
Abstract
Even though lysosomal storage disorders (LSDs) are considered to be orphan diseases, they pose a highly relevant cause for morbidity and mortality as their cumulative prevalence is estimated to be 1:4,000. This is especially important as treatment in form of enzyme replacement therapy, substrate reduction therapy or stem cell transplantation is amenable for some LSDs. It is plausible that an early start of treatment might improve the overall prognosis and, even more important, prevent irreversible damage of key organs. To get a more precise insight into the real frequency of some LSDs in the general population, we screened 40,024 samples from the Hungarian newborn screening (NBS) program in Szeged for Fabry disease (FD), Gaucher disease (GD), Pompe disease (PD), and Niemann-Pick A/B (NPB) disease using tandem mass spectrometry. Altogether, 663 samples (1.66%) were submitted for retesting. Genetic confirmation was carried out for 120 samples with abnormal screening results after retesting, which identified three cases of GD, three cases of FD, nine cases of PD, and two cases with NPB. In some cases, we detected up to now unknown mutations – one in NPB and seven in PD – which raise questions about the clinical consequences of a NBS in the sense of late-onset manifestations. Overall, we conclude that screening for LSDs by tandem MS/MS followed by a genetic workup in identified patients is a robust, easy, valid, and feasible technology in newborn screening programs. Furthermore, early diagnosis of LSDs gives a chance to early treatment, but needs more clinical long-term data especially regarding the consequence of private mutations.
Competing interests: None declared
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others


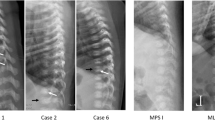
References
Amaral O, Marcão A, Sá Miranda M, Desnick RJ, Grace ME (2000) Gaucher disease: expression and characterization of mild and severe acid beta-glucosidase mutations in Portuguese type 1 patients. Eur J Hum Genet 8(2):95–102
Beutler E, Gelbart T, Balicki D, Demina A, Adusumalli J, Elsas L 2nd et al (1996) Gaucher disease: four families with previously undescribed mutations. Proc Assoc Am Physicians 108(3):179–184
Blanchard S, Sadilek M, Scott CR, Turecek F, Michael H (2008) Tandem mass spectrometry for the direct assay of lysosomal enzymes in dried blood spots: application to screening newborns for mucopolysaccharidosis I. Clin Chem 54:2067–2070
Blaydon D, Hill J, Winchester B (2001) Fabry disease: 20 novel GLA mutations in 35 families. Hum Mutat 18(5):459
Castro-Gago M, Eirís-Puñal J, Rodríguez-Núñez A, Pintos-Martínez E, Benlloch-Marín T, Barros-Angueira F (1999) Severe form of juvenile type II glycogenosis in a compound-heterozygous boy (Tyr-292–> Cys/Arg-854–>Stop)]. Rev Neurol 29:46–49
Chien YH, Lee NC, Thurberg BL, Chiang SC, Zhang XK, Keutzer J et al (2009) Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics 124:e1116–e1125
Dajnoki A, Mühl A, Fekete G, Keutzer J, Orsini J, DeJesus V et al (2008) Newborn screening for Pompe disease by measuring acid α glucosidase activity using tandem mass spectrometry. Clin Chem 54(10):1624–1629
Dajnoki A, Fekete G, Keutzer J, Orsini JJ, De Jesus VR, Chien YH et al (2010) Newborn screening for Fabry disease by measuring GLA activity using tandem mass spectrometry. Clin Chim Acta 411(19–20):1428–1431
Duffner PK, Caggana M, Orsini JJ, Wenger DA, Patterson MC, Crosley CJ et al (2009) Newborn screening for Krabbe disease: the New York State model. Pediatric Neurol 40:245–252
Eng CM, Resnick-Silverman LA, Niehaus DJ, Astrin KH, Desnick RJ (1993) Nature and frequency of mutations in the alpha-galactosidase A gene that cause Fabry disease. Am J Hum Genet 53(6):1186–1197
Eng CM, Ashley GA, Burgert TS, Enriquez AL, D'Souza M, Desnick RJ (1997) Fabry disease: thirty-five mutations in the alpha-galactosidase A gene in patients with classic and variant phenotypes. Mol Med 3(3):174–182
Finckh U, Seeman P, von Widdern OC, Rolfs A (1998) Simple PCR amplification of the entire glucocerebrosidase gene (GBA) coding region for diagnostic sequence analysis. DNA Seq 8(6):349–356
Froissart R, Guffon N, Vanier MT, Desnick RJ, Maire I (2003) Fabry disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in lasma. Mol Genet Metab 80(3):307–314
Guthrie R, Susi A (1963) A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 32:338–343
Han M, Jun SH, Song SH, Park KU, Kim JQ, Song J (2011) Use of tandem mass spectrometry for newborn screening of 6 lysosomal storage disorders in a Korean population. Korean J Lab Med 31(4):250–256
Hermans MM, De Graaff E, Kroos MA, Mohkamsing S, Eussen BJ, Joosse M, Willemsen R, Kleijer WJ, Oostra BA, Reuser AJ (1994) The effect of a single base pair deletion (delta T525) and a C1634T missense mutation (pro545leu) on the expression of lysosomal alpha-glucosidase in patients with glycogen storage disease type II. Hum Mol Genet 3(12):2213–2218
Hermans MM, van Leenen D, Kroos MA, Beesley CE, Van Der Ploeg AT, Sakuraba H, Wevers R, Kleijer W, Michelakakis H, Kirk EP, Fletcher J, Bosshard N, Basel-Vanagaite L, Besley G, Reuser AJ (2004) Twenty-two novel mutations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II. Hum Mutat 23:47–56
Hodanová K, Hrebícek M, Cervenková M, Mrázová L, Vepreková L, Zemen J (1999) Analysis of the beta-glucocerebrosidase gene in Czech and Slovak Gaucher patients: mutation profile and description of six novel mutant alleles. Blood Cells Mol Dis 25(5–6):287–298
Horowitz M, Zimran A (1994) Mutations causing Gaucher disease. Hum Mutat 3(1):1–11
Hubbard WC, Moser AB, Liu AC, Jones RO, Steinberg SJ, Lorey F et al (2009) Newborn screening for X linked adrenoleukodystrophy (X-ALD): validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol Genet Metab 97(3):212–220
Huie ML, Chen AS, Tsujino S, Shanske S, DiMauro S, Engel AG et al (1994) Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1 (-13T–>G) mutation in a majority of patients and a novel IVS10 (+1GT–>CT) mutation. Hum Mol Genet 3(12):2231–2236
Hwu WL, Chien YH, Lee NC, Dobrovolny R, Huang AC, Yeh HY et al (2009) Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c. 9361919G4A (IVS41919G4A). Hum Mut 30:1397–1405
Kasper DC, Herman J, DeJesus VR, Mechtler TP, Metz TF, Sushan B (2010) The application of multiplexed, multi-dimensional ultra-high-performance liquid chromatograph/tandem mass spectrometry to the high-throughput screening of lysosomal storage disorder in newborn dried bloodspots. Rapid Commun Mass Spectrom 24:986–994
Kroos M, Manta P, Mavridou I, Muntoni F, Halley D, Van der Helm R et al (2006) Seven cases of Pompe disease from Greece. J Inherit Metab Dis 29(4):556–563
Kwon JM, Levy PA, Miller-Horn J, Naidich TP, Pellegrino JE, Provenzale JM et al (2009) Newborn screening for Krabbe disease: the New York State model. Pediatr Neurol 40(4):245–252
LaMarca G, Casetta B, Malvgia S, Guerrini R, Zammarchi E (2009) New strategy for the screening of lysosomal storage disorders: the use of the online trapping-and cleanup liquid chromatography/mass spectromentry. Anal Chem 81:6113–6121
Legnini E, Orsini JJ, Hung C, Martin M, Showers A, Scarpa M et al (2011a) Analysis of glucocerebrosidase activity in dry blood spots using tandem mass spectrometry. Clin Chim Acta 412(3–4):343–346
Legnini E, Orsini JJ, Hung C, Martin M, Showers A, Scarpa M, Zhang XK, Keutzer J, Mühl A, Bodamer OA (2011b) Analysis of glucocerebrosidase activity in dry blood spots using tandem mass spectrometry. Clin Chim Acta 412(3–4):343–346
Li Y, Scott CR, Chamoles NA, Ghavami A, Pinto BM, Turecek F et al (2004) Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem 50:1785–1796
Marsden D, Levy H (2010) Newborn screening of lysosomal storage disorders. Clin Chem 56:1071–1079
Meikle PJ, Hopwood JJ, Clague AE, Carey WF (1999) Prevalence of lysosomal storage disorders. JAMA 281(3):249–254
Millington DS, Kodo N, Norwood DL, Roe CR (1990) Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis 13:321–324
Miocić S, Filocamo M, Dominissini S, Montalvo AL, Vlahovicek K, Deganuto M et al (2005) Identification and functional characterization of five novel mutant alleles in 58 Italian patients with Gaucher disease type 1. Hum Mutat 25(1):100
Mitsui J, Mizuta I, Toyoda A, Ashida R, Takahashi Y, Goto J et al (2009) Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol 66(5):571–576
Montfort M, Chabás A, Vilageliu L, Grinberg D (2004) Functional analysis of 13 GBA mutant alleles identified in Gaucher disease patients: pathogenic changes and "modifier" polymorphisms. Hum Mutat 23(6):567–575. Erratum in: Hum Mutat 26(3):276
Nakamura K, Hattori K, Endo F (2011) Newborn screening for lysosomal storage disorders. Am J Med Genet C Semin Med Genet 157(1):63–71
Nichols WC, Pankratz N, Marek DK, Pauciulo MW, Elsaesser VE, Halter CA, Rudolph A, Wojcieszek J, Pfeiffer RF, Foroud T; Parkinson Study Group-PROGENI Investigators (2009) Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 72(4):310–316
Orsini JJ, Morrissey NA, Slavin LN, Wojcik M, Biski C, Martin M et al (2009) Implementation of newborn screening for Krabbe disease: population study and cut off determination. Clin Biochem 42:877–884
Pavlů H, Elleder M (1997) Two novel mutations in patients with atypical phenotypes of acid sphingomyelinase deficiency. J Inherit Metab Dis 20(4):615–616
Poupetova H, Ledvinova J, Berna L, Dvorakova L, Kozich V, Elleder M (2010) The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 33:387–396
Santamaria R, Chabás A, Coll MJ, Miranda CS, Vilageliu L, Grinberg D (2006) Twenty-one novel mutations in the GLB1 gene identified in a large group of GM1-gangliosidosis and Morquio B patients: possible common origin for the prevalent p.R59H mutation among gypsies. Hum Mutat 27(10):1060
Schulze A, Lindner M, Kohlmüller D, Olgemöller K, Mayatepek E, Hoffmann GF (2003) Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics 111:1399–1406
Seeman PJ, Finckh U, Höppner J, Lakner V, Liebisch I, Grau G et al (1996) Two new missense mutations in a non-Jewish Caucasian family with type 3 Gaucher disease. Neurology 46(4):1102–1107
Shabbeer J, Yasuda M, Benson SD, Desnick RJ (2006) Fabry disease: identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum Genomics 2(5):297–309
Simonaro CM, Desnick RJ, McGovern MM, Wasserstein MP, Schuchman EH (2002) The demographics and distribution of type B Niemann-Pick disease: novel mutations lead to new genotype/phenotype correlations. Am J Hum Genet 71(6):1413–1419
Sinigerska I, Chandler D, Vaghjiani V, Hassanova I, Gooding R, Morrone A et al (2006) Founder mutation causing infantile GM1-gangliosidosis in the Gypsy population. Mol Genet Metab 88(1):93–95
Spada M, Pagliardini S, Yasuda M, Turkel T, Thiagarajan G, Sakuraba H et al (2006) Incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet 79(1):31–40
Tanislav C, Kaps M, Rolfs A, Bottcher T, Lackner K, Paschke E et al (2011) Frequency of Fabry disease in patients with small-fibre neuropathy of unknown aetiology: a pilot study. Eur J Neurol 18:631–636
Tsuji S, Martin BM, Barranger JA, Stubblefield BK, LaMarca ME, Ginns EI (1988) Genetic heterogeneity in type 1 Gaucher disease: multiple genotypes in Ashkenazic and non-Ashkenazic individuals. Proc Natl Acad Sci USA 85(7):2349–2352. Erratum in: Proc Natl Acad Sci USA 85(15):5708
Zhang XK, Elbin CS, Chuang WL, Cooper SK, Marashio CA, Beauregard C et al (2008) Multiplex enzyme assay screening of dried blood spots for lysosomal storage disorders by using tandem mass spectrometry. Clin Chem 54:1725–1728
Acknowledgments
The authors are thankful to Shire Human Genetic Therapies for an unrestricted education grant supporting the study to OB. Nicole Deinet and Mirjam Lange have contributed significantly with the ambitious analysis of the genetic samples.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Rodney Pollitt
Conflict of Interest
Conflict of Interest
No conflict of interest to declare.
Rights and permissions
Copyright information
© 2012 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Wittmann, J. et al. (2012). Newborn Screening for Lysosomal Storage Disorders in Hungary. In: JIMD Reports - Case and Research Reports, 2012/3. JIMD Reports, vol 6. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2012_130
Download citation
DOI: https://doi.org/10.1007/8904_2012_130
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-28128-0
Online ISBN: 978-3-642-28129-7
eBook Packages: MedicineMedicine (R0)