
Abstract
Animal health governance faces new challenges as the ecology of infectious livestock diseases is changing (Tomley and Shirley 2009). Environmental and climate changes, intensification of livestock production, modification in land-use and agricultural practices, globalization of human travel, the development of the trade of livestock and livestock products have created conditions for an increase in the emergence and re-emergence of infectious agents in the last decades (Weiss and McMichael 2004; Randolph and Rogers 2010; Jones et al. 2008; Gibbs 2005). The frequency of emergence of new highly pathogenic avian influenza viruses (HPAIV) has increased over the past 20 years, as well as the economic impact of associated outbreaks (Alexander and Brown 2009). Bluetongue virus serotypes have continuously increased their spatial distribution, specifically in a northern direction. Treatment-resistant strains, such as methicillin-resistant Staphylococcus aureus (MRSA), have appeared. Numerous infectious diseases such as foot-and-mouth disease (FMD) are endemic in many parts of the world, and may have a high impact on animal health and farmer livelihood. Moreover, they constrain the ability of affected countries to trade livestock and livestock-derived products. Production systems in developed countries are also vulnerable. For example, outbreaks of FMD in United Kingdom in 2001, classical swine fever in Holland in 1997/1998, and highly pathogenic avian influenza H7N7 in Holland in 2003 resulted in the loss of millions of animals, mainly as a result of culling of affected and exposed animals. Finally, infectious livestock diseases are a threat for public health: about 75% of human infectious agents that emerged in the last 25 years had an animal origin (King et al. 2006).
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Avian Influenza
- Severe Acute Respiratory Syndrome
- Avian Influenza Virus
- Severe Acute Respiratory Syndrome
- Infectious Period
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Animal health governance faces new challenges as the ecology of infectious livestock diseases is changing (Tomley and Shirley 2009). Environmental and climate changes, intensification of livestock production, modification in land-use and agricultural practices, globalization of human travel, the development of the trade of livestock and livestock products have created conditions for an increase in the emergence and re-emergence of infectious agents in the last decades (Weiss and McMichael 2004; Randolph and Rogers 2010; Jones et al. 2008; Gibbs 2005). The frequency of emergence of new highly pathogenic avian influenza viruses (HPAIV) has increased over the past 20 years, as well as the economic impact of associated outbreaks (Alexander and Brown 2009). Bluetongue virus serotypes have continuously increased their spatial distribution, specifically in a northern direction. Treatment-resistant strains, such as methicillin-resistant Staphylococcus aureus (MRSA), have appeared. Numerous infectious diseases such as foot-and-mouth disease (FMD) are endemic in many parts of the world, and may have a high impact on animal health and farmer livelihood. Moreover, they constrain the ability of affected countries to trade livestock and livestock-derived products. Production systems in developed countries are also vulnerable. For example, outbreaks of FMD in United Kingdom in 2001, classical swine fever in Holland in 1997/1998, and highly pathogenic avian influenza H7N7 in Holland in 2003 resulted in the loss of millions of animals, mainly as a result of culling of affected and exposed animals. Finally, infectious livestock diseases are a threat for public health: about 75% of human infectious agents that emerged in the last 25 years had an animal origin (King et al. 2006).
To meet these challenges, transparent scientific approaches are needed to obtain a better understanding of epidemiological patterns and to inform policy-development. Epidemiological systems are composed of multiple processes interacting nonlinearly. They are thus too complex to be represented using purely mental thought processes. The ability of humans to mentally conceptualize a system seems to be reached when more than three variables and six transitions from a state to another are involved (Klein 1998). Therefore, the study of epidemiological systems can benefit significantly from the use of quantitative modeling tools. By integrating individual animal-level knowledge of epidemiological, biological and behavioral factors, mathematical models of infectious diseases can provide insights into disease dynamics at the population-level and predictions of the impact of control strategies on outbreak outcome. Their mathematical formulation makes the underlying assumptions explicit, thereby ensuring the transparency of the approach, at least to those with the relevant understanding of the underlying mathematical methodologies. According to the Royal Society’s Infectious diseases in Livestock report (Society 2002), “Quantitative modeling is one of the essential tools both for developing strategies in preparation for an outbreak and for predicting and evaluating the effectiveness of control policies during an outbreak.”
The aim of this chapter is to present the basic concepts of mathematical modeling of infectious diseases using examples of models developed for the spread of avian influenza viruses.
Use of Mathematical Models to Study the Spread of Infectious Diseases
Mathematical models of infectious disease transmission can be defined as a set of equations conceptualizing the spread of an infectious agent in a host population. They are a simplification of a complex phenomenon, but the simplification should have a limited effect on the disease dynamics properties, which are under study (Britton and Lindenstrand 2009). Models can be used either to simulate disease spread, they are then referred to as simulation models, or to estimate epidemiological parameters. Simulation models use existing knowledge regarding the spread of infection and host population dynamics to investigate mechanisms underlying disease spread, or to predict the future trajectory of an epidemic and the impact of control strategies. Alternatively, given assumptions as to the nature of transmission, models can quantitatively estimate key transmission parameters retrospectively from existing outbreak or experimental infection data.
Simulating Infectious Disease Spread
Models can be used as an explanatory tool to improve our understanding of the dynamics of infectious diseases, and to obtain insight into the impact of interventions. They provide a rigorous framework where the complexity of disease transmission is disentangled and each aspect of the disease spread can be monitored. Allowing identification of the underlying factors that drive disease dynamics, these models can thus be used to generate and to test hypotheses. They can also help in defining priorities for epidemiological data collection by identifying which elements of the disease dynamics are most important for meaningful model experimentation.
Models can also be used to predict the outcome of a disease outbreak and to develop practical control strategies that will minimize its impact. Models then provide quantitative outputs that can be used to inform decision-making. Also, to accurately reproduce the behavior of an epidemic and to improve the reliability of the predictions, those models should ideally include all biological mechanisms and heterogeneities known to influence the disease dynamics.
To date mathematical models have played a major role in elucidating fundamental principles of infectious disease spread. This has included the description of the properties of the dynamics of an epidemic within a population (Dietz 1993; Ross and Hudson 1917; Anderson and May 1991; Kermack and McKendrick 1991; Fine 1993), the demonstration of the role of heterogeneity in driving an epidemic (Yorke et al. 1978; Woolhouse et al. 1997), and the assessment of the possible effects of different control strategies for a wide range of diseases (Ross et al. 2008; Velasco-Hernandez et al. 2002; Hallett et al. 2008; Woolhouse 1992; Grassly et al. 2006).
In recent times, inspired by events such as human disease outbreaks including variant Creutzfeldt-Jakob disease and severe acute respiratory syndrome (SARS), the spread of resistant bacteria such as MRSA and the 2009 H1N1 influenza pandemic, modeling has increasingly become a standard component of public health decision making (Temime et al. 2008; Fraser et al. 2009; Austin and Anderson 1999; Li et al. 2004; Lipsitch et al. 2003; Ghani et al. 2000). In the field of livestock diseases, models of the spread of diseases such as bovine spongiform encephalopathy, scrapie and classical swine fever have all been developed to address pertinent issues regarding ongoing epidemics such as optimal culling strategies, farming restrictions, and the impact upon human health (Anderson et al. 1996; Ferguson et al. 1998; Stringer et al. 1998; Woolhouse et al. 1998; Boender et al. 2008). The 2001 FMD epidemic in Great Britain further served to demonstrate that, following advances in computing power and given access to the best available data (Levin et al. 1997), mathematical models can play an active role in providing policy makers with vital information as to the optimum disease control strategy during a rapidly progressing epidemic (Ferguson et al. 2001; Keeling et al. 2001). However, how useful this information was is still debated, since modeling outputs might have led to an excessive slaughtering of uninfected animals (Kitching et al. 2006).
The spread of avian influenza viruses in poultry has also led to a surge in modeling associated scientific publications over the last decade. Simulation models have been developed to get a better understanding of factors driving the disease spread, such as the potential role played by environmental transmission in the maintenance of avian influenza viruses in natural ecosystems (Lebarbenchon et al. 2009; Breban et al. 2009; Rohani et al. 2009; Guberti et al. 2007; Roche et al. 2009); and the impact of poultry population management on the silent spread of HPAIV strain type H5N1 viruses in live bird markets (Fournié et al. 2010). Further models have been developed to investigate direct and indirect effects of control measures, including the hypothesis that large-scale culling campaigns may impede the evolution of avian host resistance to the virus (Shim et al. 2009), and the evolution and spread of resistance following vaccination campaigns (Iwami et al. 2009a, b). At the farm level, models allowed evaluating the impact of different mortality and morbidity thresholds for the detection of infection (Savill et al. 2006; Carpenter et al. 2004) and investigating the use of sentinel birds for detecting the emergence of HPAIV (Verdugo et al. 2009). The potential for infection to spread silently in vaccinated populations, as a result of lower levels of clinical signs and flock mortality (Savill et al. 2006) within a commercial flock, and the dynamics of waning immunity in backyard systems (Lesnoff et al. 2009) was also evaluated using models. Predictive models have been developed to investigate the potential spread of HPAIV H5N1 in the United Kingdom and to develop control strategies that could efficiently mitigate an outbreak (Dent et al. 2008; Sharkey et al. 2008; Truscott et al. 2007). A simulation model was used to explore the different pathways of infection from a commercial broiler farm in the United States (Dorea et al. 2010).
Quantifying Epidemiological Parameters
Such simulation models, whilst providing useful insight into the infection process, do, however, often require the specification of a range of model parameters. In the absence of context-specific outbreak data, sensitivity analyses can, to a certain extent, address issues associated with misspecification of parameter values. However, modelers are generally reliant upon parameter estimates available in the scientific literature or expert opinion to obtain a reasonable indication of the ranges of values such inputs may take.
In contrast, transmission models fitted to outbreak data are generally a lot simpler, requiring less parameters. This is partially because fewer parameters are easier to fit, but also because this provides parsimonious and easily interpretable measures by which to quantify the spread of infection and the incremental effect of interventions. Fitting such models allows estimation of important epidemiological parameters which, due to the nonlinearity of the transmission process, would not otherwise be possible.
In the case of avian influenza, at the level of transmission between birds, such models are often fitted to data obtained through laboratory experiments. This is because such controlled environments allow relatively detailed data collection, and often offer the opportunity to quantify differences in transmissibility between host species, viral strains, or subtypes and are straightforward to model. Such analyses have been used to estimate the transmissibility of avian influenza viruses in various host species within experimental settings, such as HPAI H7N7 viruses in chickens (van der Goot et al. 2005, 2007a), HPAI H5N1 in chickens (Bouma et al. 2009) and ducks (van der Goot et al. 2007b).
In a field setting, heterogeneity in the distribution of various risk factors such as environmental conditions, host species, poultry housing and the provision of preventative control measures complicates the dynamics of infection, and it is rarely possible to collect detailed data on these factors, or even only on the incidence of infection. As such it is more difficult to ensure that the underlying model dynamics are truly representative of those of the outbreak. However, inferring the transmissibility of infection from such “real-life” settings provides important information as to the effectiveness of ongoing interventions and the incremental effectiveness of alternative strategies necessary for achieving control (Tiensin et al. 2007; Soares Magalhaes et al. 2010; Bos et al. 2010). Moreover, such analyses can provide insight into the dynamics of infection at a larger population scale than that possible in an experiment setting. This is particularly relevant for quantifying between-flock transmission (Stegeman et al. 2004; Garske et al. 2007; Mannelli et al. 2007; Ward et al. 2009) and the geographical spread of infection (Boender et al. 2007; Le Menach et al. 2006; Walker et al. 2010).
Infectious Agents, Transmission, and Infection
Microparasites and Macroparasites
Based on the characteristics of their population biology, infectious agents can be grouped into two categories: microparasites, including bacteria, viruses, prions, protozoa, and macroparasites, including helminths, arthropods. In this chapter, we focus on microparasites. With some exceptions, the duration of microparasitic infections, such as avian influenza, is generally much lower than the host life span, and they reproduce to very high numbers within hosts (Anderson and May 1992).
Transmission
Transmission can be defined as the passing of an infectious agent from an infected host to a susceptible one. Susceptibility refers to the level of vulnerability of a host to an infectious agent. It can be reduced by prior immunization, due to former infection or vaccination. Transmission can be direct, where an infectious agent passes to a susceptible host due to close contact with an infected host, or indirect, where transmission is mediated by the environment or by animate or inanimate vectors. In the case of avian influenza viruses, while direct bird-to-bird contact is likely to be the predominant mode of within-flock transmission, indirect transmission may be crucial for transmitting viruses between farms, and for virus sustainability in natural ecosystems.
Transmission efficacy depends on the infectiousness of infectious hosts and the susceptibility of individuals yet to be infected. Infectiousness is defined by Grassly and Fraser (2008) as the characteristics of an infected host that determine the rate at which susceptible individuals become infected. Both, biological factors, such as the within-host dynamics of pathogenic agents which affect the level of shedding, and behavioral factors, which influence the rate at which contacts are made between individuals, determine the extent to which direct transmission contributes to infectiousness. In contrast, indirect transmissibility is modulated by factors such as the survival of infectious agents in the environment and the ecology of vectors.
Infection
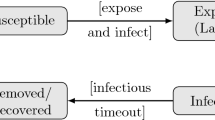
The abundance dynamics of microparasites within hosts are often simplified, and the infection is modeled as the passing of a host through several infection states. One widely used mathematical model of the infection process is known as the SEIR model where a host may move through four states, or compartments (Fig. 11.1): Susceptible, Latent (or exposed), Infectious, and Recovered (or Removed). Initially, susceptible hosts may become infected after contact with an infected host or the infected environment. In the early stages of infection, infectious agents may not be shed in sufficient quantity to transmit infection to other hosts. Individuals are then said to be in the latent (or exposed) state. Once they are able to transmit the infection, they are classified as infectious and they remain infectious for a period of time. Then, depending on the pathogenicity of the parasite and the immune system of the infected individual, the host either dies or the infection is cleared. Following clearance, the host may become immune to infection for a duration dependent upon the parasite in question.

Infection states and disease progress adapted from Keeling and Rohani (2007)
An infection is thus described according to the host’s capacity to transmit the infectious agent, the number of infection states a host can move through, and by the time individuals remain in each state. The average time infected hosts spent in the latent and infectious states are known as the latent and infectious periods, respectively. Such a model may need to be further developed to incorporate other important characteristics of a particular disease that may have a bearing on disease dynamics. For example, as in the case of HPAIV H5N1 in ducks, an infection may not always lead to clinical symptoms. It may also be the case that expression of disease signs leads to a reduction in host activity, or to its isolation, and thus lowers the contact rate and subsequently the infectiousness.
Moreover, symptomatic and infectious periods may not be synchronous. If the time from infection to the onset of symptoms, referred to as the incubation period, is longer than the latent period, an infectious host will be able to transmit an infectious agent before the onset of symptoms (Fig. 11.1). This feature may have important implications for disease control (Fraser et al. 2004).
A Simple Approach to Model Disease Spread
Modeling Transmission
In many models, transmission is assumed to follow a “mass-action” principle. This means that the rate of infection is proportional to the product of the density of infectious and susceptible individuals within a population. This makes the assumption of homogenous mixing within the population: each individual has the same probability to contact any other individual in the population. This mass-action principle can be modulated depending on selected transmission characteristics, specifically how contact probability varies with changes in population size. The two main approaches are either to define the transmission as density-dependent (pseudo mass-action), where the number of contacts increases with population size, or as frequency-dependent (true mass action), where the number of contacts remains independent of population size. The rate at which individuals get infected per unit of time, the so-called force of infection, is then equal to:
where β is the rate of transmission. I t is the number of infectious individuals at time t. For density-dependent transmission, N is equal to N 0, the initial number of susceptible individuals. For frequency-dependent transmission, N is equal to N t , the population size at time t.
The rate of transmission β refers to the infectiousness previously described. It is often defined as the product of the contact rate C, the average number of contacts made by an individual per unit of time, and the probability p c that such a contact leads to an infection (Vynnycky and White 2010):
Characterizing an Epidemic
An epidemic can then be defined as a chain of transmission, and is principally governed by two key individual-level transmission parameters: the basic reproduction number R 0 and the generation time T g (Grassly and Fraser 2008; Ferguson et al. 2003). T g is the period between the infection of a given individual and the infection of another one as a result of transmission from this individual (=secondary case), and R 0 is defined as the expected number of secondary cases of a typical infected case in a fully susceptible population (Diekmann et al. 2000). R 0 assesses the intrinsic transmissibility of an infectious agent and thus its potential to be sustained in a given population. If R 0 is greater than one, an infected host will transmit the infection to more than one individual, the epidemic can spread throughout the population. In contrast, if it is lower than one, an infected host will transmit the infection, on average, to less than one individual, and the epidemic will fade out. R 0 influences both the intensity of the peak and overall final size of the epidemic. Together, R 0 and T g determine the rate of growth in the number of infected individuals during the initial stages of an epidemic.
For simple models, R 0 can be easily derived analytically. In the case of the SEIR model presented in Box 11.1, R 0 is the product between the rate of transmission β and the duration of the infectiousness D:
During the early stages of the spread of an infectious agent in a susceptible population, the number of infectious individuals is much lower than the number of susceptible ones, and the incidence rate increases exponentially. For a closed population in which there is no supply of susceptible individuals, either through new individuals entering the population or through individuals becoming susceptible again following recovery from infection or waning immunity, the ongoing transmission leads to the depletion of the number of susceptible individuals. This causes a reduction in the average number of secondary cases infected by an infectious individual, defined as the net reproductive number R n , also referred to as the effective reproduction number. This can be calculated as the product between R 0 and the fraction s of susceptible individuals in the population, such as:
Contrary to R 0, R n varies as a function of time and informs on the evolution of the incidence. If R n is greater than one the incidence increases, if it is less than one the incidence decreases. As a result an epidemic moves to extinction when R n < 1, meaning that the proportion of susceptible individuals is lower than \( 1/{R_0} \).
In such closed populations, the average number of secondary infections produced by an infected individual eventually falls below 1 and the epidemic fades out (Fig. 11.2). The final fraction of the number of individuals that experienced infection, \( {F_{\infty }} \), is given by:
In contrast, if the susceptible fraction of the population is replenished due to demographic processes such as birth and migration, or due to the absence, or short-lived nature, of immunity, the transmission may be sustained in a population and the infection may become endemic (Fig. 11.2). In that case, the final number of infected individuals cannot be deduced from the former equation. Instead the epidemic may converge to an equilibrium prevalence or may be subject to seasonal peaks.

Course of an transmission in a closed and an open population. In a closed population, an epidemic spreads and then fades out. In an open population, the endemic transmission can be sustained
Modeling Control Strategies
As mentioned earlier, one of the essential goals of mathematical models is to assess the efficiency of interventions and possibly to optimize them. When facing an outbreak, a range of strategies can be implemented to control its magnitude. Their effect will depend on the epidemiology of the infectious agent, the host population dynamics, and the scale of the epidemic. Interventions may achieve a reduction in transmissibility by affecting one, or any combination, of the components of R 0: the contact rate, the probability that a contact leads to infection and the infectious period. Therefore, if the values of the components that an intervention acts upon and R 0 are known, the effectiveness of an intervention at reducing these components required to prevent the spread of infection can be obtained. In this section, we describe some of the most frequently used control strategies to mitigate the spread of HPAI outbreaks in poultry, and how they impact on transmission.
Main Strategies to Control Livestock Disease Outbreaks
Vaccination
By inducing a strong immune response through the production of neutralizing antibodies, vaccination aims to protect individuals from disease signs and death and to prevent outbreaks by reducing susceptibility to infection and viral shedding. The vaccination thus impacts transmissibility by lowering the probability that a contact leads to an infection. The following example assumes a fully effective vaccine, meaning that vaccinated individuals are no longer susceptible to the infection. As discussed in Section “Characterizing an Epidemic” transmission is not sustained if the value of R n remains below its threshold of one. This is achieved in a randomly mixing population if the proportion of susceptible individuals is lower than \( 1/{R_0} \). Therefore, in this case, the eradication of a disease does not require all individuals to be vaccinated as long as the fraction of the population that has been vaccinated is higher than the herd immunity threshold (Fine 1993):
Stamping Out
Stamping out is widely used to control infectious diseases in livestock. The culling of all birds in infected and exposed premises successfully contributed to the eradication of HPAI H7N7 in the Netherlands (Stegeman et al. 2004) and HPAI H7N1 in Italy (Mannelli et al. 2007). The successive outbreak waves of HPAI H5N1 occurring in Hong Kong since 1997 (Sims et al. 2003), and also during the epidemics that have affected South Korea and Japan since 2003, have also been controlled through the depopulation of poultry farms and live bird markets. As it depletes both the number of infected and susceptible individuals, stamping out operates by reducing the infectious period and bird-to-bird contact rates. The usefulness of this strategy for developed countries facing HPAI outbreaks has also been highlighted by mathematical models (Truscott et al. 2007; Le Menach et al. 2006), and was shown to highly depend on the scale of the disease spread and the early detection of infected flocks. Once disease becomes widespread, the costs of sustaining such a strategy can become prohibitive and may not lead to a sustainable reduction in transmission (Sims 2007).
Movement Restrictions
If implemented quickly after the detection of an outbreak, movement restrictions lower disease transmissibility by reducing the number of contacts between farms, and thus prevent disease spread. Importantly, in the context of the spatial spread of an epidemic, when contacts can potentially be made over long distances (such as in the case of live bird trade or indirect transmission by fomites), this may also prevent the disease from accessing regions, which have yet to experience infection. During the 2001 FMD outbreak in United Kingdom, long-range movements of sheep occurring before restrictions were put in place had spread the disease all around the country (Kao 2002).
Impact of Control Strategies on the Course of an Epidemic
As shown in Fig. 11.3, when interventions are able to reduce R n below its threshold of one, the transmission is not sustained anymore and both the number of individuals becoming infected, and the outbreak duration will be curtailed. However, reducing R n below one may be neither logistically nor economically possible, particularly for diseases with a high R 0 and/or affecting a fully susceptible host population. In the case that R n is reduced but remains higher than one, the disease may still spread in the population, but the outbreak size, the total number of individuals that experience infection, will be reduced (Fig. 11.3).

Impact of lowering R n on the epidemic curve. Three scenarios are shown for a SEIR model with similar initial parameters: No control (blue line), controls but R n remains higher than one (red line); controls with R n lower than one (green line). This figure is adapted from Ferguson et al. (2003)
In some cases, interventions may have unintended consequences on the components of R 0. If the vaccination coverage is suboptimal, or if the vaccine is not fully protective at the individual level, it may create conditions for a silent spread of the infectious agent within a host population. It was shown that insufficient vaccination coverage for HPAIV H5N1 may extend the infectious period of a poultry flock. Indeed, as the vaccination coverage rises, fewer birds become infected but outbreaks become harder to detect (Savill et al. 2006). Likewise, Walker et al. (2010) found that the time from infection to reporting was significantly increased in northern Viet Nam following vaccination campaigns. Stamping out may also have unintended effects: failing to implement effective incentive policies leads to underreporting and late detection of outbreaks. In Egypt (Meleigy 2007; Peyre et al. 2009) and Viet Nam, humans are now acting as “sentinels” for the disease in poultry (Minh et al. 2009a).
Increasing Model Complexity
The simplicity of models similar to the SEIR model has helped to elucidate major principles driving the dynamics of numerous infectious agents in a parsimonious manner. However, to extend our understanding of epidemiological patterns and improve the extent to which outcomes reflect reality, it is often necessary to incorporate a higher degree of complexity into the modeled disease dynamics. In this section, we describe various methods to achieve this for livestock diseases.
Stochasticity
So far, all considered models were deterministic: for the same initial conditions and parameter values, all simulations give the same outcome, which is expected to be the average epidemic behavior. However, even in the most controlled settings, for example during laboratory-based transmission experiments, the course of an epidemic would be expected to vary, due to chance. Incorporating stochasticity in mathematical models allow taking into account the random nature of certain epidemiological features, such as the number of transmission events and the latent and infectious periods. Therefore, contrary to deterministic models, several simulations of a stochastic model do not provide a single outcome but a probability distribution of the outcome.
There are several ways to implement stochasticity in a model. Input parameter values can be drawn from probability distributions. This is possible when the extent to which a parameter varies, as a result for instance of interindividual variability or external forces such as climate or vector density, is known. Stochasticity can also concern infection processes. The transition of individuals between infection states, for instance from susceptible to infected, while being associated with an underlying probability, can also be thought of as a random process. Several approaches exist to implement such stochastic processes in a model. One of them is described in Box 11.2. Other algorithms are presented by Keeling and Rohani (2007), and Vynnycky and White (2010).
Accounting for the random nature of disease spread can greatly impact the course of a modeled epidemic. In a deterministic model, if R 0 is higher than one, then an infectious agent will always invade a host population, resulting in a major outbreak, and possibly in its endemicity. In contrast, the infectious agent will not be transmitted if R 0 is lower than one. In reality, for some diseases, an infectious individual can recover before transmitting the infection. As a result, during the early stage of a disease invasion when the number of infectious individuals is low, the disease can become extinct before leading to a major outbreak. The probability that the outbreak avoids this “epidemic fade out” and leads on to a major outbreak following the introduction of a single infectious individual can be calculated as follows:
This equation applies to values of R 0 higher than one. If R 0 is lower than one, transmission events may occur, but they will be limited and will not lead to a major outbreak. As R 0 increases above unity, the distribution of the final epidemic size becomes bimodal (Fig. 11.4): either the epidemic fades out before invading the population, or the disease becomes established and spreads to most individuals. Outbreaks involving a large number of infected individuals become more likely with increasing values of R 0.

Probability of disease invasion and distribution of the final epidemic size for R 0 equal to 1.5 and 3. The probability of disease invasion refers to the probability that the introduction of a single infected host into a fully susceptible population leads to a major outbreak. The distribution of the final epidemic size is obtained from 1,000 simulations of the stochastic SEIR model described in Box 11.2. The density function describes the probability associated with each possible final epidemic size. The initial population size is 100
Heterogeneity
The strong assumptions involved by models described so far may limit their use when investigating infectious agents or host populations for which the heterogeneity in transmission is thought to be an important characteristic.
The rate of transmission β has been assumed to be constant over the host’s infectious period. Such an assumption is often not justifiable, especially for diseases where the infectious period lasts for a significant fraction of the host’s lifespan. In this case, the infectiousness may vary considerably with time, as is the case with HIV infection. Therefore, assuming a constant rate of transmission over the infectious period may be misleading. The infectious compartment I may then be divided into N multiple and successive sub-compartments I n (with n = 1,..,N), each being associated with a specific level of infectiousness β n .
Individuals within a population often differ in their contact patterns and can mix preferentially with a particular subgroup or even with some specific individuals. Therefore, assuming that individuals mix randomly is an oversimplifying assumption. Nonrandom mixing may have an important impact on disease dynamics and on the implementation of control strategies. In the following paragraphs, increasingly complex ways to take into account heterogeneity in mixing patterns are described. They are illustrated in Fig. 11.5.

Representing mixing patterns
Five ways to represent mixing patterns are shown. Each circle represents an individual. The red circles represent the infectious individuals and the blue circles are the susceptible ones. In randomly mixing populations, each individual has the same probability of making contact with the infectious host. The presence of a particular social structure allows the population to be divided into groups, assuming that the rate of transmission varies within and between these groups. In a metapopulation model, the population is divided into subpopulations that are linked by either contacts, or movements of individuals from one subpopulation to another. In a network, the contact patterns between all individuals are known and explicitly represented. For spatial transmission, each individual is assigned a location and the probability of disease transmission depends on the distance from an infectious individual.
Social Structure
Factors such as infectiousness, susceptibility, and the duration of latent or infectious periods may vary between hosts according to intrinsic characteristics, such as their age, gender, or species. Such heterogeneity can often be incorporated into a model by stratifying the host population into groups. A set of compartments is attributed to each group and different rates of transmission are specified within and between those groups. For example, the course of HPAIV H5N1 infection is known to vary according to poultry species. Ducks show a long infectious period, may not exhibit clinical signs and may even survive the infection, in contrast to chickens for which disease is more pathogenic and almost always causes fatality shortly following infection (Saito et al. 2009; Hulse-Post et al. 2005; Spickler et al. 2008). Therefore, simulating the spread of HPAIV H5N1 within a flock composed by chickens and ducks would require modeling the two species separately, with one set of compartments for ducks, and another for chickens. In that case, the force of infection for chickens can be formulated as follows:
where \( {\beta_{{C \to C}}} \) is the rate of transmission between chickens, and \( {\beta_{{D \to C}}} \) NC is the rate of transmission from ducks to chickens. N C is the initial size of the chicken population, I C,t and I D,t are the number of infectious chickens and ducks at time t, respectively.
Accounting for the structure of a population may have a major impact on control strategy design. Indeed, if a group of individuals is responsible for most of the transmission, interventions targeting this group may have a higher impact than interventions applied at random.
Metapopulation
A metapopulation is composed of multiple distinct subpopulations, which are separated spatially and are then coupled only by the movement of individuals between these subpopulations (Colizza et al. 2007) or by indirect contacts. This approach has been recommended when the spatial structure of the population is known to have an important role in the disease dynamics. This is generally the case for infectious diseases affecting livestock where animals are usually aggregated into farms, and the movements of animals and fomites between farms can drive the transmission. Models with such a metapopulation structure can replicate patterns of infection that other models cannot. For infectious diseases of livestock, epidemics which would eventually become extinct in a single farm can become endemic in a production system consisting of a population of farms. This can occur as a consequence of infectious agents becoming amplified within farms and then spreading to others, leading to the sustainability of transmission.
A metapopulation framework has been used to model the spread of HPAIV H5N1 through a market chain (Fournié et al. 2010). At the end of a farm production cycle, birds are sent to a wholesale market and then from a wholesale market to regional markets where they are sold and slaughtered. In addition to this movement of birds from farms to markets, the disease can also be transmitted by indirect contacts between farms or between farms and markets. Such a design was required to evaluate the impact of various interventions that are targeted in subpopulations rather than being indiscriminately applied over the entire population. These included stamping out and vaccination at the farm level and hygiene measures at the market level.
Spatial and Network Transmission
The spatial spread of an infectious agent between livestock premises is generally characterized by a combination of two types of transmission (Riley 2007):
-
Short-range transmission, for which the probability that a premise becomes infected is determined by the infectious state of the other premises in proximity. This can reflect air or vector-borne transmission, or the extent to which farms preferentially contact neighboring farms, for example through sharing equipments and movement of people. Such transmission is generally modeled either by categorizing premises into distinct spatial patches, or by assuming that the probability of transmission scales with the radial distance from the infectious source.
-
Long-range transmission, which is often determined by an underlying network of contacts. This contact network defines how individuals are connected between them through, for example, movements of animals from farm to farm or to slaughterhouses, or movements or visitors such as feed providers or traders.
During the 2003 HPAI H7N7 outbreak that occurred in Holland, the proximity of poultry farms within densely populated poultry areas is likely to have favored intense local transmission of viruses between farms, requiring the implementation of severe interventions to eradicate the disease (Alexander and Brown 2009). In contrast, during the FMD outbreak that affected the United Kingdom in 2001, in addition to local spatial transmission, the movement of infected animals in the days preceding the implementation of interventions was considered to be a major driver of the spread of infection across the country, and thus a key contributor to the large scale of the outbreak (Kao 2002).
The spread of HPAIV H5N1 between farms is often thought to be driven by these short- and long-range modes of transmission. Outbreak waves that occurred in Viet Nam between 2003 and 2007 appeared to result from a combination of short and long-range transmission (Walker et al. 2010; Minh et al. 2009b). Simulation models developed to simulate the spread of HPAIV H5N1 among poultry farms in the United Kingdom have thus taken into account local spatial spread and long-range transmission between premises, mediated, for instance, by the transport of poultry to slaughterhouses (Truscott et al. 2007). For the former, a spatial kernel function is often used to describe how the probability that any infectious premise infects any susceptible premise decays with distance. Truscott et al. (2007) used the following spatial kernel to describe the spatial spread of HPAIV H5N1 between premises:
where d is the distance between two premises, and α and γ are two constant parameters. Long-range transmission was modeled by specifying a network of contacts between premises. Le Menach et al. (2006) chose a different approach, defining three values for the rate of transmission, each one being associated to a range of distances between two premises: short, medium, and long-range.
Impact of Heterogeneity on Disease Transmission and Control
The distribution of factors that determine disease spread, such as the spatial location of farms, contact patterns, production type, and environmental factors, is likely to be heterogeneous. Because of this heterogeneity, the risk for premises to be infected or to transmit the infection follows a distribution that is likely to be highly skewed. Generally, R 0 will be higher when susceptibility and infectiousness are positively correlated (i.e., premises with a high risk of being infected are also at high risk to spread the infection to a high number of other premises) than if the same overall level of infectiousness and susceptibility is evenly distributed within a population (Diekmann et al. 2000). The role of such heterogeneity was highlighted during the SARS epidemic where “superspreading” events were identified as key factor in the spread of infection (Galvani and May 2005), as some infectious hosts infected a much higher number of individuals than the average infectious case. In the cattle movement network formed by Scottish holdings, it was also estimated that 20% holdings were responsible for 80% of the value of R 0 (Woolhouse et al. 2005). From a policy development perspective, heterogeneity can have an important impact when assessing the effectiveness of control measures. Indeed, heterogeneity may cause strong stochastic fluctuations in the early stages of an epidemic, and therefore control policies may need to be readily adaptable to changes in the observed rate of transmission. Moreover, interventions targeting potential high-risk premises may lead to effective disease control with less effort than interventions, which do not consider this level of heterogeneity.
Finally, integrating information relating to the spatial location of premises and their contact patterns increases the level of realism, and, if this can be accurately parameterized with existing data, is likely to improve the reliability of predictions. However, the number of compartments necessary to monitor the number of individuals in each disease state increases rapidly as they are stratified by each additional risk factor. Therefore, when a relatively complex level of heterogeneity is required, individual-based models, where the infection status of each individual in a population is explicitly modeled, are often preferable.
Trade-Off Between Simplicity and Complexity
Simple models, such as the SEIR model, have advantage of being transparent and the way in which the model components interact and drive the disease dynamics can be easily mathematically understood (Keeling and Rohani 2007). Such models often generate analytical solutions (formulae for the general behavior of the model can be obtained directly from the set of model equations) and can thus provide qualitative insights into fundamental principles driving the disease dynamics. However, a higher level of realism in model assumptions is generally needed when seeking quantitative predictions.
Incorporating more detail about host demography and infection processes may result in a model that cannot be solved analytically. Instead, the dynamics of such models have to be simulated in order to obtain numerical approximations of the general model behavior. Although increasing complexity of models improves their realism, it also reduces their transparency, with the mechanisms underlying the disease dynamics becoming increasingly hard to identify, as the number of assumptions and parameters rises. The high number of parameters makes the task of robustly estimating them from data, or finding reliable estimates from existing literature, increasingly arduous. Thus, parameter values or distributions often have to be assumed, subjecting the model outputs to additional and often unquantifiable uncertainty. Moreover, as the desired level of complexity of a model increases, it becomes progressively more difficult to validate any given choice of model assumptions. Therefore, complex models may well become less reliable than simpler ones.
In many cases, over-complexity is just as undesirable as over-simplification (Grassly and Fraser 2008). Models should ideally be as parsimonious as possible, including only mechanisms describing properly the epidemic patterns while ensuring that additional complexity does not alter the outcome. In practice, the level of detail represented in a model needs to be informed by the availability of demographic and disease data. For example in Viet Nam, it has been possible to model the overall dynamics of waves of outbreaks at the commune-level, because this was the geographical resolution at which outbreaks were reported (Walker et al. 2010). Moreover, model parameterization should also be complemented by uncertainty and sensitivity analyses. These involve systematically investigating how potential errors in parameter estimation and changes in parameter values are likely to affect the modeled outputs.
However, whatever their level of detail and the robustness of parameter estimates, models are always a reflection of our current understanding of mechanisms underlying disease transmission, which often remains limited. Thus, models cannot be expected to predict the exact course of an epidemic. In light of this, Medley (2001) recommends their use for relative rather than absolute predictions. Nevertheless, if absolute predictions are made, they should always be associated with a measure of the uncertainty arising from both the stochastic nature of a transmission process and our limited understanding of processes affecting disease spread.
Conclusion
Mathematical models have been widely used to simulate the spread of infectious agents within susceptible host populations, with the principle aims of understanding the fundamental mechanisms underpinning disease dynamics, predicting the course of an epidemic, and assessing the impact of control strategies. Mathematical models can also be used to assess important epidemiological parameters, such as the basic reproduction number and the generation time. Depending upon the scale at which the dynamics of transmission need to be understood, the availability of data and the ability to accurately estimate parameters, models can be developed to incorporate an increasing level of complexity to be able to more faithfully replicate the real-life behavior of epidemics. However, to do so, modelers need to carefully assess the implications this is likely to have for the reliability and transparency of modeled conclusions and predictions. As a result, in many cases, the optimal approach is to develop collections of models that have sequentially increasing complexity. Doing this can provide both general insights into qualitative aspects of disease dynamics and the factors which are most likely to affect transmission, and a robust foundation from which to generate more detailed and realistic predictions.
References
D. J. Alexander, I. H. Brown, Rev Sci Tech 28, 19 (Apr, 2009).
R. M. Anderson, R. M. May, Infectious diseases of humans: dynamics and control. (Oxford University Press, UK, 1991).
R. M. Anderson, R. M. May, Infectious diseases of humans: dynamics and control. (Oxford University Press, UK, 1992).
R. M. Anderson et al., Nature 382, 779 (Aug 29, 1996).
D. J. Austin, R. M. Anderson, Philos Trans R Soc Lond B Biol Sci 354, 721 (Apr 29, 1999).
G. J. Boender et al., Plos Comput Biol 3, 704 (Apr, 2007).
G. J. Boender, G. Nodelijk, T. J. Hagenaars, A. R. W. Elbers, M. C. M. de Jong, BMC veterinary research 4, (Feb 25, 2008).
M. E. Bos et al., Prev Vet Med 95, 297 (Jul 1, 2010).
A. Bouma et al., PLoS Pathog 5, e1000281 (Jan, 2009).
R. Breban, J. M. Drake, D. E. Stallknecht, P. Rohani, Plos Comput Biol 5, (Apr, 2009).
T. Britton, D. Lindenstrand, Math Biosci 222, 109 (Dec, 2009).
T. E. Carpenter, M. C. Thurmond, T. W. Bates, J Vet Diagn Invest 16, 11 (Jan, 2004).
V. Colizza, M. Barthelemy, A. Barrat, A. Vespignani, C R Biol 330, 364 (Apr, 2007).
J. E. Dent, R. R. Kao, I. Z. Kiss, K. Hyder, M. Arnold, BMC veterinary research 4, (Jul 23, 2008).
O. Diekmann, J. A. Heesterbeek, Mathematical Epidemiology of Infectious Diseases: Model Building, Analysis and Interpretation. W. Series, Ed., (Chichester, 2000).
K. Dietz, Stat Methods Med Res 2, 23 (1993).
F. C. Dorea, A. R. Vieira, C. Hofacre, D. Waldrip, D. J. Cole, Avian Dis 54, 713 (Mar, 2010).
N. M. Ferguson, A. C. Ghani, C. A. Donnelly, G. O. Denny, R. M. Anderson, Proceedings of the Royal Society of London Series B-Biological Sciences 265, 545 (Apr 7, 1998).
N. M. Ferguson, C. A. Donnelly, R. M. Anderson, Science 292, 1155 (May 11, 2001).
N. M. Ferguson et al., Nature 425, 681 (Oct 16, 2003).
P. E. Fine, Epidemiol Rev 15, 265 (1993).
G. Fournié, F. J. Guitian, P. Mangtani, A. C. Ghani, J R Soc Interface, (Aug 7, 2011).
C. Fraser, S. Riley, R. M. Anderson, N. M. Ferguson, Proc Natl Acad Sci U S A 101, 6146 (Apr 20, 2004).
C. Fraser et al., Science 324, 1557 (Jun 19, 2009).
A. P. Galvani, R. M. May, Nature 438, 293 (Nov 17, 2005).
T. Garske, P. Clarke, A. C. Ghani, PLoS ONE 2, e349 (2007).
A. C. Ghani, N. M. Ferguson, C. A. Donnelly, R. M. Anderson, Nature 406, 583 (Aug 10, 2000).
E. P. Gibbs, Vet Rec 157, 673 (Nov 26, 2005).
N. C. Grassly, C. Fraser, Nat Rev Microbiol 6, 477 (Jun, 2008).
N. C. Grassly et al., Science 314, 1150 (Nov 17, 2006).
V. Guberti, M. Scremin, L. Busani, L. Bonfanti, C. Terregino, Avian Dis 51, 275 (Mar, 2007).
T. B. Hallett et al., PLoS ONE 3, (May 21, 2008).
D. J. Hulse-Post et al., Proc Natl Acad Sci U S A 102, 10682 (Jul 26, 2005).
S. Iwami, T. Suzuki, Y. Takeuchi, PLoS ONE 4, (Mar 18, 2009a).
S. Iwami, Y. Takeuchi, X. N. Liu, S. Nakaoka, Journal of Theoretical Biology 259, 219 (Jul 21, 2009b).
K. E. Jones et al., Nature 451, 990 (Feb 21, 2008).
R. R. Kao, Trends Microbiol 10, 279 (Jun, 2002).
M. J. Keeling, P. Rohani, Modeling Infectious Diseases in Humans and Animals. (Princeton University Press, UK, 2007).
M. J. Keeling et al., Science 294, 813 (Oct 26, 2001).
W. O. Kermack, A. G. McKendrick, Bull Math Biol 53, 33 (1991).
D. A. King, C. Peckham, J. K. Waage, J. Brownlie, M. E. Woolhouse, Science 313, 1392 (Sep 8, 2006).
R. P. Kitching, M. V. Thrusfield, N. M. Taylor, Rev Sci Tech 25, 293 (Apr, 2006).
G. Klein, Sources of power: how people make decisions. (MIT Press, Cambridge, MA, 1998).
A. Le Menach, E. Vergu, R. F. Grais, D. L. Smith, A. Flahault, Proceedings of the Royal Society B-Biological Sciences 273, 2467 (Oct 7, 2006).
C. Lebarbenchon et al., PLoS One 4, e7289 (2009).
M. Lesnoff, M. Peyre, P. C. Duarte, J. F. Renard, J. C. Mariner, Epidemiol Infect 137, 1405 (Oct, 2009).
S. A. Levin, B. Grenfell, A. Hastings, A. S. Perelson, Science 275, 334 (Jan 17, 1997).
Y. Li et al., American journal of epidemiology 160, 719 (Oct 15, 2004).
M. Lipsitch et al., Science 300, 1966 (Jun 20, 2003).
A. Mannelli, L. Busani, M. Toson, S. Bertolini, S. Marangon, Prev Vet Med 81, 318 (Oct 16, 2007a).
A. Mannelli, L. Busani, M. Toson, S. Bertolini, S. Marangon, Preventive Veterinary Medicine 81, 318 (Oct 16, 2007b).
G. F. Medley, Science 294, 1663 (Nov 23, 2001).
M. Meleigy, Lancet 370, 553 (Aug 18, 2007).
P. Q. Minh et al., Transbound Emerg Dis 56, 311 (Oct, 2009a).
P. Q. Minh et al., Prev Vet Med 89, 16 (May 1, 2009b).
M. Peyre et al., J Mol Genet Med 3, 198 (2009).
S. E. Randolph, D. J. Rogers, Nat Rev Microbiol 8, 361 (May, 2010).
S. Riley, Science 316, 1298 (Jun 1, 2007).
B. Roche et al., Infect Genet Evol 9, 800 (Sep, 2009).
P. Rohani, R. Breban, D. E. Stallknecht, J. M. Drake, P Natl Acad Sci USA 106, 10365 (Jun 23, 2009).
R. Ross, H. P. Hudson, Proceedings of the Royal Society London A 43, 225 (1917).
A. Ross et al., PLoS ONE 3, e2661 (2008).
T. Saito et al., Vet Microbiol 133, 65 (Jan 1, 2009).
N. J. Savill, S. G. St Rose, M. J. Keeling, M. E. J. Woolhouse, Nature 442, 757 (Aug 17, 2006).
K. J. Sharkey, R. G. Bowers, K. L. Morgan, S. E. Robinson, R. M. Christley, Proceedings of the Royal Society B-Biological Sciences 275, 19 (Jan 7, 2008).
E. Shim, A. P. Galvani, PLoS ONE 4, (May 11, 2009).
L. D. Sims, Avian Dis 51, 174 (Mar, 2007).
L. D. Sims et al., Avian Dis 47, 832 (2003).
R. J. Soares Magalhaes, D. U. Pfeiffer, J. Otte, BMC Vet Res 6, 31 (2010).
Royal Society, Infectious Diseases in Livestock. (The Royal Society, London, UK, 2002).
A. R. Spickler, D. W. Trampel, J. A. Roth, Avian Pathol 37, 555 (Dec, 2008).
A. Stegeman et al., Journal of Infectious Diseases 190, 2088 (Dec 15, 2004a).
A. Stegeman et al., J Infect Dis 190, 2088 (Dec 15, 2004b).
S. M. Stringer, N. Hunter, M. E. J. Woolhouse, Math Biosci 153, 79 (Nov, 1998).
L. Temime, G. Hejblum, M. Setbon, A. J. Valleron, Epidemiol Infect 136, 289 (Mar, 2008).
T. Tiensin et al., J Infect Dis 196, 1679 (Dec 1, 2007).
F. M. Tomley, M. W. Shirley, Philos Trans R Soc Lond B Biol Sci 364, 2637 (Sep 27, 2009).
J. Truscott et al., Proc Biol Sci 274, 2287 (Sep 22, 2007).
J. A. van der Goot, G. Koch, M. C. M. de Jong, M. van Boven, P Natl Acad Sci USA 102, 18141 (Dec 13, 2005).
J. A. van der Goot, M. van Boven, G. Koch, M. C. M. de Jong, Vaccine 25, 8318 (Nov 28, 2007a).
J. A. van der Goot, M. van Boven, M. C. de Jong, G. Koch, Avian Dis 51, 323 (Mar, 2007b).
J. X. Velasco-Hernandez, H. B. Gershengorn, S. M. Blower, Lancet Infect Dis 2, 487 (Aug, 2002).
C. Verdugo, C. J. Cardona, T. E. Carpenter, Prev Vet Med 88, 109 (Feb 1, 2009).
E. Vynnycky, R. White, An Introduction to Infectious Disease Modelling. (Oxford University Press, UK, 2010).
P. G. Walker et al., PLoS Comput Biol 6, e1000683 (2010).
M. P. Ward, D. Maftei, C. Apostu, A. Suru, Epidemiol Infect 137, 219 (Feb, 2009).
R. A. Weiss, A. J. McMichael, Nat Med 10, S70 (Dec, 2004).
M. E. J. Woolhouse, Acta Tropica 50, 189 (Feb, 1992).
M. E. Woolhouse et al., Proc Natl Acad Sci U S A 94, 338 (Jan 7, 1997).
M. E. J. Woolhouse, S. M. Stringer, L. Matthews, N. Hunter, R. M. Anderson, Proceedings of the Royal Society of London Series B-Biological Sciences 265, 1205 (Jul 7, 1998).
M. E. Woolhouse et al., Biol Lett 1, 350 (Sep 22, 2005).
J. A. Yorke, H. W. Hethcote, A. Nold, Sexually transmitted diseases 5, 51 (Apr-Jun, 1978).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Food and Agriculture Organization of the United States
About this chapter
Cite this chapter
Fournié, G., Walker, P., Porphyre, T., Métras, R., Pfeiffer, D. (2012). Mathematical Models of Infectious Diseases in Livestock: Concepts and Application to the Spread of Highly Pathogenic Avian Influenza Virus Strain Type H5N1. In: Zilberman, D., Otte, J., Roland-Holst, D., Pfeiffer, D. (eds) Health and Animal Agriculture in Developing Countries. Natural Resource Management and Policy, vol 36. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-7077-0_11
Download citation
DOI: https://doi.org/10.1007/978-1-4419-7077-0_11
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-7076-3
Online ISBN: 978-1-4419-7077-0
eBook Packages: Business and EconomicsEconomics and Finance (R0)