
Abstract
Ever since the advent of Rituximab and subsequently the emergence of other compounds targeting B cells, a cornucopia of medical applications have been found for this family of compounds. After their establishment as standard of care in many conditions such as rituximab in lymphoma and rheumatoid arthritis, they have been progressively found to aid in the treatment of many other conditions. This area constituted a fertile area of research in the past 12 years. Physicians have investigated the B-cell depleting agents use in cases of autoimmune hematologic cytopenias such as immune thrombocytopenia, Evans syndrome, cold and warm autoimmune hemolytic anemia, and other thrombophilic disorders such as the antiphospholipid syndrome and thrombocytopenic purpura. This chapter presents a historical perspective reviewing the various studies that have been published in this field. In addition, it offers a current assessment of the evidence regarding the use of B-cell depleting agents in the aforementioned conditions.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others
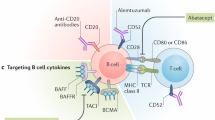


Keywords
- Autoimmune Hemolytic Anemia
- Immune Thrombocytopenia
- Common Variable Immune Deficiency
- Lymphoma Protocol
- Warm AIHA
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Ever since the advent of Rituximab and subsequently the emergence of other compounds targeting B cells, a cornucopia of medical applications have been found for this family of compounds. After their establishment as standard of care in many conditions such as rituximab in lymphoma, they have been progressively found to aid in the treatment of many other conditions. This area constituted a fertile area of research in the past 12 years (Zimmer et al. 2004). Physicians have investigated the B-cell depleting agents use in cases of autoimmune hematologic cytopenias such as ITP (immune thrombocytopenia), Evans syndrome, cold and warm autoimmune hemolytic anemia, and other thrombophilic disorders such as the antiphospholipid syndrome and TTP (thrombocytopenic purpura).
2 B-Cell Depleting Agents in Primary ITP
2.1 ITP at a Glance
Immune thrombocytopenia is an autoimmune disease of platelet destruction and subsequent thrombocytopenia. The disease has been witnessed to many changes all the way from understanding its pathophysiology to the evolution in its management options. Initially called idiopathic thrombocytopenic purpura and thus the acronym ITP, it went on to be named immune thrombocytopenic purpura, and more recently immune thrombocytopenia (Stasi et al. 1995). Changes in nomenclature emerged due to advances in understanding the disease. It was primarily thought of as a disease of peripheral platelet destruction of unknown origin. ITP’s definition progressively evolved to include peripheral platelet destruction due to autoantibodies in addition to an inadequate production by megakaryocytes through disturbed level of erythropoietin (Neunert et al. 2011; Nugent et al. 2009). ITP is classified into primary and secondary. No test has been yet developed that can accurately pinpoint the diagnosis. The primary form is usually diagnosed by exclusion of a secondary form which is usually part of another disease process such as: antiphospholipid syndrome, systemic lupus erythematosus, a hematologic or non-hematologic malignancy, common variable immune deficiency or might be due to hepatitis C or Helicobacter pylori, cytomegalovirus, varicella zoster or human immunodeficiency virus infection or to a vaccination side effect (Cines et al. 2009). Secondary ITP accounts for around 20 % of the total number of patients diagnosed with immune thrombocytopenia (Cines et al. 2009). Of relevance to this chapter is the primary form since this is where B-cell depleting agents come into play.
2.2 Standard of Care and Available Treatments
The management of ITP varies between children and adults. This stems from the fact that children have a more acute disease with a higher tendency for spontaneous remission as opposed to adults (Neunert et al. 2011; Stasi et al. 1995).
2.2.1 Therapy in Children
Initial therapy in an emergency setting for symptomatic patients mainly consists of glucocorticoids, IVIG, or alternatively anti-D in select cases (non-splenectomized, Rh-positive patients). High dose dexamethasone can also be considered in patients who do not respond to the above-mentioned treatments or as an alternative to splenectomy in chronic ITP patients. Splenectomy is exclusively a second-line therapy however, should be delayed until 12 months after the diagnosis since patients might spontaneously remit within this period (Sailer et al. 2006).
2.2.2 Therapy in Adults
Treatment is usually administered in this population when patients are bleeding or are at increased risk of bleeding, such as premenopausal women or when they have coexistent risk factors, lifestyle, preference and pros versus cons of treatment (Stasi et al. 1995; Cohen et al. 2000; Daou et al. 2008). It has been proposed that a platelet count of 30 × 109 L−1 can be adopted as a threshold for treatment as it was shown to improve mortality in newly diagnosed ITP patients (Li et al. 2005; Neylon et al. 2003; Neunert et al. 2011). Glucocorticoids, IVIG, and anti-D have all been proposed as treatment options for ITP and are given alone or in combination based on the patients’ tolerability of every treatment or the need for a quicker response in some cases (Nugent et al. 2009; Thota et al. 2012; Godeau et al. 1999; Newman et al. 2001; Zimmer et al. 2004). High dose dexamethasone has also been reported to be highly effective however, due to paucity of head to head studies with other treatments, it is not yet considered a standard of care (Cheng et al. 2003). Splenectomy, rituximab, and TPO agonists are all considered adequate second-line therapies (Provan et al. 2010; Arnold 2013; Neunert et al. 2011). Splenectomy is the option physicians have the most experience with not to mention the fact that it has the most studies to support it since it was the first and only second-line treatment option for a long time (Ghanima et al. 2012).
2.3 Rituximab in ITP
The first study exploring B depleting agents in ITP was reported in 2000. It was a retrospective one that included patients who had already been treated with steroids. It showed a 30 % response rate to rituximab when given at 375 mg/m2 weekly for 4 weeks (as per the lymphoma protocol) (Saleh et al. 2000). The first prospective trial using rituximab in ITP was reported in 2001 (Stasi et al. 2001). This study included 25 patients who had chronic ITP and had failed prior treatments. All of the patients received the 375 mg/m2 dose. The results from that trial showed an encouraging 52 % response rates in otherwise treatment refractory patients. With the minimal adverse event profile of this drug, it was found to be a safe alternative to more traditional treatments. A large systematic review exploring the efficacy of rituximab as a second-line treatment in patients older than 15 years of age with roughly half of them having undergone splenectomy found that treatment with rituximab gave a complete response rate (CR) of 46.3 % (95 % CI: 29.5–57.7) and a partial response rate (PR) of 24.0 % (95 % CI: 15.2–32.7). In that study PR and CR were defined as exceeding 50 × 109 cells/L and 150 × 109 cells/L, respectively (Arnold et al. 2007). This is different than the most recent ASH guidelines that define a CR as one that leads to a platelet count ≥100 × 109 cells/L without bleeding and a response as one that leads platelet count ≥30 × 109 cells/L accompanied with a greater than twofold increase of platelet count from baseline with absence of bleeding (Neunert et al. 2011). The median time to response and response duration in the above-mentioned review were 5.5 weeks and 10.5 months, respectively. A recent meta-analysis confirmed rituximab’s efficacy in ITP revealing an overall response and complete response of 59.7 and 45.7 %, respectively (Barcellini and Zanella 2011). Previous studies have shown that the response rate does not vary whether rituximab is used before or after splenectomy or used after a previous trial of rituximab (Auger et al. 2012). It has also been shown that the 100 mg/m2 dose can be as effective as the much higher ones used in the lymphoma protocol giving an overall response rate of 71 % (Stasi 2010). A recent systematic review recently published that included 18 observational studies with a total number 323 children with ITP concluded that CR for these patients was 39 % (CI: 30–49 %). The response rate was, however, 68 % (CI: 58–77 %). This is very similar to the response rates observed in adults; however, the definitions of CR (≥100 × 109 cells/L) and response (≥30 × 109 cells/L) differed from their adult counterparts (Liang et al. 2012). A multicenter prospective trial of chronic ITP patients, who are candidates for splenectomy and were treated with rituximab, has shown promising results. At 1 year, 40 % of patients had responded (95 % CI: 28–52 %). This group, however, decreased over time with only 6.7 % of the patients experiencing sustained response after a single course of rituximab using the lymphoma protocol. This proved that rituximab could be used to delay or prevent splenectomy (Godeau et al. 2008) A recent large meta-analysis exploring rituximab as an option prior to splenectomy demonstrated a 57 % overall response rate after rituximab treatment with younger patients responding the most (Auger et al. 2012). Another study using rituximab earlier in the course of treatment of patients with immune thrombocytopenia evaluated 103 patients that were treatment naïve. It consisted of randomizing patients to either dexamethasone alone or to dexamethasone with concomitant rituximab (lymphoma protocol). Results showed a significantly better sustained response by 27 % in patients receiving the steroid and rituximab regiment; 63 % versus 36 % in the steroid only arm. The occurrence of side effects nonetheless (grade 3 and 4) was much more pronounced however in the combined treatment group (Zaja et al. 2010). A newer trial demonstrated that there was no significant difference in the composite outcome of reaching a platelet count less than 50 × 109 cells/L, significant bleeding or need for rescue treatment, between a group of untreated ITP patients receiving their standard treatment and another receiving additional adjuvant rituximab (Arnold et al. 2012). One prohibitive factor in the use of rituximab has been the elevated cost. On average, a splenectomy would cost $20,000 whereas thrombopoietin and rituximab would each cost $2,500–$4,500 per month, and $10,000–$50,000, respectively (Ghanima et al. 2012).
2.4 Proposed Mechanism of Action of Rituximab in ITP
Three mechanisms of action of rituximab have been proposed. The first involves a decrease in macrophage phagocytosis and peripheral destruction of platelet coated with autoantibodies, when the latter are bound by rituximab. This would explain the early response (after 4 weeks) which occurs in the majority of cases after rituximab treatment. It is thought to be mediated by an inhibition of the Fc receptor portion of macrophages. The second mechanism involves B-cell depletion and accounts for the late response by decreasing the number of autoantibody producing B cells. The third mechanism is probably through T cell modulation effect of the drug since in some cases no correlation is observed between the disease severity and the antibody level (Stasi 2010).
2.5 Other B-Cell Depleting Agents in ITP
It seems that rituximab dominates the biologics field in ITP. A recent noteworthy trial has combined low rituximab with alemtuzumab yielded an impressive 100 % overall response rate and a complete response rate of 56 % (Gomez-Almaguer et al. 2010).
3 B-Cell Depleting Agents in TTP
3.1 TTP at a Glance
Thrombotic cytopenic purpura is a rare hematological disease that has an estimated annual incidence of 11.3 cases/1,000,000 people. It has been classically defined as patients presenting with the pentad of thrombocytopenia, uremia, microangiopathic hemolytic anemia, fever, and neurologic symptoms. This continues to be the classic teaching even though these symptoms are present all together in a minority of patients: 5 % of 64 patients from the Oklahoma registry of TTP patients (George 2010). Although TTP shares the same histological appearance with the hemolytic uremic syndrome, it is important to differentiate it from that latter since the management differs radically (Claus et al. 2010). It is considered to be an ancient disease that was first described as early as 1923 (Moschcowitz 2003). It has been proposed that it is due to aggregation of single von Willebrand factors units into a large multimer due to the absence of a metalloproteinase that usually breaks it down in order to keep the system in check (Galbusera et al. 1999). Later on, this metalloproteinase was discovered to be ADAMTS 13 (Zheng et al. 2001). The direct result of this abnormally large multimer is platelet aggregation, injury to RBCs, thrombosis, and subsequent end organ damage. ADAMTS 13’s deficiency was considered the culprit of the disease up until recently when some TTP patients were discovered not to have a deficiency in this metalloproteinase (Kremer Hovinga et al. 2010; Vesely et al. 2003). TTP has been classified as either congenital or acquired. Patients having the congenital form of the disease (Upshaw–Schulman Syndrome) constitute 5 % of people with TTP whereas the people with the acquired type make up the remaining 95 %. The acquired type is further subdivided into an autoimmune idiopathic subtype that makes up 70–80 % of those patients versus a secondary subtype that constitutes the rest (Rizzo et al. 2012). Some of the secondary causes that have been described are infections such as HIV, tumors, autoimmune diseases, pregnancy, and stem cell transplant recipients (Kremer Hovinga et al. 2010).
The diagnosis of the disease continues to be mainly a clinical one due to the absence of a specific lab test that could pinpoint the diagnosis. ADAMTS 13 as mentioned earlier maybe negative in a subset of patients due mainly to the variability in the testing methods however, when present, it offers supporting evidence of diagnosis (Shah and Sarode 2013; George 2010).
3.2 Standard of Care and Available Treatments
Plasma exchange is the sinequanone of treatment in TTP since untreated, this disease is usually fatal (Ghanima et al. 2012). It has been shown to be superior to plasma infusion and can reduce mortality to 20 % or less (Rock et al. 1991). Nonetheless, plasma exchange is not without risk. With a death rate that can reach 3 % not to mention fatal arrests, hypotension, catheter-related complications, and venous thrombosis, plasmapheresis should be used with caution (Cohen et al. 2000; McMinn et al. 2003). In plasma exchange non-responders glucocorticoids and other immunosuppressive agents such as vincristine, cyclosporine, cyclophosphamide, and even splenectomy have been tried with varying degrees of success. In addition, a large numbers of initial responders (>30 %) relapsed at a later time (George 2000, 2010; Sadler et al. 2004).
3.3 Rituximab in TTP
The investigation of rituximab as a potential treatment for TTP started in 2002 after two women with refractory TTP were reported to respond after addition of rituximab to their plasma exchange therapy (Chemnitz et al. 2002). Many studies later on observed the same effect of rituximab in treating refractory TTP patients in conjunction with plasmapheresis. A recent review reviewing six studies each of which included five or more patients reported clinical remission in 97 % from a total of 67 patients treated with rituximab given as per lymphoma protocol (Stasi 2010). This result should be interpreted with caution, since due to the rarity of the disease, many of the studies included represented small case series with no adequate controls (Sallah et al. 2004; Reddy et al. 2005; Heidel et al. 2007; Ling et al. 2009). Another review that included 118 patients with either refractory of relapsing TTP treated with rituximab came to a conclusion that 85 % of patients achieved remission and considered rituximab to be a safe and efficient treatment option in this subset of patients (Caramazza et al. 2010). Rituximab has also been shown to be a good first-line treatment in conjunction with plasma exchange. It has been shown to decrease hospital stay by a mean of 7 days in non-ICU admitted patients. Moreover, it markedly decreased relapse to 10 % versus 56 % in controls (Scully et al. 2011). Rituximab has successfully been used as a preemptive maintenance therapy in patients with recurrent disease and has also been shown to be equally effective in patients with long-standing and recently diagnosed TTP (Herbei and Venugopal 2006; Stasi 2010).
It has been postulated that rituximab acts not only by depleting antibodies against ADAMTS 113 but also by decreasing cytokine production. This stems from the fact that patients with normal ADAMTS 13 levels still respond to the biologic therapy (Kameda et al. 2007; Reddy et al. 2005).
The regular lymphoma dose of Rituximab has been used in most studies however, some have used lower or more numerous dosing regiments with success (Newman et al. 2001; Kivity and Agmon-Levin 2011). Furthermore, some authors advise for performing plasma exchange 24 h after rituximab infusion whereas others recommend doing it after 72 h (Boctor and Smith 2006).
3.4 Other B-Cell Depleting Agents in TTP
No other B-cell depleting agent has been investigated in TTP, but given the success of rituximab, we should be expecting increasing interest in this field in the near future.
4 B-Cell Depleting Agents in Evan’s Syndrome
4.1 Definition and Available Therapy
Evan’s syndrome is defined as autoimmune hemolytic anemia coexisting with ITP. Patients usually suffer from intermittent exacerbations and remissions in their lifetime. Its diagnosis is usually confirmed by the direct antiglobulin test which is usually positive (Norton and Roberts 2006). Corticosteroids were historically found to be the cornerstone of treatment. This poses a challenge to the treating physician especially when considering corticosteroids side effects on the long term owing to the chronicity of the disease. Research tackling second-line treatments has been scarce in this field and relied mostly on immunosuppressant such as danazol, mycophenolate mofetil, cyclosporine, or splenectomy (Norton and Roberts 2006). We have recently witnessed an emergence of studies employing rituximab as a second-line or even first-line treatment (Barcellini and Zanella 2011).
4.2 Rituximab in Evan’s Syndrome
Early reports have shown encouraging results. A response rate of 83 % was first reported which subsequently increased to 94 % in new reports (Norton and Roberts 2006; Barcellini and Zanella 2011). The problem with those numbers however is that they are based on case reports and case series with no adequate controls not to mention the eventual publication bias that predominates such studies. Most of the studies deal with relapsed patients with Evan’s syndrome but dosing has been highly variable between different patients. A recent retrospective study looked at the charts of 11 patients having received rituximab, seven for refractory ITP, three for relapsing hemolytic anemia, and one for refractory ITP and hemolytic anemia. An encouraging 82 % response rate was achieved with a 64 % long-term response rate after a 1 year mean follow-up (Michel et al. 2009).
5 B-Cell Depleting Agents in Antiphospholipid Syndrome
5.1 APS at a Glance
The antiphospholipid syndrome (APS) is an acquired autoimmune disease characterized by a hypercoagulable state that leads to arterial and/or venous thrombosis, recurrent pregnancy loss, and persistently positive antiphospholipid (aPL) antibodies, namely anticardiolipin (aCL), lupus anticoagulant (LA), and anti-β2-glycoprotein I antibodies (anti-β2GPI). The latest classification criteria for diagnosing APS are the 2006 updated Sapporo criteria that require the presence of at least one clinical manifestation and one positive laboratory criteria (Table 1) (Miyakis et al. 2006).
In a small subset of APS patients, the disease can have an accelerated progression resulting in multiorgan failure, called “catastrophic” APS (CAPS). CAPS is characterized by multiple organ involvement with histopathologic evidence of small-vessel thrombosis developing over a very short period of time, in the presence of laboratory confirmation of aPL antibodies (Asherson et al. 2003).
In 2003, Hughes and Khamashta described another group of patients who present with clinical manifestations highly suggestive of APS but with persistently negative LA, aCL, and anti-β2GPI antibodies. This group was collectively referred to as seronegative APS (SNAPS) (Hughes and Khamashta 2003; Nayfe et al. 2013).
Taking into consideration the diverse clinical manifestations of APS, it is suggested that more than one pathological process may be involved. Despite this fact, the current therapeutic approaches are mostly restricted to anticoagulation therapy, which does not happen to benefit all patients (Pierangeli et al. 1995, 1999). Moreover, recurrent thrombotic events can occur in up to 30 % of APS patients (Gharavi et al. 1999), and 2–3 % might experience bleeding complications (Pierangeli et al. 1996). The best treatment for these APS patients who are intolerant or resistant to long-term anticoagulation remains unclear. New pathogenic mechanisms in APS are under investigation by ongoing research, including aPL-induced activation of platelets, endothelial cells, monocytes, complement and coagulation cascade, leading to the discovery of potential targets and therapies for APS (Comarmond and Cacoub 2012). New data indicates a link between high titers of aPL antibodies and elevated circulating CD5+ B cells, suggesting that APS may be responsive to B cell targeted therapies (Youinou and Renaudineau 2004).
5.2 Pathogenesis of APS
The pathogenic mechanisms behind the clinical symptoms of APS are not fully understood, and many factors contribute in the aPL-induced manifestations of the disease (Comarmond and Cacoub 2012).
5.2.1 aPL Antibodies
aPL antibodies promote thrombus formation in both the venous and arterial circulation, and their thrombogenic properties have been shown in several in vitro and in vivo animal studies to be responsible for the pathogenesis of APS (Domenico Sebastiani et al. 2003; Dagenais et al. 1992; Zhou et al. 2011). Nonetheless, how these antibodies are produced and the exact mechanism by which they mediate thrombosis is not fully elucidated. aPLs comprise a heterogeneous family of autoantibodies; yet, similar HLA class alleles are identified to be consistent with APS patients, namely HLA-DR4, -DR7, and -DRw53 (Doring et al. 2010). aPL antibodies bind to their target cells (i.e., monocytes, platelets, endothelial cells, and trophoblasts) through a mediator plasma apolipoprotein called β2GPI, the main autoantigen for aPL antibodies. Consequently, up-regulation of tissue factor expression on monocytes and endothelial cells takes place leading to thrombosis and fetal loss through a series of signal transduction events (Romay-Penabad et al. 2007). Since aPLs play a central role in the pathogenesis of APS, special interest has been put on B cells, as they are the source of these pathogenic autoantibodies. Over the past 10 years, the role of autoreactive B cells in APS and the breakdown of B-cell tolerance have been extensively studied (Rand et al. 2008; Edwards et al. 1997). In addition to antibody production, B cells have other pathogenic mechanisms in APS, such as modifying their B-cell receptor specificity and acting as antigen presenting cells for self-antigens; besides differentiating into B effector cells (Be-1 and Be-2) which regulate helper T cells and their functions (Wallace 1994; Khattri et al. 2012).
5.3 Immunomodulatory Approach: B-Cell Targeted Therapies
Because accumulated data support the pathogenic role of B cells in the development and progression of APS, B-cell targeting therapies have been investigated in both human and murine APS.
5.3.1 B-Cell Depletion in APS
To date, literature review reveals only a limited number of case reports and series published regarding the use of rituximab in the treatment of APS. No randomized clinical trials were retrieved. Rituximab is a chimeric (murine/human) monoclonal antibody that targets CD20 on peripheral B cells, depleting them from the circulation and consequently decreasing disease activity (Willems et al. 2006; Youinou et al. 2009). Rituximab is FDA approved for the treatment of rheumatoid arthritis, B-cell non-Hodgkin’s lymphoma (Higashida et al. 2005) and recently for anti-neutrophilic cytoplasmic antibody (ANCA)-associated vasculitis (Cohen Tervaert 2011). In addition to these indications, rituximab is being used as off-label treatment in a number of inflammatory and systemic autoimmune diseases (Butterly et al. 2010).
No reports on other B-cell directed therapies in APS patients were found.
Rituximab was used in the treatment of 27 reported cases of APS (primary, secondary, and APS with concomitant malignancies), including 18 females and 9 male patients, whose age ranged from 3 months to 69 years (Khattri et al. 2012). Of the 27 APS patients, four suffered from concomitant lymphomas; two had non-Hodgkin’s lymphoma for which rituximab was administered as part of the R-CHOP chemotherapy regimen (Veneri et al. 2005; Erre et al. 2008), and two received rituximab for marginal zone lymphoma (Manner et al. 2008; Harner et al. 2004), one of which had Sjogren’s syndrome also (Harner et al. 2004). Five patients had SLE with secondary APS, where rituximab was used after failure of treatment with anticoagulation and/or immunosuppression for lupus (Cianciulli et al. 2008; Weide et al. 2003; Tomietto et al. 2004; Ahn et al. 2005; Anandacoomarasamy et al. 2006). One patient had Evans syndrome (Ruckert et al. 2008). The rest of the patients had primary APS (Iglesias-Jimenez et al. 2010; Nageswara Rao et al. 2009; Tsagalis et al. 2010; Adamson et al. 2008; van Wissen et al. 2008; Chalam et al. 2007; Ames et al. 2007; Rubenstein et al. 2006; Trappe et al. 2006; Binstadt et al. 2003; Erdozain et al. 2004; Asherson et al. 2008; Sciascia et al. 2011). Table 2 summarizes the clinical and serological manifestations of the reported APS patients along with their outcomes after treatment with rituximab (Khattri et al. 2012). The rituximab dosing regimen used in the majority of the cases was 375 mg/m2 body surface area, given weekly for 4 weeks. Four patients received rituximab 1,000 mg given 15 days apart. The reported patients were not treatment naïve prior to rituximab administration; most received anticoagulation unless otherwise contraindicated, eight patients were treated with cyclophosphamide earlier (Ahn et al. 2005; Anandacoomarasamy et al. 2006; Iglesias-Jimenez et al. 2010; Ames et al. 2007; Rubenstein et al. 2006; Binstadt et al. 2003; Asherson et al. 2008), and all but one case (Tsagalis et al. 2010) used corticosteroids. The treatment regimen contained other immunosuppressants too, including azathiprine, mycophenolate mofetil, dapsone, and cyclosporine (Khattri et al. 2012). Improvement in the serological markers of APS was noticed in the majority of the cases, where decrease or normalization of LA, aCL, and anti-β2GPI antibodies was seen. Furthermore, multiple systemic clinical manifestations associated with APS improved after starting rituximab regardless whether the patient suffered from primary or secondary APS. There was a uniformly good response to rituximab in primary APS and in APS associated with SLE (Weide et al. 2003; Cianciulli et al. 2008; Tomietto et al. 2004; Ahn et al. 2005; Anandacoomarasamy et al. 2006; Ruckert et al. 2008; Tsagalis et al. 2010; Adamson et al. 2008; Chalam et al. 2007; Ames et al. 2007; Rubenstein et al. 2006; Trappe et al. 2006; Binstadt et al. 2003; Erdozain et al. 2004; Sciascia et al. 2011; Vianna et al. 1994; Danowski et al. 2009) or lymphoma (Erre et al. 2008; Veneri et al. 2005; Manner et al. 2008; Harner et al. 2004) in the case reports reviewed. Moreover, six out of seven CAPS patients benefited from rituximab treatment, knowing the severity and frequent fatality faced in this entity in spite of standard treatment with anticoagulants and immunosuppressant agents (Manner et al. 2008; Iglesias-Jimenez et al. 2010; Nageswara Rao et al. 2009; van Wissen et al. 2008; Rubenstein et al. 2006; Asherson et al. 2008).
In the BIOGEAS registry, a multicenter, national registry in Spain, rituximab was shown to have beneficial therapeutic effects in APS, with 92 % response rate in 12 APS patients (Ramos-Casals et al. 2008).
Despite the promising data from case reports and the BIOGEAS registry on the beneficial effect of rituximab in APS patients, the literature is still limited on this topic as there are no clinical trial data available yet. Moreover, the above-mentioned published case reports have several limitations including their small number, besides the fact that in all the cases, other immunosuppressants were used including steroids and cyclophosphamide, which creates confusion on whether it was rituximab-induced B-cell depletion by itself or the combination of immunosuppressants used that caused improvement in APS patients. Also, it is worth mentioning that the treated population was diverse including patients with primary or secondary APS or malignancy (Khattri et al. 2012). Finally, in a pilot open-label phase II trial aimed primarily to evaluate the safety of rituximab in aPL-positive patients with non-criteria manifestations of APS, and secondarily to evaluate the effect on the aPL profile and efficacy of treatment, it was suggested that rituximab may be effective in controlling some but not all non-criteria manifestations of APS with a safety profile in aPL-positive patients consistent with that of rituximab (Erkan et al. 2013).
5.4 Conclusion
Rituximab, a chimeric anti-CD20 monoclonal antibody, is FDA approved for the treatment of rheumatoid arthritis, B-cell non-Hodgkin’s lymphoma and ANCA-associated vasculitis. The off-label use of B-cell depleting agents in several systemic autoimmune diseases has been studied. Data on the use of rituximab in the treatment of APS is limited to case reports, the BIOGEAS registry and a pilot open-label phase II trial, and suggests a beneficial role in the therapeutic approach of APS. However, well-designed randomized clinical trials are needed to evaluate the use of rituximab, alone or in combination with other immunosuppressants, in improving the clinical and serological manifestations of the disease.
6 B-Cell Depleting Agents in Autoimmune Hemolytic Anemia
6.1 AIHA at a Glance
Autoimmune hemolytic anemia (AIHA) is an uncommon disorder characterized by autoantibodies directed against self red blood cells (RBCs) (Gehrs and Friedberg 2002). Consequently, the normal 100–120 days lifetime of the RBCs is reduced to just a few days in serious cases (Sawitsky and Ozaeta 1970). AIHA can be idiopathic or secondary to infections, other autoimmune conditions or lymphoproliferative disorders, and depending on the thermal range of the autoantibodies involved, the disease can be classified into warm, cold (which includes cold agglutinin disease and paroxysmal nocturnal hemoglobinuria) or mixed (Gehrs and Friedberg 2002). Whether warm- or cold- type secondary AIHA, each can result from its own more common secondary causes. For instance, secondary warm-type AIHA mostly results from lymphoproliferative disorders (e.g., chronic lymphocytic leukemia, lymphoma) and other autoimmune disorders, including SLE, RA, scleroderma, and ulcerative colitis. Less commonly, it can be caused by neoplasms other than lymphoid and infection. Similarly, secondary cold-type AIHA is primarily caused by lymphoproliferative disorders, but also occurs secondary to infection, especially by mycoplasma, viral pneumonia, infectious mononucleosis, and other respiratory infections, and infrequently due to concomitant autoimmune disorders (Sokol et al. 1981).
6.2 Treatment of AIHA
6.2.1 Standard Treatment
In warm AIHA, the first-line therapy has been the administration of corticosteroids, where the response rate reaches 70–85 %, of which only one third remain in long-term remission after drug discontinuation, 50 % necessitate maintenance doses, and around 20–30 % require second-line therapies including immunosuppressants and splenectomy (Wahl et al. 2008). Splenectomy is probably the most effective second-line treatment with a response rate of 50 %, especially in relapsing patients on corticosteroids or those requiring the equivalent of 10–15 mg prednisone per day to maintain adequate hemoglobin levels. We have to bear in mind the surgical and infective complications, particularly gram-negative sepsis in patients above 65 (Gehrs and Friedberg 2002; Valent and Lechner 2008; Newland et al. 2005). Moreover, patients who are unresponsive to or do not fit for splenectomy, have limited options including cytotoxic or immunosuppressive medications such as azathioprine, cyclophosphamide, or cyclosporine, with a response rate of 40–60 % and associated side effects (52). On the other hand, cold-type AIHA has failed to demonstrate a convincing response to standard therapy, particularly cold hemaglutinin disease (Barcellini and Zanella 2011).
6.2.2 Rituximab: Treatment of Cases Refractory to Standard Therapy
In a recent review carried by Garvey et al. collecting data from studies using rituximab in the treatment of AIHA, rituximab (375 mg/m2 given weekly for 4 weeks) was found to be effective in treating both warm AIHA and cold hemaglutinin disease, with a median response rate of 60 % and lasting responses for more than 3 years (Garvey 2008). Furthermore, in three more recent studies (Dierickx et al. 2009; Bussone et al. 2009; Penalver et al. 2010), the response rate was higher and ranged from 77 to 93 % with a disease-free survival at 1 and 2 years in 72 and 56 % of cases, respectively (Dierickx et al. 2009). In several case series, rituximab has been shown to be effective in both patient groups with idiopathic and secondary AIHA, even in those associated with autoimmune and lymphoproliferative disorders, and bone marrow transplant (Shanafelt et al. 2003; Penalver et al. 2010; Quartier et al. 2001; Zecca et al. 2003; Narat et al. 2005; Berentsen et al. 2006; Schollkopf et al. 2006; D’Arena et al. 2007; Gupta et al. 2002; Trape et al. 2003; D’Arena et al. 2006). Moreover, rituximab proved to be effective whether used as a monotherapy or in combination with corticosteroids, immunosuppressants and interferon (Zecca et al. 2003; Narat et al. 2005; Berentsen et al. 2006; D’Arena et al. 2007; Gupta et al. 2002), and irrespective or prior therapy (Penalver et al. 2010; Shanafelt et al. 2003; Quartier et al. 2001; Zecca et al. 2003; Narat et al. 2005; Berentsen et al. 2006; Schollkopf et al. 2006; Gupta et al. 2002; Trape et al. 2003; D’Arena et al. 2006). Time to maximum response varied between the studies from quick response to weeks or even months, where in two recent studies, the median time to response was 3 and 6 weeks, respectively (Bussone et al. 2009; Penalver et al. 2010). It is noteworthy that re-treatment with rituximab is effective with both warm AIHA and cold hemaglutinin disease (Rao et al. 2008; Zecca et al. 2003; Berentsen et al. 2006; Gupta et al. 2002), with re-treatment benefiting some patients more than once (Zecca et al. 2003; Berentsen et al. 2006; Penalver et al. 2010). When comparing rituximab to the next best therapeutic regimen which includes alkylating agents with or without corticosteroids, rituximab was the only treatment able to induce a complete response in cold hemaglutinin disease with a response of 60 % (10 % complete response and 50 % partial response), compared with 16 % (all partial responses) (Berentsen et al. 2006; Berentsen et al. 2004; Barcellini and Zanella 2011). Regarding rituximab safety when given to patients with AIHA, the drug was well tolerated and no adverse events were reported in most cases, besides mild to moderate infusion-related side effects (e.g., fever, chills, hypotension, and upper airway edema) (Rao et al. 2008; Zecca et al. 2003; Schollkopf et al. 2006; Gupta et al. 2002; Trape et al. 2003). Some patients (around 7 %) experienced possible rituximab-related infections (Quartier et al. 2001; Zecca et al. 2003; Narat et al. 2005; Trape et al. 2003; Schollkopf et al. 2006), and few (roughly 2 %) had grade 4 neutropenia (Berentsen et al. 2006; Gupta et al. 2002). Moreover, low dose rituximab (100 mg × 4 weeks) was tried in patients unresponsive to standard therapy in an attempt to reduce side effects and costs, and it was found effective as a monotherapy (Provan et al. 2007) and in combination with alemtuzumab (Gomez-Almaguer et al. 2010). In a study conducted by Barcellini et al, low dose rituximab along with standard oral prednisone was able to induce an overall response rate of 86 % (complete 67 % and partial 19 %) in 21 patients with warm and cold AIHA; response was sustained (Hb >10 g/dl) in 13/14 patients at 6 months and in 11/11 evaluable patients at 1 year (BARCELLINI et al. 2010).
6.3 Conclusion
Rituximab proved to be an effective therapeutic alternative to standard therapy of splenectomy and/or chemotherapy in patients with primary or secondary warm AIHA with a significant response rate and sustained remissions. Furthermore, rituximab induced durable responses in patients with cold hemaglutinin disease, once a disease with very limited therapeutic options (Barcellini and Zanella 2011).
References
Adamson R, Sangle S, Kaul A, Hughes GR, D’Cruz DP (2008) Clinical improvement in antiphospholipid syndrome after rituximab therapy. J Clin Rheumatol 14:359–360
AHN ER, Lander G, Bidot CJ, JY W, AHN YS (2005) Long-term remission from life-threatening hypercoagulable state associated with lupus anticoagulant (LA) following rituximab therapy. Am J Hematol 78:127–129
Ames PR, Tommasino C, Fossati G, Scenna G, Brancaccio V, Ferrara F (2007) Limited effect of rituximab on thrombocytopaenia and anticardiolipin antibodies in a patient with primary antiphospholipid syndrome. Ann Hematol 86:227–228
Anandacoomarasamy A, Gibson J, Mcgill N (2006) ‘Cure’ of life-threatening antiphospholipid syndrome with rituximab. Intern Med J 36:474–475
Arnold DM (2013) Positioning new treatments in the management of immune thrombocytopenia. Pediatr Blood Cancer 60(Suppl 1):S19–S22
Arnold DM, Dentali F, Crowther MA, Meyer RM, Cook RJ, Sigouin C, Fraser GA, LIM W, Kelton JG (2007) Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 146:25–33
Arnold DM, Heddle NM, Carruthers J, Cook DJ, Crowther MA, Meyer RM, LIU Y, Cook RJ, Mcleod A, Maceachern JA, Mangel J, Anderson D, Vickars L, Tinmouth A, Schuh AC, Kelton JG (2012) A pilot randomized trial of adjuvant rituximab or placebo for nonsplenectomized patients with immune thrombocytopenia. Blood 119:1356–1362
Asherson RA, Cervera R, DE Groot PG, Erkan D, Boffa MC, Piette JC, Khamashta MA, Shoenfeld Y (2003) Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus 12:530–534
Asherson RA, Espinosa G, Menahem S, Yinh J, Bucciarelli S, Bosch X, Cervera R (2008) Relapsing catastrophic antiphospholipid syndrome: report of three cases. Semin Arthritis Rheum 37:366–372
Auger S, Duny Y, Rossi JF, Quittet P (2012) Rituximab before splenectomy in adults with primary idiopathic thrombocytopenic purpura: a meta-analysis. Br J Haematol 158:386–398
Barcellini W, Zanella A (2011) Rituximab therapy for autoimmune haematological diseases. Eur J Intern Med 22:220–229
Barcellini W, Zaja F, Zaninoni A, Imperiali FG, Battista M, Di Bona E et al (2010) Low-dose rituximab in idiopathic autoimmune haemolytic anaemia. Haematologica 95:204 (abstract 503)
Berentsen S, Ulvestad E, Gjertsen BT, Hjorth-Hansen H, Langholm R, Knutsen H, Ghanima W, Shammas FV, Tjonnfjord GE (2004) Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood 103:2925–2928
Berentsen S, Ulvestad E, Langholm R, Beiske K, Hjorth-Hansen H, Ghanima W, Sorbo JH, Tjonnfjord GE (2006) Primary chronic cold agglutinin disease: a population based clinical study of 86 patients. Haematologica 91:460–466
Binstadt BA, Caldas AM, Turvey SE, Stone KD, Weinstein HJ, Jackson J, Fuhlbrigge RC, Sundel RP (2003) Rituximab therapy for multisystem autoimmune diseases in pediatric patients. J Pediatr 143:598–604
Boctor FN, Smith JA (2006) Timing of plasma exchange and rituximab for the treatment of thrombotic thrombocytopenic purpura. Am J Clin Pathol 126:965 (author reply 965–966)
Bussone G, Ribeiro E, Dechartres A, Viallard JF, Bonnotte B, Fain O, Godeau B, Michel M (2009) Efficacy and safety of rituximab in adults’ warm antibody autoimmune haemolytic anemia: retrospective analysis of 27 cases. Am J Hematol 84:153–157
Butterly SJ, Pillans P, Horn B, Miles R, Sturtevant J (2010) Off-label use of rituximab in a tertiary Queensland hospital. Intern Med J 40:443–452
Caramazza D, Quintini G, Abbene I, Malato A, Saccullo G, Lo Coco L, Di Trapani R, Palazzolo R, Barone R, Mazzola G, Rizzo S, Ragonese P, Aridon P, Abbadessa V, Siragusa S (2010) Relapsing or refractory idiopathic thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: the role of rituximab. Transfusion 50:2753–2760
Chalam KV, Gupta SK, Agarwal S (2007) Rituximab effectively reverses papilledema associated with cerebral venous sinus thrombosis in antiphospholipid antibody syndrome. Eur J Ophthalmol 17:867–870
Chemnitz J, Draube A, Scheid C, Staib P, Schulz A, Diehl V, Sohngen D (2002) Successful treatment of severe thrombotic thrombocytopenic purpura with the monoclonal antibody rituximab. Am J Hematol 71:105–108
Cheng Y, Wong RS, Soo YO, Chui CH, Lau FY, Chan NP, Wong WS, Cheng G (2003) Initial treatment of immune thrombocytopenic purpura with high-dose dexamethasone. N Engl J Med 349:831–836
Cianciulli TF, Saccheri MC, Redruello HJ, Cosarinsky LA, Celano L, Trila CS, Parisi CE, Prezioso HA (2008) Right atrial thrombus mimicking myxoma with pulmonary embolism in a patient with systemic lupus erythematosus and secondary antiphospholipid syndrome. Tex Heart Inst J 35:454–457
Cines DB, Bussel JB, Liebman HA, Luning Prak ET (2009) The ITP syndrome: pathogenic and clinical diversity. Blood 113:6511–6521
Claus RA, Bockmeyer CL, Sossdorf M, Losche W (2010) The balance between von-Willebrand factor and its cleaving protease ADAMTS13: biomarker in systemic inflammation and development of organ failure? Curr Mol Med 10:236–248
Cohen Tervaert JW (2011) Rituximab in ANCA-associated vasculitis: a revolution? Nephrol Dial Transplant 26:3077–3079
Cohen YC, Djulbegovic B, Shamai-Lubovitz O, Mozes B (2000) The bleeding risk and natural history of idiopathic thrombocytopenic purpura in patients with persistent low platelet counts. Arch Intern Med 160:1630–1638
Comarmond C, Cacoub P (2012) Antiphospholipid syndrome: from pathogenesis to novel immunomodulatory therapies. Autoimmun Rev 12(7):752–757
D’Arena G, Califano C, Annunziata M, Tartarone A, Capalbo S, Villani O, Amendola G, Pietrantuono G, Ferrara F, Pinto A, Musto P, D’Arco AM, Cascavilla N (2007) Rituximab for warm-type idiopathic autoimmune hemolytic anemia: a retrospective study of 11 adult patients. Eur J Haematol 79:53–58
D’Arena G, Laurenti L, Capalbo S, D’Arco AM, De Filippi R, Marcacci G, Di Renzo N, Storti S, Califano C, Vigliotti ML, Tarnani M, Ferrara F, Pinto A (2006) Rituximab therapy for chronic lymphocytic leukemia-associated autoimmune hemolytic anemia. Am J Hematol 81:598–602
Dagenais P, Urowitz MB, Gladman DD, Norman CS (1992) A family study of the antiphospholipid syndrome associated with other autoimmune diseases. J Rheumatol 19:1393–1396
Danowski A, De Azevedo MN, De Souza Papi JA, Petri M (2009) Determinants of risk for venous and arterial thrombosis in primary antiphospholipid syndrome and in antiphospholipid syndrome with systemic lupus erythematosus. J Rheumatol 36:1195–1199
Daou S, Federici L, Zimmer J, Maloisel F, Serraj K, Andres E (2008) Idiopathic thrombocytopenic purpura in elderly patients: a study of 47 cases from a single reference center. Eur J Intern Med 19:447–451
Dierickx D, Verhoef G, Van Hoof A, Mineur P, Roest A, Triffet A, Kentos A, Pierre P, Boulet D, Bries G, Le PQ, Janssens A, Delannoy A (2009) Rituximab in auto-immune haemolytic anaemia and immune thrombocytopenic purpura: a Belgian retrospective multicentric study. J Intern Med 266:484–491
Domenico Sebastiani G, Minisola G, Galeazzi M (2003) HLA class II alleles and genetic predisposition to the antiphospholipid syndrome. Autoimmun Rev 2:387–394
Doring Y, Hurst J, Lorenz M, Prinz N, Clemens N, Drechsler MD, Bauer S, Chapman J, Shoenfeld Y, Blank M, Lackner KJ, Von Landenberg P (2010) Human antiphospholipid antibodies induce TNFalpha in monocytes via Toll-like receptor 8. Immunobiology 215:230–241
Edwards MH, Pierangeli S, Liu X, Barker JH, Anderson G, Harris EN (1997) Hydroxychloroquine reverses thrombogenic properties of antiphospholipid antibodies in mice. Circulation 96:4380–4384
Erdozain JG, Ruiz-Irastorza G, Egurbide MV, Aguirre C (2004) Sustained response to rituximab of autoimmune hemolytic anemia associated with antiphospholipid syndrome. Haematologica 89:ECR34
Erkan D, Vega J, Ramon G, Kozora E, Lockshin MD (2013) A pilot open-label phase II trial of rituximab for non-criteria manifestations of antiphospholipid syndrome. Arthritis Rheum 65:464–471
Erre GL, Pardini S, Faedda R, Passiu G (2008) Effect of rituximab on clinical and laboratory features of antiphospholipid syndrome: a case report and a review of literature. Lupus 17:50–55
Galbusera M, Benigni A, Paris S, Ruggenenti P, Zoja C, Rossi C, Remuzzi G (1999) Unrecognized pattern of von Willebrand factor abnormalities in hemolytic uremic syndrome and thrombotic thrombocytopenic purpura. J Am Soc Nephrol 10:1234–1241
Garvey B (2008) Rituximab in the treatment of autoimmune haematological disorders. Br J Haematol 141:149–169
Gehrs BC, Friedberg RC (2002) Autoimmune hemolytic anemia. Am J Hematol 69:258–271
George JN (2000) How I treat patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Blood 96:1223–1229
George JN (2010) How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 116:4060–4069
Ghanima W, Godeau B, Cines DB, Bussel JB (2012) How I treat immune thrombocytopenia: the choice between splenectomy or a medical therapy as a second-line treatment. Blood 120:960–969
Gharavi AE, Pierangeli SS, Colden-Stanfield M, Liu XW, Espinola RG, Harris EN (1999) GDKV-induced antiphospholipid antibodies enhance thrombosis and activate endothelial cells in vivo and in vitro. J Immunol 163:2922–2927
Godeau B, Caulier MT, Decuypere L, Rose C, Schaeffer A, Bierling P (1999) Intravenous immunoglobulin for adults with autoimmune thrombocytopenic purpura: results of a randomized trial comparing 0.5 and 1 g/kg b.w. Br J Haematol 107:716–719
Godeau B, Porcher R, Fain O, Lefrere F, Fenaux P, Cheze S, Vekhoff A, Chauveheid MP, Stirnemann J, Galicier L, Bourgeois E, Haiat S, Varet B, Leporrier M, Papo T, Khellaf M, Michel M, Bierling P (2008) Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study. Blood 112:999–1004
Gomez-Almaguer D, Solano-Genesta M, Tarin-Arzaga L, Herrera-Garza JL, Cantu-Rodriguez OG, Gutierrez-Aguirre CH, Jaime-Perez JC (2010) Low-dose rituximab and alemtuzumab combination therapy for patients with steroid-refractory autoimmune cytopenias. Blood 116:4783–4785
Gupta N, Kavuru S, Patel D, Janson D, Driscoll N, Ahmed S, Rai KR (2002) Rituximab-based chemotherapy for steroid-refractory autoimmune hemolytic anemia of chronic lymphocytic leukemia. Leukemia 16:2092–2095
Harner KC, Jackson LW, Drabick JJ (2004) Normalization of anticardiolipin antibodies following rituximab therapy for marginal zone lymphoma in a patient with Sjogren’s syndrome. Rheumatology (Oxford) 43:1309–1310
Heidel F, Lipka DB, Von Auer C, Huber C, Scharrer I, Hess G (2007) Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 97:228–233
Herbei L, Venugopal P (2006) Recurrent thrombotic thrombocytopenic purpura treated repeatedly and successfully with the monoclonal antibody rituximab. Clin Adv Hematol Oncol 4:215–217 (discussion 217–218)
Higashida J, Wun T, Schmidt S, Naguwa SM, Tuscano JM (2005) Safety and efficacy of rituximab in patients with rheumatoid arthritis refractory to disease modifying antirheumatic drugs and anti-tumor necrosis factor-alpha treatment. J Rheumatol 32:2109–2115
Hughes GR, Khamashta MA (2003) Seronegative antiphospholipid syndrome. Ann Rheum Dis 62:1127
Iglesias-Jimenez E, Camacho-Lovillo M, Falcon-Neyra D, Lirola-Cruz J, Neth O (2010) Infant with probable catastrophic antiphospholipid syndrome successfully managed with rituximab. Pediatrics 125:e1523–e1528
Kameda T, Dobashi H, Kittaka K, Susaki K, Yamaoka G, Arai K, Tokuda M, Ishida T (2007) Two cases of refractory thrombotic thrombocytopenic purpura associated with collagen vascular disease were significantly improved by rituximab treatment. Clin Rheumatol 26:2159–2162
Khattri S, Zandman-Goddard G, Peeva E (2012) B-cell directed therapies in antiphospholipid antibody syndrome–new directions based on murine and human data. Autoimmun Rev 11:717–722
Kivity S, Agmon-Levin N (2011) Rituximab for thrombotic thrombocytopenic purpura. Isr Med Assoc J 13:436–437
Kremer Hovinga JA, Vesely SK, Terrell DR, Lammle B, George JN (2010) Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood 115:1500–1511 (quiz 1662)
Li HQ, Zhang L, Zhao H, Ji LX, Yang RC (2005) Chronic idiopathic thrombocytopenic purpura in adult Chinese patients: a retrospective single-centered analysis of 1791 cases. Chin Med J (Engl) 118:34–37
Liang Y, Zhang L, Gao J, Hu D, Ai Y (2012) Rituximab for children with immune thrombocytopenia: a systematic review. PLoS One 7:e36698
Ling HT, Field JJ, Blinder MA (2009) Sustained response with rituximab in patients with thrombotic thrombocytopenic purpura: a report of 13 cases and review of the literature. Am J Hematol 84:418–421
Manner H, Jung B, Tonassi L, Hackenberg U, Plum N, Josten KM, Kirchmaier CM, Frickhofen N (2008) Successful treatment of catastrophic antiphospholipid antibody syndrome (CAPS) associated with splenic marginal-zone lymphoma with low-molecular weight heparin, rituximab and bendamustine. Am J Med Sci 335:394–397
Mcminn JR Jr, Thomas IA, Terrell DR, Duvall D, Vesely SK, George JN (2003) Complications of plasma exchange in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: a study of 78 additional patients. Transfusion 43:415–416
Michel M, Chanet V, Dechartres A, Morin AS, Piette JC, Cirasino L, Emilia G, Zaja F, Ruggeri M, Andres E, Bierling P, Godeau B, Rodeghiero F (2009) The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood 114:3167–3172
Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, PG DEG, Koike T, Meroni PL, Reber G, Shoenfeld Y, Tincani A, Vlachoyiannopoulos PG, Krilis SA (2006) International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 4:295–306
Moschcowitz E (2003) An acute febrile pleiochromic anemia with hyaline thrombosis of the terminal arterioles and capillaries: an undescribed disease. 1925. Mt Sinai J Med 70:352–355
Nageswara RAO AA, Arteaga GM, Reed AM, Gloor JM, Rodriguez V (2009) Rituximab for successful management of probable pediatric catastrophic antiphospholipid syndrome. Pediatr Blood Cancer 52:536–538
Narat S, Gandla J, Hoffbrand AV, Hughes RG, Mehta AB (2005) Rituximab in the treatment of refractory autoimmune cytopenias in adults. Haematologica 90:1273–1274
Nayfe R, Uthman I, Aoun J, Saad Aldin E, Merashli M, Khamashta MA (2013) Seronegative antiphospholipid syndrome. Rheumatology (Oxford) 52(8):1358–1367
Neunert C, Lim W, Crowther M, Cohen A, Solberg L Jr, Crowther MA (2011) The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 117:4190–4207
Newland A, Provan D, Myint S (2005) Preventing severe infection after splenectomy. BMJ 331:417–418
Newman GC, Novoa MV, Fodero EM, Lesser ML, Woloski BM, Bussel JB (2001) A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 112:1076–1078
Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR (2003) Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 122:966–974
Norton A, Roberts I (2006) Management of Evans syndrome. Br J Haematol 132:125–137
Nugent D, Mcmillan R, Nichol JL, Slichter SJ (2009) Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. Br J Haematol 146:585–596
Penalver FJ, Alvarez-Larran A, Diez-Martin JL, Gallur L, Jarque I, Caballero D, Diaz-Mediavilla J, Bustelos R, Fernandez-Acenero MJ, Cabrera JR (2010) Rituximab is an effective and safe therapeutic alternative in adults with refractory and severe autoimmune hemolytic anemia. Ann Hematol 89:1073–1080
Pierangeli SS, Colden-Stanfield M, Liu X, Barker JH, Anderson GL, Harris EN (1999) Antiphospholipid antibodies from antiphospholipid syndrome patients activate endothelial cells in vitro and in vivo. Circulation 99:1997–2002
Pierangeli SS, Liu SW, Anderson G, Barker JH, Harris EN (1996) Thrombogenic properties of murine anti-cardiolipin antibodies induced by beta 2 glycoprotein 1 and human immunoglobulin G antiphospholipid antibodies. Circulation 94:1746–1751
Pierangeli SS, Liu XW, Barker JH, Anderson G, Harris EN (1995) Induction of thrombosis in a mouse model by IgG, IgM and IgA immunoglobulins from patients with the antiphospholipid syndrome. Thromb Haemost 74:1361–1367
Provan D, Butler T, Evangelista ML, Amadori S, Newland AC, Stasi R (2007) Activity and safety profile of low-dose rituximab for the treatment of autoimmune cytopenias in adults. Haematologica 92:1695–1698
Provan D, Stasi R, Newland AC, Blanchette VS, Bolton-Maggs P, Bussel JB, Chong BH, Cines DB, Gernsheimer TB, Godeau B, Grainger J, Greer I, Hunt BJ, Imbach PA, Lyons G, Mcmillan R, Rodeghiero F, Sanz MA, Tarantino M, Watson S, Young J, Kuter DJ (2010) International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 115:168–186
Quartier P, Brethon B, Philippet P, Landman-Parker J, LE Deist F, Fischer A (2001) Treatment of childhood autoimmune haemolytic anaemia with rituximab. Lancet 358:1511–1513
Ramos-Casals M, Brito-Zeron P, Munoz S, Soto MJ (2008) A systematic review of the off-label use of biological therapies in systemic autoimmune diseases. Med (Baltimore) 87:345–364
Rand JH, Wu XX, Quinn AS, Chen PP, Hathcock JJ, Taatjes DJ (2008) Hydroxychloroquine directly reduces the binding of antiphospholipid antibody-beta2-glycoprotein I complexes to phospholipid bilayers. Blood 112:1687–1695
Rao A, Kelly M, Musselman M, Ramadas J, Wilson D, Grossman W, Shenoy S (2008) Safety, efficacy, and immune reconstitution after rituximab therapy in pediatric patients with chronic or refractory hematologic autoimmune cytopenias. Pediatr Blood Cancer 50:822–825
Reddy PS, Deauna-Limayo D, Cook JD, Ganguly SS, Blecke C, Bodensteiner DC, Skikne BS, Sahud MA (2005) Rituximab in the treatment of relapsed thrombotic thrombocytopenic purpura. Ann Hematol 84:232–235
Rizzo C, Rizzo S, Scire E, Di Bona D, Ingrassia C, Franco G, Bono R, Quintini G, Caruso C (2012) Thrombotic thrombocytopenic purpura: a review of the literature in the light of our experience with plasma exchange. Blood Transfus 10:521–532
Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, Spasoff RA (1991) Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian apheresis study group. N Engl J Med 325:393–397
Romay-Penabad Z, Liu XX, Montiel-Manzano G, Papalardo De Martinez E, Pierangeli SS (2007) C5a receptor-deficient mice are protected from thrombophilia and endothelial cell activation induced by some antiphospholipid antibodies. Ann N Y Acad Sci 1108:554–566
Rubenstein E, Arkfeld DG, Metyas S, Shinada S, Ehresmann S, Liebman HA (2006) Rituximab treatment for resistant antiphospholipid syndrome. J Rheumatol 33:355–357
Ruckert A, Glimm H, Lubbert M, Grullich C (2008) Successful treatment of life-threatening Evans syndrome due to antiphospholipid antibody syndrome by rituximab-based regimen: a case with long-term follow-up. Lupus 17:757–760
Sadler JE, Moake JL, Miyata T, George JN (2004) Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 407–23
Sailer T, Lechner K, Panzer S, Kyrle PA, Pabinger I (2006) The course of severe autoimmune thrombocytopenia in patients not undergoing splenectomy. Haematologica 91:1041–1045
Saleh MN, Gutheil J, Moore M, Bunch PW, Butler J, Kunkel L, Grillo-Lopez AJ, Lobuglio AF (2000) A pilot study of the anti-CD20 monoclonal antibody rituximab in patients with refractory immune thrombocytopenia. Semin Oncol 27:99–103
Sallah S, Husain A, Wan JY, Nguyen NP (2004) Rituximab in patients with refractory thrombotic thrombocytopenic purpura. J Thromb Haemost 2:834–836
Sawitsky A, Ozaeta PB Jr (1970) Disease-associated autoimmune hemolytic anemia. Bull N Y Acad Med 46:411–426
Schollkopf C, Kjeldsen L, Bjerrum OW, Mourits-Andersen HT, Nielsen JL, Christensen BE, Jensen BA, Pedersen BB, Taaning EB, Klausen TW, Birgens H (2006) Rituximab in chronic cold agglutinin disease: a prospective study of 20 patients. Leuk Lymphoma 47:253–260
Sciascia S, Naretto C, Rossi D, Bazzan M, Roccatello D (2011) Treatment-induced downregulation of antiphospholipid antibodies: effect of rituximab alone on clinical and laboratory features of antiphospholipid syndrome. Lupus 20:1106–1108
Scully M, Mcdonald V, Cavenagh J, Hunt BJ, Longair I, Cohen H, Machin SJ (2011) A phase 2 study of the safety and efficacy of rituximab with plasma exchange in acute acquired thrombotic thrombocytopenic purpura. Blood 118:1746–1753
Shah N, Sarode R (2013) Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 28:30–35
Shanafelt TD, Madueme HL, Wolf RC, Tefferi A (2003) Rituximab for immune cytopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 78:1340–1346
Sokol RJ, Hewitt S, Stamps BK (1981) Autoimmune haemolysis: an 18-year study of 865 cases referred to a regional transfusion centre. Br Med J (Clin Res Ed) 282:2023–2027
Stasi R (2010) Rituximab in autoimmune hematologic diseases: not just a matter of B cells. Semin Hematol 47:170–179
Stasi R, Pagano A, Stipa E, Amadori S (2001) Rituximab chimeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood 98:952–957
Stasi R, Stipa E, Masi M, Cecconi M, Scimo MT, Oliva F, Sciarra A, Perrotti AP, Adomo G, Amadori S et al (1995) Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 98:436–442
Thota S, Kistangari G, Daw H, Spiro T (2012) Immune thrombocytopenia in adults: an update. Cleve Clin J Med 79:641–650
Tomietto P, Gremese E, Tolusso B, Venturini P, De Vita S, Ferraccioli G (2004) B cell depletion may lead to normalization of anti-platelet, anti-erythrocyte and antiphospholipid antibodies in systemic lupus erythematosus. Thromb Haemost 92:1150–1153
Trape G, Fianchi L, Lai M, Laurenti L, Piscitelli R, Leone G, Pagano L (2003) Rituximab chimeric anti-CD20 monoclonal antibody treatment for refractory hemolytic anemia in patients with lymphoproliferative disorders. Haematologica 88:223–225
Trappe R, Loew A, Thuss-Patience P, Dorken B, Riess H (2006) Successful treatment of thrombocytopenia in primary antiphospholipid antibody syndrome with the anti-CD20 antibody rituximab–monitoring of antiphospholipid and anti-GP antibodies: a case report. Ann Hematol 85:134–135
Tsagalis G, Psimenou E, Nakopoulou L, Laggouranis A (2010) Effective treatment of antiphospholipid syndrome with plasmapheresis and rituximab. Hippokratia 14:215–216
Valent P, Lechner K (2008) Diagnosis and treatment of autoimmune haemolytic anaemias in adults: a clinical review. Wien Klin Wochenschr 120:136–151
Van Wissen S, Bastiaansen BA, Stroobants AK, Van Den Dool EJ, Idu MM, Levi M, Stroes ES (2008) Catastrophic antiphospholipid syndrome mimicking a malignant pancreatic tumour–a case report. Lupus 17:586–590
Veneri D, Ambrosetti A, Franchini M, Mosna F, Poli G, Pizzolo G (2005) Remission of severe antiphospholipid syndrome associated with non-Hodgkin’s B-cell lymphoma after combined treatment with rituximab and chemotherapy. Haematologica 90(Suppl):ECR37
Vesely SK, George JN, Lammle B, Studt JD, Alberio L, El-Harake MA, Raskob GE (2003) ADAMTS13 activity in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients. Blood 102:60–68
Vianna JL, Khamashta MA, Ordi-Ros J, Font J, Cervera R, Lopez-Soto A, Tolosa C, Franz J, Selva A, Ingelmo M et al (1994) Comparison of the primary and secondary antiphospholipid syndrome: a European Multicenter Study of 114 patients. Am J Med 96:3–9
Wahl D, Lecompte T, Bounameaux H (2008) Need for additional trials of primary prophylaxis in patients with high-risk antiphospholipid antibody profiles: comment on the article by Erkan et al. Arthritis Rheum 58:635–636 (author reply 636)
Wallace DJ (1994) Antimalarial agents and lupus. Rheum Dis Clin North Am 20:243–263
Weide R, Heymanns J, Pandorf A, Koppler H (2003) Successful long-term treatment of systemic lupus erythematosus with rituximab maintenance therapy. Lupus 12:779–782
Willems M, Haddad E, Niaudet P, Kone-Paut I, Bensman A, Cochat P, Deschenes G, Fakhouri F, Leblanc T, Llanas B, Loirat C, Pillet P, Ranchin B, Salomon R, Ulinski T, Bader-Meunier B (2006) Rituximab therapy for childhood-onset systemic lupus erythematosus. J Pediatr 148:623–627
Youinou P, Renaudineau Y (2004) The antiphospholipid syndrome as a model for B cell-induced autoimmune diseases. Thromb Res 114:363–369
Youinou P, Taher TE, Pers JO, Mageed RA, Renaudineau Y (2009) B lymphocyte cytokines and rheumatic autoimmune disease. Arthritis Rheum 60:1873–1880
Zaja F, Baccarani M, Mazza P, Bocchia M, Gugliotta L, Zaccaria A, Vianelli N, Defina M, Tieghi A, Amadori S, Campagna S, Ferrara F, Angelucci E, Usala E, Cantoni S, Visani G, Fornaro A, Rizzi R, De Stefano V, Casulli F, Battista ML, Isola M, Soldano F, Gamba E, Fanin R (2010) Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 115:2755–2762
Zecca M, Nobili B, Ramenghi U, Perrotta S, Amendola G, Rosito P, Jankovic M, Pierani P, De Stefano P, Bonora MR, Locatelli F (2003) Rituximab for the treatment of refractory autoimmune hemolytic anemia in children. Blood 101:3857–3861
Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K (2001) Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem 276:41059–41063
Zhou H, Yan Y, Xu G, Zhou B, Wen H, Guo D, Zhou F, Wang H (2011) Toll-like receptor (TLR)-4 mediates anti-beta2GPI/beta2GPI-induced tissue factor expression in THP-1 cells. Clin Exp Immunol 163:189–198
Zimmer J, Andres E, Noel E, Koumarianou A, Blickle JF, Maloisel F (2004) Current management of adult idiopathic thrombocytopenic purpura in practice: a cohort study of 201 patients from a single center. Clin Lab Haematol 26:137–142
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Basel
About this chapter
Cite this chapter
Matta, B.N., Nayfe, R., Taher, A.T., Uthman, I. (2014). B-Cell Targeted Therapies in Autoimmune Cytopenias and Thrombosis. In: Bosch, X., Ramos-Casals, M., Khamashta, M. (eds) Drugs Targeting B-Cells in Autoimmune Diseases. Milestones in Drug Therapy. Springer, Basel. https://doi.org/10.1007/978-3-0348-0706-7_11
Download citation
DOI: https://doi.org/10.1007/978-3-0348-0706-7_11
Published:
Publisher Name: Springer, Basel
Print ISBN: 978-3-0348-0705-0
Online ISBN: 978-3-0348-0706-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)