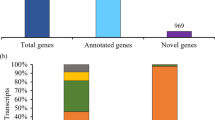
We attempted to identify standard transcripts in common wheat from the EST data; our data for 805,544 ESTs, including those for FLcDNAs, were assembled in each of the ten strains of common wheat. After assembly with MIRS (http://chevreux.org/projects_mira.html), 191,988 contigs and 326,851 singlets were obtained. The resultant contigs and singlets were clustered by the CD HIT software by setting the criteria at an identity of 99.5 % to obtain homoeologs (6×), and 95 % to obtain homologs (2×). Consequently, 162,874 gene clusters (2×) were grouped. Out of these clusters, 81,694 clusters were matched to expressed genes of barley (Matsumoto et al. 2011), Brachypodium (Mochida et al. 2013), and rice (http://ricegaas.dna.affrc.go.jp/rgadb/). By removing redundant results, 27,943 gene clusters that had homologies with Poaceae transcripts could be established at the diploid level; 81,180 clusters had no homologs among cereal genomes. Therefore, we conducted a “blat search” against 5× draft wheat genome sequences (Brenchley et al. 2012); 54,446 clusters were found to have homologies with the wheat genome sequences. By grouping these clusters at 80 % identity, 13,060 wheat-specific genes were obtained. From these analyses, 41,003 gene groups were clustered in wheat. Out of the 41,003 genes, 27,943 wheat genes had common homologies to Poaceae-expressed genes.
Chromosome Assignment of Expressed Genes in Common Wheat
We assigned these expressed homoeologs to each chromosome based on the “Survey sequence” organized by the International Wheat Genome Sequence Consortium (http://www.wheatgenome.org/). From this rough assignment, we can estimate the number of expressed genes from each chromosome and/or chromosome arm (Fig. 10.3). Chromosome 3B and 5BL were highly expressed. Conversely, genes located on 6B, which is the Japanese sequencing target, were less commonly expressed.

Chromosome assignments of wheat-expressed genes by using the Chinese Wheat “Survey-sequences” of IWGSC
Conclusion
Snapshots of gene expression profiles in the wheat life cycle and/or in response to environmental stresses were developed; 22,519 sequence-verified full-length cDNAs were obtained. Of these, 77.4 % were assigned into the A, B, and D genomes with the RNAseq data from their diploid ancestors. We obtained 284,822 homoeo-clusters (6×) from 805,544 ESTs of common wheat. These homoeo-clusters were classified into 27,943 (Poaceae common) and 13,060 (wheat specific) gene groups (2×). In total, 41,003 wheat transcripts (2×) were classified; 284,822 homoeo-clusters were assigned into 21 chromosomes using “Survey-sequences” from the IWGSC.
Thus, the words of Prof. Kihara (1946), “The history of the earth is recorded in the layers of its crust. The history of all organisms is inscribed in the chromosomes” hold true even now, in the genomics era.
References
Brenchley R et al (2012) Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491:705–710
Kawaura K et al (2009) Assessment of adaptive evolution between wheat and rice as deduced from the full-length cDNA sequence data and the expression patterns of common wheat. BMC Genomics 10:271
Kikuchi S et al (2003) Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science 301:376–379
Manickavelu A et al (2010) Comparative gene expression analysis of susceptible and resistant near-isogenic lines in common wheat infected by Puccinia triticina. DNA Res 17:211–222
Manickavelu A et al (2012) Comprehensive functional analyses of expressed sequence tags in common wheat (Triticum aestivum). DNA Res 19:165–177
Matsumoto T et al (2011) Comprehensive sequence analysis of 24,783 barley full-length cDNAs derived from 12 clone libraries. Plant Physiol 156:20–28
Mochida K et al (2013) Large-scale collection and analysis of full-length cDNAs from Brachypodium distachyon and integration with Pooideae sequence resources. PLoS One 8:e75265
Yano K, Imai K, Shimizu A, Hanashita T (2006) A new method for gene discovery in large-scale microarray data. Nucleic Acids Res 34:1532–1539
Acknowledgments
We are grateful to all members of KOMUGI, Wheat Genomics Consortium of Japan. Without their contributions, this project could not have been carried out. This work was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan and the Ministry of Agriculture, Forestry and Fisheries of Japan.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this paper
Cite this paper
Ogihara, Y. et al. (2015). Comprehensive Functional Analyses of Expressed Sequence Tags in Common Wheat. In: Ogihara, Y., Takumi, S., Handa, H. (eds) Advances in Wheat Genetics: From Genome to Field. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55675-6_10
Download citation
DOI: https://doi.org/10.1007/978-4-431-55675-6_10
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55674-9
Online ISBN: 978-4-431-55675-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)