
Abstract
Influenza viruses pose a substantial threat to human and animal health worldwide. Recent studies in mouse models have revealed an indispensable role for the innate immune system in defense against influenza virus. Recognition of the virus by innate immune receptors in a multitude of cell types activates intricate signaling networks, functioning to restrict viral replication. Downstream effector mechanisms include activation of innate immune cells and, induction and regulation of adaptive immunity. However, uncontrolled innate responses are associated with exaggerated disease, especially in pandemic influenza virus infection. Despite advances in the understanding of innate response to influenza in the mouse model, there is a large knowledge gap in humans, particularly in immunocompromised groups such as infants and the elderly. We propose here, the need for further studies in humans to decipher the role of innate immunity to influenza virus, particularly at the site of infection. These studies will complement the existing work in mice and facilitate the quest to design improved vaccines and therapeutic strategies against influenza.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Influenza viruses pose a substantial threat to human and animal health worldwide. Based on the antigenic specificity of their envelope proteins, influenza viruses are classified into three distinct types: A, B, and C. Influenza A virus is the major type that circulates in humans, birds, horses, dogs, and pigs. Influenza virus has a negative sense, single-stranded RNA genome composed of eight segments, which encode up to 13 proteins (Wright et al. 2013). Influenza A virus is further classified into different subtypes based on the antigenic properties of two glycoproteins, the hemagglutinin (HA) and neuraminidase (NA). Influenza virus causes annual epidemics that result in millions of human infections due to the emergence of virus strains arising from high frequency of point mutations (antigenic drift). Furthermore, it also has high potential to cause pandemics, owing to the generation of novel subtypes, especially in animal reservoirs, following the gene reassortments between different influenza viruses (antigenic shift) (Medina and Garcia-Sastre 2011; Tscherne and Garcia-Sastre 2011). Such new variants of influenza virus possess an array of strategies to disarm the host immune system, and enable productive invasion of host cells.
Influenza virus infection in humans can result in a wide range of disease symptoms, from an asymptomatic infection to a severe form of febrile respiratory disease. (Taubenberger and Morens 2008). These acute symptoms may last for 7–10 days, but in most cases influenza virus infection is self-limiting due to the induction of protective immune response (Valkenburg et al. 2011). Enhanced disease severity associated with high mortality is very common in infants, but also in the elderly and in immunocompromised populations (Taubenberger and Morens 2008). Therefore, a key challenge is learning how to induce protective immunity in populations at the extremes of age and in immune compromised subjects. This in turn requires a much deeper understanding of the nature of the innate and adaptive immune systems in these populations.
Although virus is restricted to the respiratory tract and cleared efficiently in most cases, acute fatal infections are associated with systemic spread of virus, particularly in pandemic influenza virus infections (Taubenberger and Morens 2008; Tse et al. 2011). In the United States, more than 36,000 deaths and 200,000 hospitalizations with a total health care cost of $10 billion are associated with influenza infections every year (Mao et al. 2012; Molinari et al. 2007; Taubenberger and Morens 2008). Because of the potentially devastating consequences of influenza pandemics and epidemics, several control measures, such as annual vaccination with either inactivated (IIV) or live attenuated (LAIV) influenza vaccines, are used to prevent the likelihood of future outbreaks. These vaccines incorporate the circulating strains of influenza A/H1, A/H3, and B types, which are expected to emerge in the upcoming season, based on epidemiological data obtained from around the globe (Centers for Disease Control and Prevention 2011). Immunization with influenza vaccines, similar to virus infection, primarily induces virus-specific antibody responses, in the blood as well as the local respiratory tissues (Clements et al. 1986). Although vaccination induce a protective B cell memory response, the numbers of antibody secreting cells wane rapidly over time (Sasaki et al. 2007), leading to reduced antibody titers, which may not be sufficient to prevent virus infection (Castilla et al. 2013; Song et al. 2010). In addition, the potential for emergence of new strains and particularly novel subtypes of influenza virus against which there is no immunological memory in the population, poses major threats to human health.
Based on extensive studies in mouse models and influenza-infected patients, it is becoming clear that the innate immune response is critical for virus control and plays a key role in the induction and regulation of adaptive immune responses. Paradoxically, evidence is also emerging that an exaggerated innate immune response can lead to enhanced pathophysiology including influenza-induced acute respiratory distress syndrome (ARDS) in individuals with secondary complications such as chronic respiratory or cardiac illness, or diabetes. In this review, we discuss recent advances in our understanding of innate immunity to influenza virus, with particular emphasis on the different subsets of innate immune cells involved, the nature of the innate receptors that sense the virus, and the antiviral effector molecules. Furthermore, we highlight the contrasting roles played by the innate immune system in mediating protective antiviral immunity to influenza, versus enhanced pathophysiology and disease severity. Most of these insights have resulted from the mechanistic studies in animal models, in which specific subsets of cells or specific receptors have been ablated using gene targeting. However, the extent to which such insights can be translated into the human model is poorly understood. Thus, we review the knowledge gap about the roles of the various subsets of antigen (Ag) presenting cells and other subpopulations of innate immune cells during influenza infection in humans. We emphasize here, the paramount need for future studies deciphering the role of innate immune response to influenza virus at the site of infection in humans. We believe that these studies will facilitate better understanding of the mechanisms that mediate pathogenesis of disease, and in designing improved vaccine and therapeutic strategies against influenza.
2 Innate Immunity to Influenza Virus
Influenza virus primarily targets the respiratory epithelial cells after breaking through the first innate barrier, the mucous layer. In the majority of individuals infected with the seasonal influenza virus, the virus replication is restricted to the upper respiratory tract (nose, pharynx, and larynx). However, in some cases, the virus can also reach the lower respiratory tract (trachea, bronchi, and lung alveoli) in infections with pandemic strains, especially in children and the elderly. Avian influenza virus, such as H5N1 and H7N9, can even reach the blood circulation to infect cells in distant tissues (Taubenberger and Morens 2008; Tse et al. 2011). Attachment to cells via virus-binding receptors enables endocytic uptake resulting in recognition of the virus through innate receptors, which trigger intricate signaling networks to produce antiviral effector molecules that are capable of conferring the protective immunity.
3 Cells of the Innate Immune System Involved in Immunity to Influenza
3.1 Respiratory Epithelial Cells
It has been demonstrated in fatal human cases as well as in mouse model that the epithelial cells of alveoli (type I and II pneumocytes), trachea, bronchi, nose, pharynx and larynx, and sub mucosal glands are infected by influenza virus (Manicassamy et al. 2010; Nakajima et al. 2012, 2013; Pan et al. 2013). Influenza virus causes productive infection of these epithelial cells resulting in the release of large numbers of infectious virus progeny. Notably, the temperature of nose and pharynx is 30–34 °C, which is relatively lower than the tracheal and internal body temperature (36–37 °C). Human influenza viruses, but not avian viruses, can replicate efficiently at this lower temperature of upper respiratory tract, similar to that at 37 °C (Boonnak et al. 2012; Pelletier et al. 2011). Although different innate immune cells are found in nasal and pharyngeal cavity, the functional features of innate immunity against pathogens at this lower temperature is not known.
In vitro, primary human type II alveolar cells produced type III IFNs in response to human seasonal influenza virus (Wang et al. 2009), whereas differentiated/polarized human bronchial epithelial cells upregulated the expression of type I IFNs upon infection with human influenza virus, but not avian H5N1 influenza virus (Chan et al. 2010; Gerlach et al. 2013; Hsu et al. 2012; Zeng et al. 2007). These findings are consistent with the idea that the activation of type I IFN response in respiratory epithelial cells is crucial for limiting the initial viral infection, since the impaired type I IFN production by H5N1 virus is associated with severe virulence.
In addition to the initial IFN-mediated antiviral response, epithelial cells secrete various cytokines and chemokines such as IL-6, TNF-α, IL-8/CXCL8, CXCL10, CCL2, CCL5 (Chan et al. 2005, 2009; Vareille et al. 2011; Wang et al. 2011; Yu et al. 2011). Furthermore, influenza virus-infected epithelial cells trigger recruitment of an array of innate immune cells, which participate in the protective immune response (Table 1). Interestingly, a recent study in mice showed that expression of GM-CSF by influenza virus-infected alveolar epithelium is essential for effective viral clearance mediated by CD103+ DC-induced CD8+ T cells (Unkel et al. 2012).
3.2 Neutrophils
Neutrophils arrive very early at the site of infection, and together with the tissue-resident macrophages are among the first line of cellular defense against pathogens. Although influenza viruses are phagocytized by neutrophils in humans and mice (Brandes et al. 2013), neutrophils are not permissible for productive infection in vitro (Tate et al. 2011). Neutrophils are shown to be important for controlling replication and spread of influenza virus in mouse models (Fujisawa 2001). Lethal dose infection of mice depleted of neutrophils results in increased virus titers in the lungs and in extrapulmonary sites with increased mortality (Tate et al. 2008, 2009, 2011; Tumpey et al. 2005) (Table 1). Consistent with this, in an in vitro culture of human bronchoalveolar lavage fluid (BALF), addition of neutrophils, particularly activated neutrophils, resulted in significantly greater clearance of influenza virus (White et al. 2007). As phagocytic cells, neutrophils can uptake influenza virus-infected apoptotic cells in the lungs to augment clearance of virus (Hashimoto et al. 2007; Watanabe et al. 2005). Despite neutrophils being important participants in the antiviral response in mouse models, the phenotype and functional relevance of large number of neutrophils that accumulate at the site of infection in influenza virus-infected patients, and their mechanisms of anti viral immunity, are poorly understood.
3.3 Macrophages (MΦ)
These include both tissue-resident alveolar macrophages (aMΦ) as well as recently recruited MΦ derived from circulating monocytes (moMΦ) (Table 1). In mouse models, during the course of influenza virus infection, aMΦ are outnumbered by the migrant monocytes, which differentiate into MΦ with an activated phenotype (Herold et al. 2008). Because of their efficient phagocytic capacity, like neutrophils MΦ are critical for clearance of virus-infected and apoptotic cells (Hashimoto et al. 2007; Hoeve et al. 2012; Watanabe et al. 2005). In the absence of MΦ, influenza virus replication is enhanced, leading to greater disease severity and mortality, in mice and pigs (Ito et al. 2011; Kim et al. 2008, 2013; Tate et al. 2010; Tumpey et al. 2005). In humans, most of the studies were done employing in vitro culture of aMΦ obtained from BALF or lung specimens and blood monocyte-derived MΦ (moMΦ). Compared to moMΦ, aMΦ are considered to be resistant to productive infection with the seasonal human influenza virus strains in vitro. Consistent with this, influenza virus replication and induction of proinflammatory cytokine responses were much poorer in aMΦ when compared with moMΦ (van Riel et al. 2011). In the case of avian influenza, however, the virus is able to productively infect both moMΦ and aMΦ (reviewed in Short et al. 2012). Notably, highly pathogenic H5N1 viruses are found to induce more potent proinflammatory cytokine responses, and IFN production than seasonal human viruses (Cheung et al. 2002; Geiler et al. 2011; Lee et al. 2009; Perrone et al. 2008; Yu et al. 2011; Zhou et al. 2006). Moreover, aMΦ are believed to be the main producers of type I IFN, since they are shown to produce significantly more type I IFNs than DCs during influenza virus infection (Helft et al. 2012). Thus, MΦ appear to play critical role in the early innate response to influenza virus infection.
3.4 Monocytes
There are three monocyte subsets identified in the blood, which includes classical monocytes (CD14++CD16− in humans), intermediate monocytes (CD14++CD16+ in humans), and the so-called patrolling monocytes (CD14loCD16++ in humans) (Cros et al. 2010; Ziegler-Heitbrock et al. 2010) (Table 1).
Increased number of monocytes are found in the nasal mucosae, the first site of infection (Gill et al. 2005, 2008; Oshansky et al. 2013) as well as in the peripheral blood of influenza virus-infected patients (Giamarellos-Bourboulis et al. 2009; Gill et al. 2005, 2008; McClain et al. 2013; Oshansky et al. 2013). Interestingly, in influenza virus-infected mice, type I IFN-signaling was found to augment the differentiation of stem cells into CCL2-producing monocytes, which mediate the recruitment of additional monocytes (Brandes et al. 2013; Seo et al. 2011). Consistent with this, influenza virus infection of human monocytes induces the release of CCL2 and CXCL10 (Hoeve et al. 2012; Maddur and Pulendran, unpublished data), but inhibits the responsiveness to chemokines, such as CCL2, CCL3, CCL4, and CCL5 by downregulation of their respective chemokine receptors (Salentin et al. 2003), presumably to retain these cells at the site of infection.
As discussed earlier, in addition to transformation to moMΦ, monocytes that migrate to influenza virus-infected tissues can also differentiate into monocyte-derived dendritic cells (moDCs) (Cao et al. 2012; Lin et al. 2008; Unkel et al. 2012). In vitro studies have shown that productive influenza virus infection of human monocytes induces secretion of TNF-α and GM-CSF, and triggers rapid transformation into cells with phenotypic features of DCs (Cao et al. 2012; Hou et al. 2012; Qu et al. 2003). In contrast, we found that infection of monocytes with influenza virus did not induce activation and TNF-α secretion, but merely induces the loss of CD14 resulting in DC-like phenotype (CD11c+CD14−). Despite the phenotypic similarity to DCs, these CD14− cells lack the functional properties of DCs (Maddur and Pulendran unpublished data). Mice with reduced numbers of monocytes at the site of infection, such as CCR2−/− mice or mice treated with a CCR2 antagonist did not show increased viral loads, raising the question of the direct role of monocytes in virus clearance (Lin et al. 2008, 2011). Furthermore, the relevance of these findings of mouse models and in vitro studies to influenza human patients is not clear and is worth investigating for understanding the role of monocytes in influenza.
3.5 Dendritic Cells (DCs)
DCs are rare but widely distributed throughout the body, and function as key orchestrators of the immune response (Banchereau and Steinman 1998; Manicassamy and Pulendran 2011; Pulendran et al. 2010b; Steinman and Banchereau 2007).
3.5.1 DC Subsets
In mice, DCs can be broadly classified as CD11chi conventional DCs (cDCs) and CD11clo B220+ plasmacytoid DCs (pDCs). In the steady-state respiratory tract, the epithelial layer of conducting airways is lined by CD103+CD11blo cDCs (langerin+), which extends their long dendrites into the airway lumen. The lamina propria, which is beneath the epithelial layer, contains CD103−CD11bhi cDCs (langerin−) as well as CD11clo pDCs. Further, all the three DC subsets are found in the alveolar septa of lung parenchyma. Under inflammatory conditions, additional CD11cloCD11b+Ly6C+ moDCs are recruited to the conducting airways and lung parenchyma (Guilliams et al. 2013; Helft et al. 2010; Lambrecht and Hammad 2009, 2012). Of note, in the lungs, MΦ subsets, which are CD11chi, must be distinguished from cDCs based on other markers (Table 1). The respiratory tract draining lymphoid tissues contain resident DC subsets such as, CD8α+ cDCs, and CD11b+ cDCs as well as CD11clo pDCs, in addition to migratory DCs (Haniffa et al. 2013; Helft et al. 2010).
In influenza virus-infected mice, there is an increased accumulation of CD103−CD11bhi cDCs as well as CD103+CD11blo cDCs in the trachea and lung interstitial tissue, with a transient increase in CD11clo pDCs, all of which display an activated phenotype (GeurtsvanKessel et al. 2008; Ho et al. 2011; McGill et al. 2008). In addition, CD11cloCD11b+Ly6C+ moDCs also infiltrate the lungs in high number (Guilliams et al. 2013; Lin et al. 2008, 2011). pDCs are considered to be less susceptible to influenza virus infection, compared to the highest susceptibility of CD103+ cDCs and CD11b+Ly6C+ moDCs, and that of intermediate susceptibility of CD11bhi cDCs (Hao et al. 2008; Hargadon et al. 2011). Of relevance, murine DCs are considered to be less susceptible to productive influenza virus infection than human DCs in vitro (Hartmann et al. 2013; Ioannidis et al. 2012).
In addition to secretion of inflammatory cytokines and chemokines, DCs efficiently migrated to the regional mediastinal LN (MLN) and induce protective adaptive immune responses to augment viral clearance (see later sections) (GeurtsvanKessel et al. 2008; Helft et al. 2012; Ho et al. 2011; Kim and Braciale 2009; McGill et al. 2008; Unkel et al. 2012). Consistent with these findings, an effective reduction in influenza virus replication in the lungs and enhanced survival of infected mice was observed following adoptive transfer of CD11chi cDCs 1 day before infection (GeurtsvanKessel et al. 2008) or influenza virus-activated bone marrow-derived DCs (BMDCs) 1 day after infection (Boonnak et al. 2013). Conversely, removal or loss of pDCs and/or migratory cDCs in mice prior to influenza virus infection resulted in higher viral load in the lungs with increased mortality, supporting the contribution of DCs in viral control (GeurtsvanKessel et al. 2008; Kaminski et al. 2012; McGill et al. 2008).
In humans, similar to mice, two lineages of DCs are identified, which include CD11c+CD123lo myeloid DCs (mDCs) and CD11c−CD123hi pDCs. The respiratory tract has both mDCs and pDCs. Further, CD1c+ mDC1 (CD1a+langerin−/+), which resemble Langerhans cells and CD141+ mDC2 (CD1a−langerin−) subsets of mDCs identified in the human lungs are proposed to correspond to CD103−CD11bhi cDC and CD103+CD11blolangerin+ cDC subsets in mice, respectively (Haniffa et al. 2012; Yu et al. 2013) (Table 1). The draining lymphoid tissue contains pDCs and CD1c+ mDC1 as well as CD141+ mDC2 (Segura et al. 2012).
Studies have observed an increased number of mDCs and pDCs in the nasal mucosa, but a decreased number in the peripheral blood of influenza virus-infected patients (Gill et al. 2005, 2008; Huang et al. 2013), supporting the recruitment of circulating DCs to the site of infection. Furthermore, influenza virus infection induced activation of human DC subsets including CD1c+ mDC1, pDC and moDCs characterized by upregulation of expression of HLA-DR, HLA-ABC, CD80, CD86, CD40, and CCR7 (Fonteneau et al. 2003; Larsson et al. 2000; le Nouen et al. 2011; Osterlund et al. 2005; Piqueras et al. 2006; Smed-Sorensen et al. 2012). Interestingly, although enhanced production of IL-6, IL-8, and CCL2 was observed in pandemic H1N1 2009 influenza virus-infected patients (Lee et al. 2011), in vitro virus infection failed to enhance cytokine secretion in human moDCs (Osterlund et al. 2010). Consistent with this, influenza virus infection of moDCs failed to induce maturation and production of IFN-α, TNF-α, and IL-6, due to the suppressive effects of NS1 protein (Fernandez-Sesma et al. 2006). Notably, exposure to type I IFNs prior to influenza virus infection was found to enhance the activation and cytokine secretion of mDCs and pDCs, by partially overcoming the inhibition by IFN antagonist NS1 protein of influenza virus (Fernandez-Sesma et al. 2006; Phipps-Yonas et al. 2008).
3.6 Natural Killer (NK) Cells
NK cells possess unique natural cytotoxicity receptors (NCRs) such as NCR1 in mice, and NKp30, NKp44, and NKp46 in humans, involved in recognition of viral- and tumor-associated molecules and activation of NK cells (Jost and Altfeld 2013). Influenza virus productively infects human NK cells in vitro (Mao et al. 2009, 2010) as well as mice NK cells in vivo (Guo et al. 2009). In mouse models of influenza virus infection, there is a substantial increase in the population of activated NK cells expressing CD107a and IFN-γ in the lungs, which can lyse influenza virus-infected cells through granzyme B and perforin, and contribute to the virus control (Ge et al. 2012; He et al. 2004; Hwang et al. 2012; Verbist et al. 2012). Accordingly, NK cell–depletion (Ge et al. 2012) or defects in NK cell activity (Gazit et al. 2006) resulted in delayed virus clearance from the lungs with worsen disease in mouse models of sublethal influenza virus infection.
Consistent with this, in humans, NKp46 and NKG2D-mediated recognition of HA on influenza virus-infected cells induced NK cell-mediated cytolysis of target cells (Draghi et al. 2007; Mandelboim et al. 2001). However, in contrast to these in vitro findings, virus infection was associated with transient deficiency of circulating NK cells, particularly CD56+++ NK cells, and downregulation of NK cell activity, especially with pandemic H1N1 2009 influenza patients (Denney et al. 2010; Fox et al. 2012; Guo et al. 2011; Heltzer et al. 2009). It is not clear whether the reduced number of NK cells in peripheral blood is a reflection of augmented recruitment of NK cells to the site of infection, the respiratory tract. However, the fatal cases of influenza virus infections showed reduced number or absence of NK cells in lung inflammatory infiltrate (Denney et al. 2010; Welliver et al. 2007). Consistent with this, studies have found that influenza virus-infection of NK cells inhibits their functions of cytotoxicity and cytokine and chemokine secretion in humans (Mao et al. 2010) as well as in mice (Guo et al. 2009).
3.7 Natural Killer T (NKT) Cells
These are a heterogeneous group of T cells that share properties of both T cells and natural killer (NK) cells. Many of these cells recognize the non-polymorphic CD1d molecule, an Ag-presenting molecule that binds self- and foreign lipids and glycolipids (reviewed in Bendelac et al. 2007).
In mice, influenza virus-activated invariant NKT cells were found to reduce viral load and the immune-pathology during lethal influenza virus infection by different mechanisms mediated by IFN-γ and IL-22 (Kok et al. 2012; Paget et al. 2011). Interestingly, in influenza virus-infected mice as well as in humans, activated iNKT cells were found to diminish the immunosuppressive effect of influenza virus-induced myeloid-derived suppressor cells (MDSCs) through CD1d- and CD40-mediated interactions (de Santo et al. 2008). Despite these striking observation, the role of NKT cells in humans, particularly at the site of infection is unexplored.
3.8 Innate Lymphoid Cells (ILCs)
In mice as well as in humans, ILCs include three groups of cells, (1) IFN-γ-producing NK cells and ILC1, (2) IL-4/IL-5/IL-13-producing ILC2, and (3) IL-17/IL-22-producing ILC3 and LTi (lymphoid tissue-induce) cells (Spits et al. 2013). Recent studies have revealed the diverse role of ILC2 in influenza virus infection. In mice lacking T cells and B cells, ILC2 cells were found to accumulate in the lungs following sublethal influenza virus infection, and were critical for sustaining lung epithelial barrier and remodeling of respiratory tissue through secretion of amphiregulin (Monticelli et al. 2011). In contrast, sublethal influenza virus infection triggered airway hyper-reactivity (AHR) is shown to be mediated by IL-5 and IL-13–producing ILC2 (natural helper cells) that are activated by IL-33 secreted by aMΦ and NKT cells (Chang et al. 2011; Gorski et al. 2013). The significance of these cells during human influenza needs to be determined.
3.9 Other Innate Immune Cells
In spite of the presence of other innate immune cells like mast cells, eosinophils and basophils in the lungs and airways, the interaction of these cells with influenza virus is not fully explored.
4 Virus Binding Surface Receptors
4.1 Sialic Acid-Containing Receptors
Sialic acid (SA, N-acetylneuraminic acid) is identified as the primary attachment site on the cell surface that interacts with the receptor-binding site within the globular head of HA of influenza viruses (Skehel and Wiley 2000; Wilson and Cox 1990). Following the interaction of virus with SA-containing receptors , entry into the cell might involve clathrin-mediated endocytosis or caveolin and clathrin independent mechanism (de Vries et al. 2011; Lakadamyali et al. 2006).
Sialic acid is added to surface proteins as part of post-translational modification. SA consists of nine carbon sugar frequently attached to underlying terminal galactose residue of glycoproteins or glycolipids of cell surface receptors by either α2,3 (SAα2,3Gal) or α2,6 (SAα2,6Gal) linkage (Wilson and Cox 1990). The SA and its linkage is critical for facilitating influenza virus infection of epithelial and immune cells, since enzymatic switching of SA linkage or removal of cell-surface SA can alter susceptibility or confer resistance to influenza virus infection. It has been observed that human influenza virus strains usually bind SAα2,6Gal, whereas avian influenza virus strains have preference for SAα2,3Gal linkage, (reviewed in Londrigan et al. 2012). Thus, SA is considered to be an important determinant of virus tropism and contributes to viral pathogenesis and induction of immune response.
In the human respiratory tract, using lectins specific for SA linkage, epithelial cells of the nasal mucosa, paranasal sinuses, the pharynx, the trachea, and the bronchi and bronchioles were found to predominantly express SAα2,6Gal, with SAα2,3Gal expression being rare (Shinya et al. 2006). However, the cells of the lower respiratory tract, including non-ciliated cuboidal bronchiolar cells and the type II pneumocytes of alveoli predominantly expressed SAα2,3Gal (Ibricevic et al. 2006; Shinya et al. 2006; Thompson et al. 2006). Although a similar pattern of SAα2,3Gal expression is observed in mouse respiratory tract, SAα2,6Gal is not expressed (Ibricevic et al. 2006) (Table 1). In the human innate immune cells, cell surface expression of SAα2,6Gal is found to be predominant compared to SAα2,3Gal (Corral et al. 1990; Hartshorn et al. 1995; Nicholls et al. 2007; Ramos et al. 2011; Sakabe et al. 2011; Videira et al. 2008) (Table 1).
Despite these observations of differential expression of SA-linkages on cells and its significance in virus attachment, the identity of SA-containing receptors is unexplored. Further, it is also not clear whether virus binding SA-containing receptors trigger intracellular signals for activation and cytokine production in innate cells. However, study using UV-inactivated genetically modified human influenza viruses showed that virus binding to SAα2,3Gal induced higher levels of proinflammatory cytokines and IFN-inducible genes in DCs and MΦ compared to influenza virus with SAα2,6Gal binding specificity (Ramos et al. 2011), suggesting a viral replication-independent induction of innate response. These finding indicate that the binding of SA-containing receptors to HA can induce differential innate antiviral responses. Further, inactivated influenza vaccine (IIV) used in humans is a split vaccine mainly containing HA, and hence, the interaction of IIV with SA-containing receptors is likely to play role in the vaccine-induced immunity. Therefore, deciphering the identity, structure, and functional features (like cytoplasmic signaling network) of SA-containing receptors involved in influenza is warranted.
Although SA is critical for virus binding and tropism, the cells lacking surface SA were found to be permissive to virus entry and infection, although to lesser extent (Thompson et al. 2006). Further, it is believed that SA enhances the binding of influenza virus to cell surface to facilitate the interaction with other receptors required for virus entry (Londrigan et al. 2012). Consistent with this, several cell surface carbohydrate-recognizing receptors are also proposed in binding to sugar residues within the surface glycoproteins of influenza virus to augment virus uptake. They include C-type lectin receptors (CLRs), which are the innate recognition receptors of host (discussed below).
5 Virus Sensing Receptors
Influenza virus is sensed by different pattern-recognition receptors (PRRs). Detection of viral components by PRRs triggers intracellular signaling cascades responsible for secretion of type I IFNs, proinflammatory cytokines, and chemokines, and acquisition of activation status. Recent studies have shown that influenza virus is sensed by PRRs such as, CLRs, Toll-like receptors (TLRs ), retinoic acid-inducible gene I (RIG-I )-like receptors (RLRs), and nucleotide oligomerization domain (NOD)-like receptors (NLRs ).
5.1 C-Type Lectin Receptors (CLRs)
These are a diverse family of transmembrane proteins that contain one or more carbohydrate recognition domains (CRDs), but do not bind only carbohydrate structures (Geijtenbeek and Gringhuis 2009; Sancho and Reis e Sousa 2012). Interaction between influenza virus and CLRs specific to mannose and galactose, which serve as the receptor for virus attachment and infection of cells, has been observed. These CLRs include (i) macrophage mannose receptor (MMR, CD206, which binds to mannose, fucose, and sulphated sugars), (ii) macrophage galactose-type lectin (MGL, CD301, which binds mainly to terminal GalNAc residues, but also to galactose and Lewis-X structures) and (iii) DC-specific ICAM3-grabbing non-integrin (DC-SIGN, CD209, which binds mannose-rich glycans) (Geijtenbeek and Gringhuis 2009; Sancho and Reis e Sousa 2012).
These CLRs are primarily expressed on monocytes, MΦ, and mDCs (Table 1). They can mediate influenza virus binding to augment the SA-dependent virus up take by cells resulting in enhanced susceptibility to infection. However, these receptors alone do not mediate efficient infection in the absence of SA (Londrigan et al. 2011; Wang et al. 2008). Furthermore, these CLRs, which are known to bind influenza virus lack both activating-ITAM as well as inhibitory-ITIM cytoplasmic signaling motifs, but possess tyrosine motifs involved in endocytosis (Geijtenbeek and Gringhuis 2009; Sancho and Reis e Sousa 2012). Hence, based on the recent findings, it is speculated that virus-bound CLRs can employ the endocytic equipment to direct the captured viral antigenic cargo for processing and cross-presentation to T cells, especially MMR and MGL (Sancho and Reis e Sousa 2012). Furthermore, despite being unable to induce myeloid cell activation by themselves, these CLRs, particularly DC-SIGN, are found to modulate the outcome of signaling by other PRRs (Geijtenbeek and Gringhuis 2009; Sancho and Reis e Sousa 2012). It is therefore important to determine the effect of binding of influenza virus to these CLRs on the activation and function of myeloid cells induced by other PRRs, and also the ensuing CD8+ T cell immune response.
5.2 Toll-Like Receptors (TLRs)
TLRs have emerged as key sensors of innate immunity to viruses recognizing their PAMPs. TLR2 and TLR4 on cell surface detect the envelope glyco/lipoproteins and that of intracellular/endosomal TLR3, TLR7, TLR8, and TLR9 sense viral nucleic acids (Finberg et al. 2007; Kawai and Akira 2011).
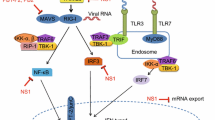
All TLRs recruit a specific set of adaptor molecules that harbor TIR (Toll-IL-1 receptor) domain, such as MyD88, TIRAP, TRIF, or TRAM, a combination of which decides the response to ligand. MyD88 is employed by all TLRs, except TLR3, which uses TRIF. Whereas TLR4 can utilize TIRAP or TRAM to recruit MyD88 or TRIF, TLR2 uses TIRAP to recruit MyD88. Further, MyD88-dependent and TRIF-dependent signaling pathways activate NF-kB, interferon regulatory factor7 (IRF7) or IRF3 through IRAKs, TRAF6, TAK1, and IKK complex, resulting in induction of antiviral status and secretion of cytokines. Interestingly, TLRs cooperate with other PRRs like NLRs and RLRs to induce innate immunity to pathogens including influenza (reviewed in Kawai and Akira 2011) (Fig. 1). Studies have shown that influenza virus is recognized by different TLRs, such as TLR7/8 that bind ssRNA and TLR3, which senses the dsRNA in the endosomes. In addition, TLR4 can detect the damage-associated molecular patterns (DAMPs) released from virus-infected cells (Table 2).

Recognition of influenza virus infection by pattern-recognition receptors. Activation of TLRs upon detection of viral RNA (TLR3 and TLR7/8) or binding of death-associated molecular patterns (DAMPS; TLR4) recruits adaptor molecules (MyD88 and TRIF) triggering distinct signaling pathways that activates nuclear translocation of transcription factors (IRF3/7 and NF-kB) to induce production of type I interferons (I IFNs) and inflammatory cytokines (IL-6, TNF and pro-IL-1β and -IL-18). Recognition of 5’ppp-RNA by RIG-I activates recruitment of MAVS on mitochondrion, which in turn induces the production of cytokines through IRF3/IRF7. Of the NLRs, NOD2 detects ssRNA to activate translocation of MAPK and IRF3/IRF7 by recruiting adaptor molecules, RIPK2 and MAVS, respectively, to induce cytokine production. Activation of NLRP3 mediated by diverse stimuli, dependent on ionic channel M2 protein of influenza virus, recruits ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), which in turn interact with pro-caspase-1 to form NLRP3 inflammasome. Autoactivation of caspase-1 cleaves pro-IL-1β/IL-18 to mature IL-1β/IL-18 for their secretion
5.2.1 TLR3
Although absent in pDCs, monocytes, and neutrophils, low levels of TLR3 is expressed in MΦ, mDCs, moDCs (Kadowaki et al. 2001) and primary respiratory epithelial cells of mice (le Goffic et al. 2006) and humans (Guillot et al. 2005; Ioannidis et al. 2013) (Table 1). It is important to note that due to the action of RNA helicase DDX39B, dsRNA is not generated during replication of influenza virus (Pichlmair et al. 2006; Wisskirchen et al. 2011). It is therefore important to determine whether TLR3 plays a significant role in antiviral immunity during influenza virus infections and to identify the ligands for TLR3 within the influenza-infected cells.
Pretreatment of human moDCs with TLR3 ligand (poly I:C) conferred resistance to infection with H5N1 influenza virus (Thitithanyanont et al. 2007). Consistent with this, intranasal pretreatment of mice with poly I:C provided high level of protection against lethal challenge with influenza virus (Wong et al. 2009). Furthermore, lethal dose influenza virus-infected mice showed TLR3-mediated enhancement of inflammatory reaction and CD8+ T cell response, associated with augmented viral clearance, compared to TLR3−/− mice (le Goffic et al. 2006) (Table 2). However, TLR3−/− mice survived longer than control mice (le Goffic et al. 2006), suggesting that TLR3-triggered innate response inhibits viral spread, but the ensuing adaptive immunity is detrimental to the host.
5.2.2 TLR7/8
In humans, the highest levels of TLR7 is expressed in pDCs (Kadowaki et al. 2001), whereas mDC1, mDC2, LCs, and NK cells lack TLR7 (Kadowaki et al. 2001; Merad et al. 2013). However, TLR8 is expressed at low levels by moMΦ (Hui et al. 2011) and mDCs, but not pDCs (Kadowaki et al. 2001). In mice, CD8α+ cDCs lack TLR7, but express TLR3, whereas CD8α- cDCs, pDCs, and BMDCs (Edwards et al. 2003), and also BMMΦ (Kawai and Akira 2011) express TLR7 (Table 1).
In response to live or inactivated influenza virus in vitro, pDCs produced high levels of IFN-α, and also inflammatory cytokines in a TLR7-and MyD88-dependent manner through recognition of viral ssRNA (Diebold et al. 2004; Koyama et al. 2007; Lund et al. 2004) (Fig. 1). TLR7-mediated sensing of inactivated-influenza virus by pDCs is required to confer protective primary adaptive immune response in mice (Geeraedts et al. 2008; Koyama et al. 2007, 2010). In contrast, cytokine production in the lungs following live influenza virus infection did not require TLR7-signaling in pDCs (Koyama et al. 2010). Despite the uncontrolled viral load and mortality observed in TLR7/MyD88-deficit mice infected with lethal dose of influenza virus in a previous study (Seo et al. 2010), the lack of TLR7 did not markedly alter the viral load, disease pathology and inflammatory cytokine response in following lethal dose (Jeisy-Scott et al. 2011) or sublethal dose (Pang et al. 2013b). Consistent with this finding, TLR7-induced type I IFN in pDCs was dispensable for induction of protective response to influenza virus in mice vaccinated with live-virus vaccine (Koyama et al. 2010). Furthermore, IFN-α secretion by murine mDC/BMDCs in response to influenza virus is found to be dependent on live virus replication, but not on TLR7/MyD88- signaling (Barchet et al. 2005; Koyama et al. 2007). Nevertheless, in murine BMDCs, TLR7-signaling was required for the induction of pro-IL-1β and secretion of mature IL-1β after influenza virus infection (Ichinohe et al. 2010). These results suggest a cell-specific role of TLR7 in the induction of innate immune response to influenza virus infection as well as vaccination.
5.2.3 TLR4
TLR4 is expressed mainly on myeloid cells including, neutrophils, monocytes, mDCs, moDCs, and MΦ (Table 2). Although the ligand for TLR4 in influenza virus is not known, a DAMP molecule, S100A9 released in influenza virus-infected lungs was found to trigger TLR4-MyD88-signaling pathway in MΦ to induce exaggerating proinflammatory response, cell-death, and virus pathogenesis following lethal infection (Tsai et al. 2014) (Fig. 1). Furthermore, similar to TLR4 deficient mice (Nhu et al. 2010), treatment of mice with a TLR4 antagonist, Eritoran, was found to protect from lethal influenza infection by alleviating lung pathology, clinical symptoms, cytokine, and oxidized phospholipid expression, as well as by controlling viral loads, a process dependent on CD14 and TLR2 expression (Shirey et al. 2013). Activation of TLR4-signaling during influenza infection seems to induce an exaggerated inflammatory response.
5.3 RIG-I-Like Receptors (RLRs)
These are the RNA-sensing PRRs expressed in the cytosol of majority of the mammalian cells. RLRs represent a family of RNA helicases, which includes three members, RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology-2 (LGP-2) (Takeuchi and Akira 2009). In addition to the RNA helicase domain, RIG-I and MDA5 contain two N-terminal caspase recruitment domains (CARDs), whereas LGP2 lacks a CARD component, and functions as a negative regulator of RIG-I/MDA5-signaling (Loo and Gale 2011). RIG-I recognizes RNA containing 5’-triphosphate with panhandle-like secondary structures, whereas MDA5 preferentially senses long dsRNA (>2 kb) (Takeuchi and Akira 2009). RLRs signal through a common adaptor IFN-β promoter stimulator-1 (IPS-1), also known as mitochondrial antiviral signaling (MAVS) leading to phosphorylation of IRF7/3 and NF-kB , which in turn induce type I IFNs, and proinflammatory cytokines and chemokines, respectively (Loo and Gale 2011). The recruitment of the adaptor MAVS is dependent on E3-ligase tripartite motif containing 25 (TRIM25)- and Riplet-dependent ubiquitination of RIG-I (Loo and Gale 2011). Under steady state, RLR is expressed at low levels, but is greatly increased in response to IFN and after virus infection (Loo and Gale 2011) (Fig. 1).
In influenza virus-infected cells, RIG-I is the RLR that senses the virus in cytosol by recognizing the 5’-triphosphate -RNA sequence motifs along RNA containing some dsRNA part (panhandle), which is generated by active viral replication within the cells (Kato et al. 2006; Pichlmair et al. 2006; Rehwinkel et al. 2010). Of note, a recent study revealed that influenza virus lacking NS1 induces antiviral stress granules that contain viral RNA together with RIG-I and antiviral proteins including protein kinase R (PKR). These antiviral stress granules are shown to serve as the site of 5’ppp RNA-induced activation of RIG-I-signaling (Onomoto et al. 2012). Consistent with this, influenza virus infection boosted an early transient expression and activation of RIG-I in respiratory epithelial cells (Crotta et al. 2013; le Goffic et al. 2007), MΦ (Ohman et al. 2009; Wang et al. 2012), BMDCs (Koyama et al. 2007) and mast cells (Graham et al. 2013). Although dispensable for viral control following sublethal dose, signaling from RIG-I and TLR7 was required for survival and restricting the virus growth after lethal dose influenza infection in mice (Koyama et al. 2007; Pang et al. 2013b). Furthermore, expression on RIG-I and MDA5 were enhanced in peripheral blood cells (Lee et al. 2013) and patients who exhibit a polymorphism resulting in the expression of a nonfunctional variant of RIG-I were severely attenuated in antiviral responses against influenza virus (Pothlichet et al. 2009). These findings support an important role for the RIG-I-mediated responses in restraining the influenza virus.
5.4 NOD-Like Receptors (NLRs)
The NLR family comprises more than 20 receptors that are expressed intracellularly in the cytosol, and respond to various PAMPs to trigger inflammatory response. NLRs can trigger several signaling pathways including MAPK, NF-kB, and MAVS-IRF3 to induce production of IL-6, TNF-α, pro- IL-1β/IL-18, and also type I IFNs, respectively (Kanneganti 2010) (Fig. 1). Importantly, some of the NLRs can recruit the adaptor ASC (apoptosis-associated speck-like protein containing a CARD), which in turn interact with the pro-caspase-1 to form the "inflammasome " (Franchi et al. 2009). Assembly of inflammasome leads to autoactivation of caspase-1, which cleaves pro-IL-1β/IL-18 to mature IL-1β/IL-18 for secretion (Yu and Finlay 2008) (Fig. 1).
Studies have shown that NLRP3 (NLR family PYD-containing protein 3/Cryopyrin), NLRC2 (NLR family CARD-containing protein 2), and NLRX1 are the NLRs responding to influenza virus (Kanneganti 2010). Whereas NLRP3 inflammasome induces the secretion of mature IL-1β/IL-18, NLRC2/NOD2, and NLRX1 signals the production of type I IFNs in response to influenza virus.
Influenza virus-induced NLRP3-mediated IL-1β and IL-18 production involve two steps that include, enhancing the transcription of genes encoding pro-IL- 1β and pro-IL-18 and NLRP3 (signal 1) and activating NLRP3 inflammasome (signal 2). Signal 1 is initiated by the detection of viral RNA by TLR7, which activates NF-kB. Although initial studies showed that NLRP3 detects influenza virus via recognition of viral RNA (Allen et al. 2009; Thomas et al. 2009), it is now clear that many sources contribute to signal 2 either in combination or alone, and all depend on the newly synthesized viral M2 protein. These include, (i) ionic imbalance of the trans-Golgi pH, (ii) potassium efflux through the P2X7 receptor, an ATP-gated cation channel, (iii) lysosomal maturation and release of cathepsin B and (iv) cellular reactive oxygen species (Ichinohe et al. 2010; Lietzen et al. 2011) (Fig. 1). Recently, influenza virus virulence protein polymerase basic protein 1-frame 2 (PB1-F2) alone in aggregated form was found to be sufficient to activate NLRP3 inflammasome-induced IL-1β (McAuley et al. 2013). In addition, recent evidences support the role of commensal microbiota derived PAMPs as signal 1 for NLRP3 inflammasome activation (Ichinohe et al. 2011). Despite these findings, the specific PAMP in influenza virus interacting with NLRP3 is not yet known. Apart from the cleavage of pro-IL-1β and pro-IL-18, NLRP3-inflammasome activation also results in the initiation of a proinflammatory form of cell death known as pyroptosis (Schroder and Tschopp 2010).
Studies have shown that live influenza virus infection induces the expression and activation of NLRP3 inflammasome components (NLRP3, ASC, and Caspase-1) to mediate IL-1β and IL-18 production in different cell types in vitro; such as mice BMDCs and BMMΦ, human MΦ, nasal airway epithelial cell line and monocytic cell line THP-1 (Allen et al. 2009; Ichinohe et al. 2009; Kanneganti et al. 2006; Thomas et al. 2009). Accordingly, mice lacking any of the NLRP3 inflammasome components did not produce IL-1β and IL-18 following the high-lethal dose influenza virus infection (Allen et al. 2009; Thomas et al. 2009). Furthermore, NLRP3-deficient mice showed a reduced protective inflammation including the suppressed accumulation of neutrophils and monocytes to the lungs and airways upon influenza virus infection, which resulted in higher mortality (Allen et al. 2009; Thomas et al. 2009). Strikingly, NLRP3-deficient mice showed collagen deposits in lungs suggesting the delayed resolution of lung injury due to the absence of pro-fibrotic role of IL-1β (Thomas et al. 2009). Notably, the inflammasome complex was found to be dispensable for early clearance (up to 6 days) of the virus (Thomas et al. 2009), but was essential for reducing the viral load in later stage of infection (Allen et al. 2009; Ichinohe et al. 2009). These findings suggested that NLRP3-inflammasome induced inflammatory response, rather than direct viral control, mediates the protective immunity to influenza virus infection, possibly via adaptive immune responses.
NLRC2 (or NOD2) is believed to recognize the viral genomic ssRNA to recruit MAVS adaptor protein to activate IRF3-mediated type I IFN production in DCs and MΦ in response to influenza virus (Fig. 1). In agreement, NOD2-deficient mice showed decreased type I IFNs and DC activation, and exhibited enhanced susceptibility to lethal dose virus-induced pathogenesis (Lupfer et al. 2014; Sabbah et al. 2009). Further, NLRX1, NLR located in mitochondria, binds to viral protein PB1-F2. NLRX-signaling prevents virus-induced MΦ apoptosis and promotes both MΦ survival as well as type I IFN signaling in mice infection with lethal dose (Jaworska et al. 2014). On contrary, NOD2 and NLRX1 are found to negatively regulate the NLRP3 and RIG-induced inflammatory response to lethal dose influenza virus, respectively, and control the immunopathology (Allen et al. 2011; Lupfer et al. 2013). Together, these findings suggest that NLRs execute differential role in response to influenza virus infection to obtain a balanced innate immunity.
6 Effector Molecules of Innate Immunity
6.1 Cytokines and Chemokines
These mainly activate and attract various immune cells to the site of infection. Type I IFNs are the principal antiviral effectors to inhibit viral replication, and also promote greater activation of innate immune cells, particularly DCs, to facilitate the induction of adaptive immunity.
6.2 Soluble Innate Mediators
These function mainly by direct interaction with the virus outside the cells, resulting in either inhibition of viral binding to target cells, or in disruption of viral membranes (reviewed in Tripathi et al. 2013). Some of these soluble innate mediators, such as mucins, surfactant protein A (SP-A), glycoprotein-340, pentraxins, ficolins- inhibit the attachment of influenza virus to cells by presenting SA to the viral HA. Whereas other mediators, such as SP-D, mannose binding lectin (MBL), H-ficolins possess a lectin activity, and can interact with glycans on viral HA to form aggregate, which prevents the binding of virus to cells. In addition, there are antimicrobial peptides, such as α-defensins (human neutrophil peptides) and β-defensins, which cause viral aggregates, and also LL-37 that causes disruption of viral membrane. Furthermore, complement proteins activated by either soluble innate mediator, MBL, or natural IgM are also shown to exert beneficial role in influenza virus infection (Tripathi et al. 2013).
6.3 Intrinsic Antiviral Factors
These factors directly interact with the virus inside host cells to restrict the entry, replication, and assembly of virus, thereby rendering the cells nonpermissive to virus. This form of immunity to virus is referred as intrinsic antiviral immunity (Yan and Chen 2012). These factors are preexistent, but can be further enhanced by viral infection and type I IFNs, the principal mediator of antiviral innate response.
Type I IFNs activate the JAK/STAT pathway upon binding to its receptor, IFNAR. In addition to upregulating the innate recognition receptors (discussed earlier), IFNAR-signaling results in transcriptional upregulation of interferon-stimulated genes (ISGs) , which in turn restrict the viral replication.
6.3.1 Mx
The myxovirus resistance gene, or Mx, was the first ISG found to restrict influenza virus replication. The human MxA and MxB, and the murine Mx2 are cytoplasmic proteins, whereas the murine Mx1 is localized within the nucleus (Haller et al. 2009). In mice, Mx1 inhibits the influenza virus infection, but Mx2 does not. In humans, MxA inhibits influenza as well as other viruses, but MxB has no effect on influenza infection. Mx proteins are reported to interact with viral NP and RNA helicases involved in the transport of viral RNA to the nucleus, which is the site of viral transcription and replication, resulting in the inhibition of viral growth (von der Malsburg et al. 2011). Different strains of influenza virus vary in their sensitivity to these proteins (Zimmermann et al. 2011). Of note, since most of the inbred mice strains are devoid of functional Mx proteins, the extrapolation of mouse data to humans has to be done with extreme caution.
6.3.2 Protein Kinase R (PKR)
It is an IFN inducible protein kinase that becomes activated upon binding to dsRNA in cytosol. In case of influenza virus, this is shown to be mediated by the panhandle secondary structure formed by 5’ppp end of RNA (Dauber et al. 2009). While there have been multiple substrates identified for PKR, most of the antiviral activity of PKR is due to phosphorylation of eIF2α, which results in a general translational block, limiting viral replication (Pindel and Sadler 2011). NS1 of influenza A virus inhibits the activity of PKR (Li et al. 2006), and PKR-KO mice are highly susceptible to infection with NS1 defective influenza A virus (Bergmann et al. 2000), highlighting the contribution of PKR in restricting influenza virus replication. In addition to its role in inhibiting the translation of viral RNA, PKR activation also initiates signal transduction via NF-kB leading to cell growth arrest and autophagy, which result in an enhanced anti-viral immunity (Sadler and Williams 2008).
6.3.3 OAS/RNAseL
OAS (oligoadenylate synthetase) and RNase L is one of the first interferon-induced antiviral pathways discovered. Similar to PKR, OAS requires binding to dsRNA for activation of its enzymatic activity in cytosol. Upon activation, OAS generates 2’–5’ oligoadenylates that act as a cofactor for a latent cytoplasmic RNAse, RNAseL. Activated RNAseL cleaves viral and cellular RNA stopping the viral replication (Chakrabarti et al. 2011).
6.3.4 ISG15
ISG15 is a 17kDa protein present in cytosol that has structural resemblance to two covalently linked ubiquitins. Like ubiquitin, ISG15 is conjugated to proteins through lysine residues. While the outcome of ISGylation are still unclear, ISG15-KO mice are more prone to infection by several viruses, including influenza A and B viruses, supporting the antiviral activity of this molecule (Lenschow et al. 2007). Importantly, NS1 protein of influenza A virus was shown to be conjugated by ISG15 resulting in the inhibition of NS1 function, and is believed to restrict the influenza virus replication (reviewed in Garcia-Sastre 2011).
6.3.5 Viperin and Tetherin
These are the recently discovered ISGs expressed on cell surface that exhibit the ability to inhibit influenza virus infection. Viperin is localized in the endoplasmic reticulum. It interferes with the enzymatic process of membrane fluidity and membrane microdomains to inhibit the efficient budding of influenza virus from infected cells (Wang et al. 2007). Tetherin (also known as BST2), like viperin, also restricts viral budding. By retaining newly assembled virions attached to the plasma membrane, tetherin restricts the formation of influenza virus like particles (reviewed in Garcia-Sastre 2011).
6.3.6 IFITM and IFIT
IFITM (IFN-inducible transmembrane protein) are ISGs that restrict viral entry (Brass et al. 2009; Everitt et al. 2012). IFITM3 has been identified as an important host restriction factor for influenza virus. IFITM3 proteins block infection early during cytosolic entry of viruses that utilize the endosomal pathway (Feeley et al. 2011), suggesting that they affect the function of viral proteins involved in viral fusion in the endosome (Garcia-Sastre 2011). Infection of IFITM3 deficient mice with low virulence influenza A virus resulted in a severe form of disease, similar to that caused by high virulence virus (Brass et al. 2009; Everitt et al. 2012). Interestingly, avian cells do not seem to express a homolog to IFITM3, which raises the possibility that IFITM proteins might influence viral tropism (reviewed in Yan and Chen 2012).
The IFIT family (interferon-induced proteins with tetratricopeptide repeats) includes four members, IFIT1, 2, 3, and 5, which are the cytoplasmic proteins that recognize viral RNA with 5’triphosphate or without 2’-O-methylation. IFIT1 is found to inhibit cellular translation by binding to the eIF3 initiation factor to suppress the viral translation and replication (Yan and Chen 2012).
7 Innate Control of Adaptive Immunity to Influenza
7.1 Innate Immune Cells
At the cellular level, innate immune cells, particularly DCs, which sense the viral invasion through unique innate receptors, are also endowed with the ability to prime adaptive immune cells, such as T cells and B cells, to induce a virus-specific long-lasting immunity (Braciale et al. 2012; Iwasaki and Medzhitov 2010; Manicassamy and Pulendran 2009, 2011; Pulendran et al. 2010b).
7.1.1 DCs
DCs are well known for their critical role in the initiation of Ag-specific response owing to their ability to uptake and process Ags, and to migrate to the lymphoid tissues for presentation to naïve Ag-specific T cells. Furthermore, DCs in airways are believed to derive viral Ag for presentation via two unique ways, endogenous Ag following direct infection or exogenous Ag by uptake of infected dead cells.
Extensive studies have shown that respiratory DC subsets, such as CD103+CD11blocDCs and CD11bhiCD103− cDCs acquire a mature phenotype in the presence of type I IFNs and migrate to the regional LN in CCR7-dependent manner (Fig. 2). Among the DCs in LN, respiratory CD103+CD11blo cDCs are found to be the only DCs that prime virus-specific naïve CD8+ T cells in the LN to differentiate into effector cells (GeurtsvanKessel et al. 2008; Helft et al. 2012; Ho et al. 2011; Kim and Braciale 2009; Kim et al. 2014; Unkel et al. 2012). This is because, type I IFN-dependent antiviral status of CD103+CD11blo cDCs restrains their productive infection, and they preferentially uptake influenza virus-infected apoptotic cells in the lungs (Desch et al. 2011; Helft et al. 2012). This results in an influx of high number of activated CD103+CD11blocDCs that carry viral Ags into LN for cross-priming of virus-specific CD8+ T cells , early during the infection (Albert et al. 1998; Desch et al. 2011; Helft et al. 2012; Ho et al. 2011) (Fig. 2).

Innate control of adaptive immunity to influenza. Innate immune cells, particularly dendritic cells (DCs) in the respiratory tissues acquire antigens either through direct infection or by uptake of influenza-infected dead cells and undergo maturation process triggered by TLR7 or RIG-I-signaling, under the influence of type I IFNs produced by macrophages and pDCs. Respiratory DC subsets (CD103+ cDCs, CD11b+ cDCs and pDCs) migrate to the draining lymph node (LN), where they can transfer influenza antigens (Ag) to LN-resident CD8α+ cDC. In the LN, respiratory CD103+ cDCs together with CD8α+ cDCs stimulate the naïve CD8+ T cells to proliferate and differentiate into cytotoxic effector CD8+ T cells, in a CD24-dependent manner. On the other hand, CD11b+ cDCs drive the activation of CD8+ T cells, mainly effector T cells at later stage of infection, to induce memory CD8+ T cells. Interaction of naïve CD4+ T cells with cDCs generates IFN-γ-producing Th1 cells, which in turn facilitates the differentiation of effector B cells in a TLR7-dependent manner. These effector cells migrate from LN to respiratory tissues, where they have second interaction with Ag-bearing innate immune cells to undergo further activation and differentiation to terminal effector cells that secrete effector molecules to control virus spread
On the contrary, CD11bhiCD103− cDCs, which is the major migratory DC subset in LN at the peak of infection, are found to drive the generation of central memory CD8+T cells (Kim et al. 2014) (Fig. 2). The differential function of cDC subsets is attributed to an enhanced expression of CD24 on CD103+CD11blocDCs, which regulate CD8+ T cell activation through HMGB1-mediated engagement of T cell RAGE (Kim et al. 2014). Although CD11clo pDCs and CD11b+Ly6C+ moDCs migrate to the LN in high number, and are shown to carry viral Ags, they were inefficient in activating naïve CD8+T cells, compared to CD103+ cDCs and CD11bhi cDCs (Ballesteros-Tato et al. 2010; GeurtsvanKessel et al. 2008; Kim and Braciale 2009).
In addition to migratory respiratory DCs, some studies suggested that LN-resident CD8α+ cDCs are also able to activate naïve CD8+ T cells by cross-presentation of Ag acquired from migratory DCs (Belz et al. 2007; Waithman et al. 2013). Notably, together with neutrophils and MΦ (Hufford et al. 2012; Kohlmeier et al. 2010; Tate et al. 2012), respiratory DCs at the site of infection were essential for the survival of effector T cells in Ag (cross-) presentation, IL-15 trans-presentation- and lymphotoxin (LT) β-dependent manner (McGill et al. 2008, 2010) (Fig. 2). Thus, the non-redundant role of DC subsets facilitates rapid generation and maintenance of the effector T cells needed to clear acute infection, followed by slower development of the cells needed for sustained memory.
Furthermore, both CD103+CD11blo cDCs and CD11bhiCD103− cDCs are found to efficiently activate naïve virus-specific CD4+ T cells in the LN (Fig. 2), compared to other migratory DC subsets, in mice exposed to infectious as well as inactivated influenza virus (GeurtsvanKessel et al. 2008; Kim and Braciale 2009). However, blood monocyte-derived moDC recruited to LN are reported to stimulate IL-12p70-mediated Th1 response in mice (Nakano et al. 2009). Recent studies in influenza-infected mice (Leon et al. 2014) and influenza-vaccinated humans (Bentebibel et al. 2013) showed that follicular CD4+ T (Tfh) cells are present in LN and circulation, respectively, and are essential for germinal center (GC) reactions and antibody production. Notably, pDCs were found to be essential for enhancement of virus-specific primary antibody response following influenza infection (GeurtsvanKessel et al. 2008; McGill et al. 2008) and vaccination (Koyama et al. 2010). Further, CD11chi cDCs in the lungs were crucial for maintenance of GC reactions in tertiary lymphoid structures, and to sustain virus-specific antibodies (GeurtsvanKessel et al. 2009). Despite these observations, the role of DC subsets in the generation of Tfh cells and primary antibody response is not clearly known.
In humans, consistent with the high susceptibility to productive infection in vitro, influenza virus-exposed mDCs were impaired in Ag (cross-) presentation to CD8+ T cells (Smed-Sorensen et al. 2012) and CD4+ T cells (Fernandez-Sesma et al. 2006). However, mDCs exposed to inactivated virus or infected dead cells were highly efficient in Ag (cross-) presentation to activate CD8+ T cells (Smed-Sorensen et al. 2012). Furthermore, when LAIV was administered to humanized mice, the lung-resident CD1c+ mDC1, but not CD141+ mDC2, are found to drive the expansion of influenza virus-specific CD103-expressing mucosal CD8+ T cells through membrane-bound TGF-β-dependent mechanisms (Yu et al. 2013).
Recent studies showed that pDCs are less efficient than mDCs, in both cross- as well as direct presentation of influenza Ag to T cells in vitro (Lui et al. 2009; Smed-Sorensen et al. 2012). Strikingly, influenza virus-activated pDCs were able to induce a strong Th1 polarization through synergistic effect of IL-12 and type I IFNs (Cella et al. 2000). Furthermore, influenza virus triggered secretion of type I IFNs and IL-6 from pDCs induced differentiation of plasma cells and virus-specific antibody production from B cells activated by T cells (Jego et al. 2003). These findings indicate that human mDCs are likely important for the induction of CD8+ T cell response and that of pDCs in CD4+ T cell-dependent virus-specific antibody response.
Despite these studies in mouse models and in vitro human studies on how influenza virus-exposed mDCs and pDCs influence different aspects of adaptive immunity; the phenotype, activation status and functional role of DC subsets in influenza virus-infected patients, particularly at the site of infection, is not known.
7.2 Virus Sensing Receptors
7.2.1 TLRs and RLRs
Although a previous study implicated TLR3 in the enhancement of CD8+ T cell response in lethal dose influenza virus-infected mice (le Goffic et al. 2006), later studies found that TLR3 and its associated adapter molecule, TRIF, do not play a significant role in the development of influenza virus-specific CD4+ or CD8+ T cell or B cell responses following sublethal infection (Heer et al. 2007; Koyama et al. 2007; Seo et al. 2010) (Table 2). Surprisingly, TLR7/MyD88 and RIG-I-signaling are also found to play negligible roles in CD8+ T cell activation and effector functions in sublethal dose influenza virus-infected mice (Heer et al. 2007; Koyama et al. 2007; Pang et al. 2013a). However, CD4+ T cell response, the number of antibody-secreting cells in secondary lymphoid organs, and the production of virus-specific antibodies following sublethal intranasal infection were dependent on TLR7 signaling, but not RIG-I signaling (Jeisy-Scott et al. 2012; Koyama et al. 2007). Furthermore, TLR7-signaling in pDCs, was essential for protective antibody response induced by virion RNA-containing split vaccine (Jeisy-Scott et al. 2012) and inactivated whole virus vaccine (Koyama et al. 2007, 2010) (Table 2). Here, along with stimulation of TLRs on B cells, TLR7-mediated induction of type I IFNs in pDCs was found to be critical for T cell-dependent antibody response following infection and vaccination (Heer et al. 2007; Koyama et al. 2010). Based on these findings, it appears that TLRs rather than RLRs contribute to the induction of effective T cell-dependent antibody response to influenza virus, whereas TLR-independent and RLR-independent mechanisms might exist with regards to CD8+ T cell responses. In this context, our recent study showed that activation of induced general control nonderepressible 2 kinase (GCN2) in DCs by the yellow fever vaccine (YF-17D) is crucial for generation of CD8+ T cell response through autophagy and enhanced Ag presentation (Ravindran et al. 2014). Interestingly, induction of CD8+ T cell responses to the LAIV was also dependent on GCN2 (Ravindran et al. 2014).
7.2.2 NLRs and Caspase-1
Mice deficient of caspase-1, ASC, and NLRP3, which lacked IL-1β and that of IL-1R-deficient mice showed a failure in the activation of virus-specific IFN-γ-secreting CD4+ and CD8+ T cells, and to generate nasal IgA and serum IgG response following sublethal dose of influenza virus infection (Ichinohe et al. 2009; Pang et al. 2013a) (Table 2). Furthermore, microbiota-mediated NLRP3-inflammasome-caspase-1 activation-induced IL-1β is believed to be essential for activation and migration of DCs from the lungs to the LN for T cell priming during sublethal dose influenza virus infection in mice (Ichinohe et al. 2011). Supporting these previous findings, a recent study showed that signaling through the IL-1R (by IL-1β/IL-1α) in uninfected DCs carrying viral Ag was required and sufficient for productive priming of CD8+ T cells, but signaling through TLR7 and RIG-I was dispensable (Pang et al. 2013a). In contrast, a previous study found that antibody production, as well as the influenza virus-specific CD8+ T cell number in the BAL, of both Nlrp3 and caspase-1-deficient mice was similar to wild type mice infected with sublethal dose of influenza virus between 7 and 11 days of infection (Thomas et al. 2009) (Table 2). While several studies favored the role of influenza virus-triggered caspase-1 activation in CD8+ T cell response, further studies are needed to clarify these findings. A recent study revealed that NOD2-deficiency causes reduced generation of virus-specific CD8+ T cell response following lethal infection in mice (Lupfer et al. 2014) (Table 2).
In summary, despite a unique indispensable role of innate immune system in antiviral immunity, cooperation with CD8+ T cell and CD4+ T cell-dependent virus-neutralizing antibody response, is essential for protection, especially heterosubtypic influenza virus immunity.
8 Pathogenic Role of Innate Immunity to Influenza Virus Infection
A major form of innate immune-mediated pathology following influenza virus infection is viral pneumonia, which leads to ARDS resulting in multiorgan failure and a high mortality rate (Short et al. 2014). In addition to virus-induced cell injury, ARDS is attributed to hyperactivation of innate immune cells, such as neutrophils, monocytes, and NK cells. These cells induce excessive inflammatory responses involving reactive oxygen species, TNF-related apoptosis-inducing ligand (TRAIL), inducible nitric oxide synthase (iNOS2) and proinflammatory cytokines (Herold et al. 2008; Hogner et al. 2013; Lin et al. 2008; Short et al. 2014).
Increased accumulation of innate cells through a chemokine-mediated feed-forward loop is observed in the lung lesions of high dose lethal infection associated with poor innate control of influenza virus (Aldridge et al. 2009; Brandes et al. 2013; Lin et al. 2008; Narasaraju et al. 2011; Seo et al. 2011). Consistent with this, interfering with the massive infiltration of these innate cells through chemokine antagonism (CCR2 for monocytes) or partial cell depletion (Aldridge et al. 2009; Brandes et al. 2013; Lin et al. 2011) was found to alleviate the immune-mediated pathology.
9 Systems Vaccinology of Influenza Vaccines
System vaccinology employs a number of high-throughput technologies like DNA microarrays, protein arrays, deep sequencing, and mass spectrometry to generate system-wide unbiased molecular measurements to reconstruct the events in an immune response (Pulendran et al. 2010a). Over the last few years, several studies have used systems biology approaches to obtain a global picture of the immune responses to vaccination, and to identify molecular signatures that can be used to predict vaccine immunity in humans, and also to understand the mechanisms involved in the vaccine-induced immunity (Li et al. 2013, 2014; Nakaya et al. 2011, 2012; Pulendran 2009; Pulendran et al. 2010a; Querec et al. 2009; Ravindran et al. 2014; Tsang et al. 2014).
A comparative study of immune response to trivalent inactivated influenza vaccine (TIV) and live attenuated influenza vaccine (LAIV) using systems biology approach revealed salient common as well as contrasting features between them. While LAIV induced the expression of several interferon-related genes, which are similar to live viral vaccines, the TIV induced a signature composed of genes highly expressed in plasma B cells (Nakaya et al. 2011). For TIV, of the 44 genes identified to accurately predict the outcome of immunization as either high or low antibody titers, one gene-CAMKIV- had no known function in regulating immunity, but was negatively correlated with antibody titers. Consistent with this, CAMKIV-deficit mice developed high antibody titers after vaccination (Nakaya et al. 2011). Furthermore, the expression of TLR5 few days after TIV administration highly correlated with the antibody titers 4 weeks post vaccination. However, TIV did not activate TLR5 signaling per se (Jason and Pulendran unpublished data). TLR5 is sensor of bacterial flagellin, which could be derived from commensal flora. In this context, perturbation of gut microbiota was found to influence the host immune response involved in the clearance of virus from lungs in influenza infected mice (Ichinohe et al. 2011). Consistent with this, our preliminary results support the significant influence of intestinal flora on TIV-induced antibody response in mouse model (Jason and Pulendran unpublished data).
In addition, studies are also being performed in influenza virus-infected patients and animal models for better understanding of the disease pathogenesis that might help in efficient control (Huang et al. 2011; Korth et al. 2013; Woods et al. 2013; Zaslavsky et al. 2013). A temporal pattern of host molecular responses was identified by systems biology approach in the peripheral blood of influenza virus-infected human volunteers, which differentiated symptomatic from asymptomatic infections of influenza virus strains (Huang et al. 2011; Woods et al. 2013). Further, while symptomatic patients showed multiple PRR-mediated antiviral and inflammatory responses, asymptomatic individuals revealed a highly regulated antiviral responses together with enhanced cell-mediated and antioxidant responses (Huang et al. 2011; Woods et al. 2013). Similarly, differential induction of inflammatory gene expression is observed in the mouse lungs following infection with influenza virus causing mild or severe respiratory disease, which is largely accounted by neutrophils (Brandes et al. 2013; Kash et al. 2006; Korth et al. 2013). Furthermore, by integrating the large-scale lipid measurements with targeted gene expression, a recent study in influenza mouse model and human patients showed that 5-lipoxygenase metabolites correlate with the pathogenic phase of the infection, whereas 12/15-lipoxygenase metabolites associate with the resolution phase (Tam et al. 2013).
10 Conclusions and Perspectives
In this review, we summarized our current knowledge of the innate immune responses to influenza. At the cellular level, respiratory epithelial cells being the primary target support the viral replication to release progeny viruses; but together with DCs and MΦ, trigger trafficking of increased number of immune cells to the site of infection. Uptake of influenza virus mainly through the SA-containing receptors and recognition of viral components by the innate receptors trigger activation and upregulation of antiviral program in respiratory epithelial as well as immune cells recruited to the airways. Antiviral innate response mainly includes type I and III IFNs and the IFN-stimulated genes, in addition to various inflammatory cytokines and chemokines. Although innate immunity seems to achieve the protective viral clearance in concert with adaptive immune response, damaging role of innate immunity in the pathogenesis of influenza is also emerging with the recent studies. Of note, variation in the amplitude of innate immune response has been linked to both virus dose and strains as well as host factors.
There is an impressive amount of knowledge emerging on innate immunity to influenza virus from experimental studies in mice. However, there is a paucity of knowledge about the mechanisms that mediate innate and adaptive immunity to influenza in humans, particularly among those populations that show enhanced morbidity and mortality to infection, such as the infants and the elderly. Thus, there is an imperative to study innate immunity in humans using cutting edge tools like systems biology, in order to acquire a deeper understanding for devising rational prophylactic and therapeutic strategies for the control of influenza.
References
Albert ML, Sauter B, Bhardwaj N (1998) Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 392:86–89. doi:10.1038/32183
Aldridge JR Jr, Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, Brown SA, Doherty PC, Webster RG, Thomas PG (2009) TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Nat Acad Sci U.S.A 106:5306–5311. doi:10.1073/pnas.0900655106
Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, Ranjan P, Monroe KM, Pickles RJ, Sambhara S, Ting JP (2011) NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity 34:854–865. doi:10.1016/j.immuni.2011.03.026 S1074-7613(11)00226-3 [PII]
Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30:556–565. doi:10.1016/j.immuni.2009.02.005
Ballesteros-Tato A, Leon B, Lund FE, Randall TD (2010) Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat Immunol 11:216–224. doi:10.1038/ni.1838
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392:245–252. doi:10.1038/32588
Barchet W, Krug A, Cella M, Newby C, Fischer JA, Dzionek A, Pekosz A, Colonna M (2005) Dendritic cells respond to influenza virus through TLR7- and PKR-independent pathways. Eur J Immunol 35:236–242. doi:10.1002/eji.200425583
Belz GT, Bedoui S, Kupresanin F, Carbone FR, Heath WR (2007) Minimal activation of memory CD8+ T cell by tissue-derived dendritic cells favors the stimulation of naive CD8+ T cells. Nat Immunol 8:1060–1066. doi:10.1038/ni1505
Bendelac A, Savage PB, Teyton L (2007) The biology of NKT cells. Ann Rev Immunol 25:297–336. doi:10.1146/annurev.immunol.25.022106.141711
Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, Flano E, Mejias A, Albrecht RA, Blankenship D, Xu H, Pascual V, Banchereau J, Garcia-Sastre A, Palucka AK, Ramilo O, Ueno H (2013) Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med 5:176ra32. doi: 10.1126/scitranslmed.3005191 5/176/176ra32 [PII]
Bergmann M, Garcia-Sastre A, Carnero E, Pehamberger H, Wolff K, Palese P, Muster T (2000) Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol 74:6203–6206
Boonnak K, Paskel M, Matsuoka Y, Vogel L, Subbarao K (2012) Evaluation of replication, immunogenicity and protective efficacy of a live attenuated cold-adapted pandemic H1N1 influenza virus vaccine in non-human primates. Vaccine 30:5603–5610. doi:10.1016/j.vaccine.2012.06.088
Boonnak K, Vogel L, Orandle M, Zimmerman D, Talor E, Subbarao K (2013) Antigen-activated dendritic cells ameliorate influenza A infections. J Clin Invest 123:2850–2861. doi:10.1172/JCI67550
Braciale TJ, Sun J, Kim TS (2012) Regulating the adaptive immune response to respiratory virus infection. Nat Rev Immunol 12:295–305. doi:10.1038/nri3166
Brandes M, Klauschen F, Kuchen S, Germain RN (2013) A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154:197–212. doi:10.1016/j.cell.2013.06.013
Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ (2009) The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254. doi:10.1016/j.cell.2009.12.017
Cao W, Taylor AK, Biber RE, Davis WG, Kim JH, Reber AJ, Chirkova T, de la Cruz JA, Pandey A, Ranjan P, Katz JM, Gangappa S, Sambhara S (2012) Rapid differentiation of monocytes into type I IFN-producing myeloid dendritic cells as an antiviral strategy against influenza virus infection. J Immunol 189:2257–2265. doi:10.4049/jimmunol.1200168
Castilla J, Martinez-Baz I, Martinez-Artola V, Reina G, Pozo F, Garcia Cenoz M, Guevara M, Moran J, Irisarri F, Arriazu M, Albeniz E, Ezpeleta C, Barricarte A (2013) Decline in influenza vaccine effectiveness with time after vaccination, Navarre, Spain, season 2011/12. Euro Surveill 18:1–8. http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20388
Cella M, Facchetti F, Lanzavecchia A, Colonna M (2000) Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol 1:305–310. doi:10.1038/79747
Centers for Disease Control and Prevention US (2011) Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2011. MMWR, vol 60, US Centers for Disease Control and Prevention, pp 1128–1132
Chakrabarti A, Jha BK, Silverman RH (2011) New insights into the role of RNase L in innate immunity. J Interferon Cytokine Res 31:49–57. doi:10.1089/jir.2010.0120
Chan MC, Chan RW, Yu WC, Ho CC, Chui WH, Lo CK, Yuen KM, Guan YI, Nicholls JM, Peiris JS (2009) Influenza H5N1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Respir Res 10:102. doi:10.1186/1465-9921-10-102
Chan MC, Cheung CY, Chui WH, Tsao SW, Nicholls JM, Chan YO, Chan RW, Long HT, Poon LL, Guan Y, Peiris JS (2005) Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir Res 6:135. doi:10.1186/1465-9921-6-135
Chan RW, Yuen KM, Yu WC, Ho CC, Nicholls JM, Peiris JS, Chan MC (2010) Influenza H5N1 and H1N1 virus replication and innate immune responses in bronchial epithelial cells are influenced by the state of differentiation. PLoS ONE 5:e8713. doi:10.1371/journal.pone.0008713
Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, Dekruyff RH, Umetsu DT (2011) Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol 12:631–638. doi:10.1038/ni.2045
Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, Gordon S, Guan Y, Peiris JS (2002) Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360:1831–1837. doi:10.1016/S0140-6736(02)11772-7 S0140673602117727 [PII]
Clements ML, Betts RF, Tierney EL, Murphy BR (1986) Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J Clin Microbiol 24:157–160
Corral L, Singer MS, Macher BA, Rosen SD (1990) Requirement for sialic acid on neutrophils in a GMP-140 (PADGEM) mediated adhesive interaction with activated platelets. Biochem Biophys Res Commun 172:1349–1356
Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, Puel A, Biswas SK, Moshous D, Picard C, Jais JP, D’Cruz D, Casanova JL, Trouillet C, Geissmann F (2010) Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 33:375–386. doi:10.1016/j.immuni.2010.08.012 S1074-7613(10)00317-1 [PII]
Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, Staeheli P, Wack A (2013) Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog 9:e1003773. doi:10.1371/journal.ppat.1003773
Dauber B, Martinez-Sobrido L, Schneider J, Hai R, Waibler Z, Kalinke U, Garcia-Sastre A, Wolff T (2009) Influenza B virus ribonucleoprotein is a potent activator of the antiviral kinase PKR. PLoS Pathog 5:e1000473. doi:10.1371/journal.ppat.1000473
de Santo C, Salio M, Masri SH, Lee LY, Dong T, Speak AO, Porubsky S, Booth S, Veerapen N, Besra GS, Grone HJ, Platt FM, Zambon M, Cerundolo V (2008) Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest 118:4036–4048. doi:10.1172/JCI36264
de Vries E, Tscherne DM, Wienholts MJ, Cobos-Jimenez V, Scholte F, Garcia-Sastre A, Rottier PJ, de Haan CA (2011) Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog 7:e1001329. doi:10.1371/journal.ppat.1001329
Denney L, Aitken C, Li CK, Wilson-Davies E, Kok WL, Clelland C, Rooney K, Young D, Dong T, McMichael AJ, Carman WF, Ho LP (2010) Reduction of natural killer but not effector CD8 T lymphocytes in three consecutive cases of severe/lethal H1N1/09 influenza A virus infection. PLoS ONE 5:e10675. doi:10.1371/journal.pone.0010675
Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH, Caminschi I, Shortman K, Henson PM, Jakubzick CV (2011) CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J Exp Med 208:1789–1797. doi:10.1084/jem.20110538
Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529–1531. doi:10.1126/science.1093616
Draghi M, Pashine A, Sanjanwala B, Gendzekhadze K, Cantoni C, Cosman D, Moretta A, Valiante NM, Parham P (2007) NKp46 and NKG2D recognition of infected dendritic cells is necessary for NK cell activation in the human response to influenza infection. J Immunol 178:2688–2698
Edwards AD, Diebold SS, Slack EM, Tomizawa H, Hemmi H, Kaisho T, Akira S, Reis e Sousa C (2003) Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol 33:827–833. doi:10.1002/eji.200323797
Everitt AR, Clare S, Pertel T, John SP, Wash RS, Smith SE, Chin CR, Feeley EM, Sims JS, Adams DJ, Wise HM, Kane L, Goulding D, Digard P, Anttila V, Baillie JK, Walsh TS, Hume DA, Palotie A, Xue Y, Colonna V, Tyler-Smith C, Dunning J, Gordon SB, Smyth RL, Openshaw PJ, Dougan G, Brass AL, Kellam P (2012) IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484:519–523. doi:10.1038/nature10921
Feeley EM, Sims JS, John SP, Chin CR, Pertel T, Chen LM, Gaiha GD, Ryan BJ, Donis RO, Elledge SJ, Brass AL (2011) IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog 7:e1002337. doi:10.1371/journal.ppat.1002337
Fernandez-Sesma A, Marukian S, Ebersole BJ, Kaminski D, Park MS, Yuen T, Sealfon SC, Garcia-Sastre A, Moran TM (2006) Influenza virus evades innate and adaptive immunity via the NS1 protein. J Virol 80:6295–6304. doi:10.1128/JVI.02381-05
Finberg RW, Wang JP, Kurt-Jones EA (2007) Toll like receptors and viruses. Rev Med Virol 17:35–43. doi:10.1002/rmv.525
Fonteneau JF, Gilliet M, Larsson M, Dasilva I, Munz C, Liu YJ, Bhardwaj N (2003) Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood 101:3520–3526. doi:10.1182/blood-2002-10-3063-10-3063
Fox A, Le NM, Horby P, van Doorn HR, Nguyen VT, Nguyen HH, Nguyen TC, Vu DP, Nguyen MH, Diep NT, Bich VT, Huong HT, Taylor WR, Farrar J, Wertheim H, Nguyen VK (2012) Severe pandemic H1N1 2009 infection is associated with transient NK and T deficiency and aberrant CD8 responses. PLoS ONE 7:e31535. doi:10.1371/journal.pone.0031535
Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10:241–247. doi:10.1038/ni.1703
Fujisawa H (2001) Inhibitory role of neutrophils on influenza virus multiplication in the lungs of mice. Microbiol Immunol 45:679–688
Garcia-Sastre A (2011) Induction and evasion of type I interferon responses by influenza viruses. Virus Res 162:12–18. doi:10.1016/j.virusres.2011.10.017 S0168-1702(11)00411-4 [PII]
Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, Hanna J, Qimron U, Landau G, Greenbaum E, Zakay-Rones Z, Porgador A, Mandelboim O (2006) Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol 7:517–523. doi:10.1038/ni1322
Ge MQ, Ho AW, Tang Y, Wong KH, Chua BY, Gasser S, Kemeny DM (2012) NK cells regulate CD8+ T cell priming and dendritic cell migration during influenza A infection by IFN-gamma and perforin-dependent mechanisms. J Immunol 189:2099–2109. doi:10.4049/jimmunol.1103474
Geeraedts F, Goutagny N, Hornung V, Severa M, de Haan A, Pool J, Wilschut J, Fitzgerald KA, Huckriede A (2008) Superior immunogenicity of inactivated whole virus H5N1 influenza vaccine is primarily controlled by toll-like receptor signalling. PLoS Pathog 4:e1000138. doi:10.1371/journal.ppat.1000138
Geijtenbeek TB, Gringhuis SI (2009) Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 9:465–479. doi:10.1038/nri2569
Geiler J, Michaelis M, Sithisarn P, Cinatl J Jr (2011) Comparison of pro-inflammatory cytokine expression and cellular signal transduction in human macrophages infected with different influenza A viruses. Med Microbiol Immunol 200:53–60. doi:10.1007/s00430-010-0173-y
Gerlach RL, Camp JV, Chu YK, Jonsson CB (2013) Early host responses of seasonal and pandemic influenza a viruses in primary well-differentiated human lung epithelial cells. PLoS ONE 8:e78912. doi:10.1371/journal.pone.0078912 PONE-D-13-28042s [PII]
GeurtsvanKessel CH, Willart MA, Bergen IM, van Rijt LS, Muskens F, Elewaut D, Osterhaus AD, Hendriks R, Rimmelzwaan GF, Lambrecht BN (2009) Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J Exp Med 206:2339–2349. doi:10.1084/jem.20090410
GeurtsvanKessel CH, Willart MA, van Rijt LS, Muskens F, Kool M, Baas C, Thielemans K, Bennett C, Clausen BE, Hoogsteden HC, Osterhaus AD, Rimmelzwaan GF, Lambrecht BN (2008) Clearance of influenza virus from the lung depends on migratory langerin+CD11b− but not plasmacytoid dendritic cells. J Exp Med 205:1621–1634. doi:10.1084/jem.20071365jem.20071365
Giamarellos-Bourboulis EJ, Raftogiannis M, Antonopoulou A, Baziaka F, Koutoukas P, Savva A, Kanni T, Georgitsi M, Pistiki A, Tsaganos T, Pelekanos N, Athanassia S, Galani L, Giannitsioti E, Kavatha D, Kontopidou F, Mouktaroudi M, Poulakou G, Sakka V, Panagopoulos P, Papadopoulos A, Kanellakopoulou K, Giamarellou H (2009) Effect of the novel influenza A (H1N1) virus in the human immune system. PLoS ONE 4:e8393. doi:10.1371/journal.pone.0008393
Gill MA, Long K, Kwon T, Muniz L, Mejias A, Connolly J, Roy L, Banchereau J, Ramilo O (2008) Differential recruitment of dendritic cells and monocytes to respiratory mucosal sites in children with influenza virus or respiratory syncytial virus infection. J Infect Dis 198:1667–1676. doi:10.1086/593018
Gill MA, Palucka AK, Barton T, Ghaffar F, Jafri H, Banchereau J, Ramilo O (2005) Mobilization of plasmacytoid and myeloid dendritic cells to mucosal sites in children with respiratory syncytial virus and other viral respiratory infections. J Infect Dis 191:1105–1115. doi:10.1086/428589 JID33373 [PII]
Gorski SA, Hahn YS, Braciale TJ (2013) Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog 9:e1003615. doi:10.1371/journal.ppat.1003615
Graham AC, Hilmer KM, Zickovich JM, Obar JJ (2013) Inflammatory response of mast cells during influenza A virus infection is mediated by active infection and RIG-I signaling. J Immunol 190:4676–4684. doi:10.4049/jimmunol.1202096
Guilliams M, Lambrecht BN, Hammad H (2013) Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal Immunol 6:464–473. doi:10.1038/mi.2013.14
Guillot L, le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M (2005) Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem 280:5571–5580. doi:10.1074/jbc.M410592200
Guo H, Kumar P, Moran TM, Garcia-Sastre A, Zhou Y, Malarkannan S (2009) The functional impairment of natural killer cells during influenza virus infection. Immunol Cell Biol 87:579–589. doi:10.1038/icb.2009.60
Guo X, Chen Y, Li X, Kong H, Yang S, Ye B, Cui D, Wu W, Li L (2011) Dynamic variations in the peripheral blood lymphocyte subgroups of patients with 2009 pandemic H1N1 swine-origin influenza A virus infection. Virol J 8:215. doi:10.1186/1743-422X-8-215
Haller O, Staeheli P, Kochs G (2009) Protective role of interferon-induced Mx GTPases against influenza viruses. Rev Sci Tech 28:219–231
Haniffa M, Collin M, Ginhoux F (2013) Ontogeny and functional specialization of dendritic cells in human and mouse. Adv Immunol 120:1–49. doi:10.1016/B978-0-12-417028-5.00001-6
Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, Wasan PS, Wang XN, Malinarich F, Malleret B, Larbi A, Tan P, Zhao H, Poidinger M, Pagan S, Cookson S, Dickinson R, Dimmick I, Jarrett RF, Renia L, Tam J, Song C, Connolly J, Chan JK, Gehring A, Bertoletti A, Collin M, Ginhoux F (2012) Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 37:60–73. doi:10.1016/j.immuni.2012.04.012 S1074-7613(12)00279-8 [PII]
Hao X, Kim TS, Braciale TJ (2008) Differential response of respiratory dendritic cell subsets to influenza virus infection. J Virol 82:4908–4919. doi:10.1128/JVI.02367-07
Hargadon KM, Zhou H, Albrecht RA, Dodd HA, Garcia-Sastre A, Braciale TJ (2011) Major histocompatibility complex class II expression and hemagglutinin subtype influence the infectivity of type A influenza virus for respiratory dendritic cells. J Virol 85:11955–11963. doi:10.1128/JVI.05830-11
Hartmann BM, Li W, Jia J, Patil S, Marjanovic N, Martinez-Romero C, Albrecht RA, Hayot F, Garcia-Sastre A, Wetmur JG, Moran TM, Sealfon SC (2013) Mouse dendritic cell (DC) influenza virus infectivity is much lower than that for human DCs and is hemagglutinin subtype dependent. J Virol 87:1916–1918. doi:10.1128/JVI.02980-12
Hartshorn KL, Liou LS, White MR, Kazhdan MM, Tauber JL, Tauber AI (1995) Neutrophil deactivation by influenza A virus. Role of hemagglutinin binding to specific sialic acid-bearing cellular proteins. J Immunol 154:3952–3960
Hashimoto Y, Moki T, Takizawa T, Shiratsuchi A, Nakanishi Y (2007) Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol 178:2448–2457. doi:10.4049/jimmunol.178.4.2448
He XS, Draghi M, Mahmood K, Holmes TH, Kemble GW, Dekker CL, Arvin AM, Parham P, Greenberg HB (2004) T cell-dependent production of IFN-gamma by NK cells in response to influenza A virus. J Clin Invest 114:1812–1819. doi:10.1172/JCI22797
Heer AK, Shamshiev A, Donda A, Uematsu S, Akira S, Kopf M, Marsland BJ (2007) TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J Immunol 178:2182–2191. doi:10.4049/jimmunol.178.4.2182
Helft J, Ginhoux F, Bogunovic M, Merad M (2010) Origin and functional heterogeneity of non-lymphoid tissue dendritic cells in mice. Immunol Rev 234:55–75. doi:10.1111/j.0105-2896.2009.00885.x
Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, Brown BD, Schmolke M, Miller JC, Leboeuf M, Murphy KM, Garcia-Sastre A, Merad M (2012) Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J Clin Invest 122:4037–4047. doi:10.1172/JCI60659
Heltzer ML, Coffin SE, Maurer K, Bagashev A, Zhang Z, Orange JS, Sullivan KE (2009) Immune dysregulation in severe influenza. J Leukoc Biol 85:1036–1043. doi:10.1189/jlb.1108710
Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, Mack M, Kuziel WA, Corazza N, Brunner T, Seeger W, Lohmeyer J (2008) Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med 205:3065–3077. doi:10.1084/jem.20080201
Ho AW, Prabhu N, Betts RJ, Ge MQ, Dai X, Hutchinson PE, Lew FC, Wong KL, Hanson BJ, Macary PA, Kemeny DM (2011) Lung CD103+ dendritic cells efficiently transport influenza virus to the lymph node and load viral antigen onto MHC class I for presentation to CD8 T cells. J Immunol 187:6011–6021. doi:10.4049/jimmunol.1100987
Hoeve MA, Nash AA, Jackson D, Randall RE, Dransfield I (2012) Influenza virus A infection of human monocyte and macrophage subpopulations reveals increased susceptibility associated with cell differentiation. PLoS ONE 7:e29443. doi:10.1371/journal.pone.0029443 PONE-D-11-16347 [PII]
Hogner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, Walmrath HD, Bodner J, Gattenlohner S, Lewe-Schlosser P, Matrosovich M, Seeger W, Lohmeyer J, Herold S (2013) Macrophage-expressed IFN-beta contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog 9:e1003188. doi:10.1371/journal.ppat.1003188 PPATHOGENS-D-12-00914
Hou W, Gibbs JS, Lu X, Brooke CB, Roy D, Modlin RL, Bennink JR, Yewdell JW (2012) Viral infection triggers rapid differentiation of human blood monocytes into dendritic cells. Blood 119:3128–3131. doi:10.1182/blood-2011-09-379479
Hsu AC, Parsons K, Barr I, Lowther S, Middleton D, Hansbro PM, Wark PA (2012) Critical role of constitutive type I interferon response in bronchial epithelial cell to influenza infection. PLoS One 7:e32947. doi:10.1371/journal.pone.0032947 PONE-D-11-20761 [PII]
Huang Y, Zaas AK, Rao A, Dobigeon N, Woolf PJ, Veldman T, Oien NC, McClain MT, Varkey JB, Nicholson B, Carin L, Kingsmore S, Woods CW, Ginsburg GS, Hero AO 3rd (2011) Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet 7:e1002234. doi:10.1371/journal.pgen.1002234
Huang Y, Zhu W, Zeng X, Li S, Li X, Lu C (2013) Innate and adaptive immune responses in patients with pandemic influenza A(H1N1)pdm09. Arch Virol 158:2267–2272. doi:10.1007/s00705-013-1692-9
Hufford MM, Richardson G, Zhou H, Manicassamy B, Garcia-Sastre A, Enelow RI, Braciale TJ (2012) Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8(+) T cells. PLoS ONE 7:e46581. doi:10.1371/journal.pone.0046581 PONE-D-12-19398 [PII]
Hui KP, Lee SM, Cheung CY, Mao H, Lai AK, Chan RW, Chan MC, Tu W, Guan Y, Lau YL, Peiris JS (2011) H5N1 influenza virus-induced mediators upregulate RIG-I in uninfected cells by paracrine effects contributing to amplified cytokine cascades. J Infect Dis 204:1866–1878. doi:10.1093/infdis/jir665
Hwang I, Scott JM, Kakarla T, Duriancik DM, Choi S, Cho C, Lee T, Park H, French AR, Beli E, Gardner E, Kim S (2012) Activation mechanisms of natural killer cells during influenza virus infection. PLoS ONE 7:e51858. doi:10.1371/journal.pone.0051858 PONE-D-12-23463 [PII]
Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, Holtzman MJ, Brody SL (2006) Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol 80:7469–7480. doi:10.1128/JVI.02677-05
Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A (2009) Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 206:79–87. doi:10.1084/jem.20081667
Ichinohe T, Pang IK, Iwasaki A (2010) Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11:404–410. doi:10.1038/ni.1861
Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A (2011) Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A 108:5354–5359. doi:10.1073/pnas.1019378108
Ioannidis I, Ye F, McNally B, Willette M, Flano E (2013) Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J Virol 87:3261–3270. doi:10.1128/JVI.01956-12
Ioannidis LJ, Verity EE, Crawford S, Rockman SP, Brown LE (2012) Abortive replication of influenza virus in mouse dendritic cells. J Virol 86:5922–5925. doi:10.1128/JVI.07060-11
Ito T, Allen RM, Carson WFt, Schaller M, Cavassani KA, Hogaboam CM, Lukacs NW, Matsukawa A, Kunkel SL (2011) The critical role of Notch ligand Delta-like 1 in the pathogenesis of influenza A virus (H1N1) infection. PLoS Pathog 7:e1002341. doi:10.1371/journal.ppat.1002341 PPATHOGENS-D-11-00960 [PII]
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327:291–295. doi:10.1126/science.1183021
Jaworska J, Coulombe F, Downey J, Tzelepis F, Shalaby K, Tattoli I, Berube J, Rousseau S, Martin JG, Girardin SE, McCullers JA, Divangahi M (2014) NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein. Proc Nat Acad Sci U.S.A 111:E2110–E2119. doi:10.1073/pnas.1322118111
Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J (2003) Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19:225–234
Jeisy-Scott V, Davis WG, Patel JR, Bowzard JB, Shieh WJ, Zaki SR, Katz JM, Sambhara S (2011) Increased MDSC accumulation and Th2 biased response to influenza A virus infection in the absence of TLR7 in mice. PLoS ONE 6:e25242. doi:10.1371/journal.pone.0025242 PONE-D-11-14981 [PII]
Jeisy-Scott V, Kim JH, Davis WG, Cao W, Katz JM, Sambhara S (2012) TLR7 recognition is dispensable for influenza virus A infection but important for the induction of hemagglutinin-specific antibodies in response to the 2009 pandemic split vaccine in mice. J Virol 86:10988–10998. doi:10.1128/JVI.01064-12
Jost S, Altfeld M (2013) Control of human viral infections by natural killer cells. Annu Rev Immunol 31:163–194. doi:10.1146/annurev-immunol-032712-100001
Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, Liu YJ (2001) Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med 194:863–869
Kaminski MM, Ohnemus A, Cornitescu M, Staeheli P (2012) Plasmacytoid dendritic cells and toll-like receptor 7-dependent signalling promote efficient protection of mice against highly virulent influenza A virus. J Gen Virol 93:555–559. doi:10.1099/vir.0.039065-0
Kanneganti TD (2010) Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol 10:688–698. doi:10.1038/nri2851
Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Nunez G (2006) Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem 281:36560–36568. doi:10.1074/jbc.M607594200
Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A, Swayne DE, Katze MG (2006) Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443:578–581. doi:10.1038/nature05181
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi:10.1038/nature04734
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650. doi:10.1016/j.immuni.2011.05.006
Kim HM, Kang YM, Ku KB, Park EH, Yum J, Kim JC, Jin SY, Lee JS, Kim HS, Seo SH (2013) The severe pathogenicity of alveolar macrophage-depleted ferrets infected with 2009 pandemic H1N1 influenza virus. Virology 444:394–403. doi:10.1016/j.virol.2013.07.006 S0042-6822(13)00408-X [PII]
Kim HM, Lee YW, Lee KJ, Kim HS, Cho SW, van Rooijen N, Guan Y, Seo SH (2008) Alveolar macrophages are indispensable for controlling influenza viruses in lungs of pigs. J Virol 82:4265–4274. doi:10.1128/JVI.02602-07
Kim TS, Braciale TJ (2009) Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS ONE 4:e4204. doi:10.1371/journal.pone.0004204
Kim TS, Gorski SA, Hahn S, Murphy KM, Braciale TJ (2014) Distinct dendritic cell subsets dictate the fate decision between effector and memory CD8(+) T cell differentiation by a CD24-dependent mechanism. Immunity 40:400–413. doi:10.1016/j.immuni.2014.02.004
Kohlmeier JE, Cookenham T, Roberts AD, Miller SC, Woodland DL (2010) Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity 33:96–105. doi:10.1016/j.immuni.2010.06.016 S1074-7613(10)00242-6 [PII]
Kok WL, Denney L, Benam K, Cole S, Clelland C, McMichael AJ, Ho LP (2012) Pivotal advance: invariant NKT cells reduce accumulation of inflammatory monocytes in the lungs and decrease immune-pathology during severe influenza A virus infection. J Leukoc Biol 91:357–368. doi:10.1189/jlb.0411184
Korth MJ, Tchitchek N, Benecke AG, Katze MG (2013) Systems approaches to influenza-virus host interactions and the pathogenesis of highly virulent and pandemic viruses. Semin Immunol 25:228–239. doi:10.1016/j.smim.2012.11.001
Koyama S, Aoshi T, Tanimoto T, Kumagai Y, Kobiyama K, Tougan T, Sakurai K, Coban C, Horii T, Akira S, Ishii KJ (2010) Plasmacytoid dendritic cells delineate immunogenicity of influenza vaccine subtypes. Sci Transl Med 2:25ra24. doi: 10.1126/scitranslmed.3000759
Koyama S, Ishii KJ, Kumar H, Tanimoto T, Coban C, Uematsu S, Kawai T, Akira S (2007) Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J Immunol 179:4711–4720. doi:10.3410/f.1091005.544378 179/7/4711 [PII]
Lakadamyali M, Rust MJ, Zhuang X (2006) Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 124:997–1009. doi:10.1016/j.cell.2005.12.038 S0092-8674(06)00123-1 [PII]
Lambrecht BN, Hammad H (2009) Biology of lung dendritic cells at the origin of asthma. Immunity 31:412–424. doi:10.1016/j.immuni.2009.08.008
Lambrecht BN, Hammad H (2012) Lung dendritic cells in respiratory viral infection and asthma: from protection to immunopathology. Ann Rev Immunol 30:243–270. doi:10.1146/annurev-immunol-020711-075021
Larsson M, Messmer D, Somersan S, Fonteneau JF, Donahoe SM, Lee M, Dunbar PR, Cerundolo V, Julkunen I, Nixon DF, Bhardwaj N (2000) Requirement of mature dendritic cells for efficient activation of influenza A-specific memory CD8+ T cells. J Immunol 165:1182–1190
le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M (2006) Detrimental contribution of the toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog 2:e53. doi:10.1371/journal.ppat.0020053
le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M (2007) Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol 178:3368–3372 178/6/3368 [PII]
le Nouen C, Hillyer P, Winter CC, McCarty T, Rabin RL, Collins PL, Buchholz UJ (2011) Low CCR7-mediated migration of human monocyte derived dendritic cells in response to human respiratory syncytial virus and human metapneumovirus. PLoS Pathog 7:e1002105. doi:10.1371/journal.ppat.1002105 PPATHOGENS-D-10-00578 [PII]
Lee N, Wong CK, Chan PK, Chan MC, Wong RY, Lun SW, Ngai KL, Lui GC, Wong BC, Lee SK, Choi KW, Hui DS (2011) Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PLoS ONE 6:e26050. doi:10.1371/journal.pone.0026050
Lee N, Wong CK, Hui DS, Lee SK, Wong RY, Ngai KL, Chan MC, Chu YJ, Ho AW, Lui GC, Wong BC, Wong SH, Yip SP, Chan PK (2013) Role of human toll-like receptors in naturally occurring influenza A infections. Influenza Other Respir Viruses 7:666–675. doi:10.1111/irv.12109
Lee SM, Gardy JL, Cheung CY, Cheung TK, Hui KP, Ip NY, Guan Y, Hancock RE, Peiris JS (2009) Systems-level comparison of host-responses elicited by avian H5N1 and seasonal H1N1 influenza viruses in primary human macrophages. PLoS ONE 4:e8072. doi:10.1371/journal.pone.0008072
Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, Garcia-Sastre A, Leib DA, Pekosz A, Knobeloch KP, Horak I, Virgin HWt (2007) IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Nat Acad Sci U.S.A 104:1371–1376. doi:10.1073/pnas.0607038104
Leon B, Bradley JE, Lund FE, Randall TD, Ballesteros-Tato A (2014) FoxP3+ regulatory T cells promote influenza-specific Tfh responses by controlling IL-2 availability. Nat Commun 5:3495. doi:10.1038/ncomms4495
Li S, Min JY, Krug RM, Sen GC (2006) Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349:13–21. doi:10.1016/j.virol.2006.01.005 S0042-6822(06)00008-0 [PII]
Li S, Nakaya HI, Kazmin DA, Oh JZ, Pulendran B (2013) Systems biological approaches to measure and understand vaccine immunity in humans. Semin Immunol 25:209–218. doi:10.1016/j.smim.2013.05.003 S1044-5323(13)00032-8 [PII]
Li S, Rouphael N, Duraisingham S, Romero-Steiner S, Presnell S, Davis C, Schmidt DS, Johnson SE, Milton A, Rajam G, Kasturi S, Carlone GM, Quinn C, Chaussabel D, Palucka AK, Mulligan MJ, Ahmed R, Stephens DS, Nakaya HI, Pulendran B (2014) Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat Immunol 15:195–204. doi:10.1038/ni.2789
Lietzen N, Ohman T, Rintahaka J, Julkunen I, Aittokallio T, Matikainen S, Nyman TA (2011) Quantitative subcellular proteome and secretome profiling of influenza A virus-infected human primary macrophages. PLoS Pathog 7:e1001340. doi:10.1371/journal.ppat.1001340 10-PLPA-RA-4245R2 [PII]
Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD (2008) CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol 180:2562–2572. doi:10.4049/jimmunol.180.4.2562 180/4/2562 [PII]
Lin KL, Sweeney S, Kang BD, Ramsburg E, Gunn MD (2011) CCR2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. J Immunol 186:508–515. doi:10.4049/jimmunol.1001002
Londrigan SL, Tate MD, Brooks AG, Reading PC (2012) Cell-surface receptors on macrophages and dendritic cells for attachment and entry of influenza virus. J Leukoc Biol 92:97–106. doi:10.1189/jlb.1011492
Londrigan SL, Turville SG, Tate MD, Deng YM, Brooks AG, Reading PC (2011) N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J Virol 85:2990–3000. doi:10.1128/JVI.01705-10
Loo YM, Gale M Jr (2011) Immune signaling by RIG-I-like receptors. Immunity 34:680–692. doi:10.1016/j.immuni.2011.05.003 S1074-7613(11)00187-7 [PII]
Lui G, Manches O, Angel J, Molens JP, Chaperot L, Plumas J (2009) Plasmacytoid dendritic cells capture and cross-present viral antigens from influenza-virus exposed cells. PLoS ONE 4:e7111. doi:10.1371/journal.pone.0007111
Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA (2004) Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Nat Acad Sci U.S.A 101:5598–5603. doi:10.1073/pnas.0400937101
Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, Xavier RJ, Green DR, Lamkanfi M, Dinarello CA, Doherty PC, Kanneganti TD (2013) Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 14:480–488. doi:10.1038/ni.2563
Lupfer C, Thomas PG, Kanneganti TD (2014) NOD2 dependent DC activation is necessary for innate immunity and optimal CD8+ T cell responses to influenza A virus infection. J Virol. doi:10.1128/JVI.01110-14
Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, Davis DM, Strominger JL, Yewdell JW, Porgador A (2001) Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 409:1055–1060. doi:10.1038/35059110
Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A (2010) Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Nat Acad Sci U.S.A 107:11531–11536. doi:10.1073/pnas.0914994107
Manicassamy S, Pulendran B (2009) Modulation of adaptive immunity with toll-like receptors. Semin Immunol 21:185–193. doi:10.1016/j.smim.2009.05.005 S1044-5323(09)00048-7 [PII]
Manicassamy S, Pulendran B (2011) Dendritic cell control of tolerogenic responses. Immunol Rev 241:206–227. doi:10.1111/j.1600-065X.2011.01015.x
Mao H, Tu W, Liu Y, Qin G, Zheng J, Chan PL, Lam KT, Peiris JS, Lau YL (2010) Inhibition of human natural killer cell activity by influenza virions and hemagglutinin. J Virol 84:4148–4157. doi:10.1128/JVI.02340-09
Mao H, Tu W, Qin G, Law HK, Sia SF, Chan PL, Liu Y, Lam KT, Zheng J, Peiris M, Lau YL (2009) Influenza virus directly infects human natural killer cells and induces cell apoptosis. J Virol 83:9215–9222. doi:10.1128/JVI.00805-09
Mao L, Yang Y, Qiu Y, Yang Y (2012) Annual economic impacts of seasonal influenza on US counties: spatial heterogeneity and patterns. Int J Health Geogr 11:16. doi:10.1186/1476-072X-11-16
McAuley JL, Tate MD, MacKenzie-Kludas CJ, Pinar A, Zeng W, Stutz A, Latz E, Brown LE, Mansell A (2013) Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog 9:e1003392. doi:10.1371/journal.ppat.1003392
McClain MT, Park LP, Nicholson B, Veldman T, Zaas AK, Turner R, Lambkin-Williams R, Gilbert AS, Ginsburg GS, Woods CW (2013) Longitudinal analysis of leukocyte differentials in peripheral blood of patients with acute respiratory viral infections. J Clin Virol 58:689–695. doi:10.1016/j.jcv.2013.09.015 S1386-6532(13)00406-X [PII]
McGill J, van Rooijen N, Legge KL (2008) Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J Exp Med 205:1635–1646. doi:10.1084/jem.20080314
McGill J, van Rooijen N, Legge KL (2010) IL-15 trans-presentation by pulmonary dendritic cells promotes effector CD8 T cell survival during influenza virus infection. J Exp Med 207:521–534. doi:10.1084/jem.20091711
Medina RA, Garcia-Sastre A (2011) Influenza A viruses: new research developments. Nat Rev Microbiol 9:590–603. doi:10.1038/nrmicro2613
Merad M, Sathe P, Helft J, Miller J, Mortha A (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Ann Rev Immunol 31:563–604. doi:10.1146/annurev-immunol-020711-074950
Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB (2007) The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 25:5086–5096. doi:10.1016/j.vaccine.2007.03.046
Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Kubota M, Turner D, Diamond JM, Goldrath AW, Farber DL, Collman RG, Wherry EJ, Artis D (2011) Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12:1045–1054. doi:10.1031/ni.2131
Nakajima N, Sato Y, Katano H, Hasegawa H, Kumasaka T, Hata S, Tanaka S, Amano T, Kasai T, Chong JM, Iizuka T, Nakazato I, Hino Y, Hamamatsu A, Horiguchi H, Tanaka T, Hasegawa A, Kanaya Y, Oku R, Oya T, Sata T (2012) Histopathological and immunohistochemical findings of 20 autopsy cases with 2009 H1N1 virus infection. Mod Pathol 25:1–13. doi:10.1038/modpathol.2011.125
Nakajima N, van Tin N, Sato Y, Thach HN, Katano H, Diep PH, Kumasaka T, Thuy NT, Hasegawa H, San LT, Kawachi S, Liem NT, Suzuki K, Sata T (2013) Pathological study of archival lung tissues from five fatal cases of avian H5N1 influenza in Vietnam. Mod Pathol 26:357–369. doi:10.1038/modpathol.2012.193
Nakano H, Lin KL, Yanagita M, Charbonneau C, Cook DN, Kakiuchi T, Gunn MD (2009) Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol 10:394–402. doi:10.1038/ni.1707
Nakaya HI, Li S, Pulendran B (2012) Systems vaccinology: learning to compute the behavior of vaccine induced immunity. Wiley Interdiscip Rev Syst Biol Med 4:193–205. doi:10.1002/wsbm.163
Nakaya HI, Wrammert J, Lee EK, Racioppi L, Marie-Kunze S, Haining WN, Means AR, Kasturi SP, Khan N, Li GM, McCausland M, Kanchan V, Kokko KE, Li S, Elbein R, Mehta AK, Aderem A, Subbarao K, Ahmed R, Pulendran B (2011) Systems biology of vaccination for seasonal influenza in humans. Nat Immunol 12:786–795. doi:10.1038/ni.2067
Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT (2011) Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179:199–210. doi:10.1016/j.ajpath.2011.03.013 S0002-9440(11)00329-4 [PII]
Nhu QM, Shirey K, Teijaro JR, Farber DL, Netzel-Arnett S, Antalis TM, Fasano A, Vogel SN (2010) Novel signaling interactions between proteinase-activated receptor 2 and toll-like receptors in vitro and in vivo. Mucosal Immunol 3:29–39. doi:10.1038/mi.2009.120
Nicholls JM, Chan MC, Chan WY, Wong HK, Cheung CY, Kwong DL, Wong MP, Chui WH, Poon LL, Tsao SW, Guan Y, Peiris JS (2007) Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat Med 13:147–149. doi:10.1038/nm1529
Ohman T, Rintahaka J, Kalkkinen N, Matikainen S, Nyman TA (2009) Actin and RIG-I/MAVS signaling components translocate to mitochondria upon influenza A virus infection of human primary macrophages. J Immunol 182:5682–5692. doi:10.4049/jimmunol.0803093
Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T (2012) Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 7:e43031. doi:10.1371/journal.pone.0043031
Oshansky CM, Gartland AJ, Wong SS, Jeevan T, Wang D, Roddam PL, Caniza MA, Hertz T, Devincenzo JP, Webby RJ, Thomas PG (2013) Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am J Respir Crit Care Med. doi:10.1164/rccm.201309-1616OC
Osterlund P, Pirhonen J, Ikonen N, Ronkko E, Strengell M, Makela SM, Broman M, Hamming OJ, Hartmann R, Ziegler T, Julkunen I (2010) Pandemic H1N1 2009 influenza A virus induces weak cytokine responses in human macrophages and dendritic cells and is highly sensitive to the antiviral actions of interferons. J Virol 84:1414–1422. doi:10.1128/JVI.01619-09
Osterlund P, Veckman V, Siren J, Klucher KM, Hiscott J, Matikainen S, Julkunen I (2005) Gene expression and antiviral activity of alpha/beta interferons and interleukin-29 in virus-infected human myeloid dendritic cells. J Virol 79:9608–9617. doi:10.1128/JVI.79.15.9608-9617.2005
Paget C, Ivanov S, Fontaine J, Blanc F, Pichavant M, Renneson J, Bialecki E, Pothlichet J, Vendeville C, Barba-Spaeth G, Huerre MR, Faveeuw C, Si-Tahar M, Trottein F (2011) Potential role of invariant NKT cells in the control of pulmonary inflammation and CD8+ T cell response during acute influenza A virus H3N2 pneumonia. J Immunol 186:5590–5602. doi:10.4049/jimmunol.1002348
Pan W, Dong Z, Li F, Meng W, Feng L, Niu X, Li C, Luo Q, Li Z, Sun C, Chen L (2013) Visualizing influenza virus infection in living mice. Nat Commun 4:2369. doi:10.1038/ncomms3369
Pang IK, Ichinohe T, Iwasaki A (2013a) IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat Immunol 14:246–253. doi:10.1038/ni.2514
Pang IK, Pillai PS, Iwasaki A (2013b) Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc Nat Acad Sci U.S.A 110:13910–13915. doi:10.1073/pnas.1303275110
Pelletier I, Rousset D, Enouf V, Colbere-Garapin F, van der Werf S, Naffakh N (2011) Highly heterogeneous temperature sensitivity of 2009 pandemic influenza A(H1N1) viral isolates, northern France. Euro Surveill 16:1–7. http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19999
Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM (2008) H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog 4:e1000115. doi:10.1371/journal.ppat.1000115
Phipps-Yonas H, Seto J, Sealfon SC, Moran TM, Fernandez-Sesma A (2008) Interferon-beta pretreatment of conventional and plasmacytoid human dendritic cells enhances their activation by influenza virus. PLoS Pathog 4:e1000193. doi:10.1371/journal.ppat.1000193
Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C (2006) RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 314:997–1001. doi:10.1126/science.1132998
Pindel A, Sadler A (2011) The role of protein kinase R in the interferon response. J Interferon Cytokine Res 31:59–70. doi:10.1089/jir.2010.0099
Piqueras B, Connolly J, Freitas H, Palucka AK, Banchereau J (2006) Upon viral exposure, myeloid and plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood 107:2613–2618. doi:10.1182/blood-2005-07-2965
Pothlichet J, Burtey A, Kubarenko AV, Caignard G, Solhonne B, Tangy F, Ben-Ali M, Quintana-Murci L, Heinzmann A, Chiche JD, Vidalain PO, Weber AN, Chignard M, Si-Tahar M (2009) Study of human RIG-I polymorphisms identifies two variants with an opposite impact on the antiviral immune response. PLoS ONE 4:e7582. doi:10.1371/journal.pone.0007582
Pulendran B (2009) Learning immunology from the yellow fever vaccine: innate immunity to systems vaccinology. Nat Rev Immunol 9:741–747. doi:10.1038/nri2629
Pulendran B, Li S, Nakaya HI (2010a) Systems vaccinology. Immunity 33:516–529. doi:10.1016/j.immuni.2010.10.006 S1074-7613(10)00366-3 [PII]
Pulendran B, Tang H, Manicassamy S (2010b) Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol 11:647–655. doi:10.1038/ni.1894
Qu C, Moran TM, Randolph GJ (2003) Autocrine type I IFN and contact with endothelium promote the presentation of influenza A virus by monocyte-derived APC. J Immunol 170:1010–1018
Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, Kennedy K, Wu H, Bennouna S, Oluoch H, Miller J, Vencio RZ, Mulligan M, Aderem A, Ahmed R, Pulendran B (2009) Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol 10:116–125. doi:10.1038/ni.1688
Ramos I, Bernal-Rubio D, Durham N, Belicha-Villanueva A, Lowen AC, Steel J, Fernandez-Sesma A (2011) Effects of receptor binding specificity of avian influenza virus on the human innate immune response. J Virol 85:4421–4431. doi:10.1128/JVI.02356-10
Ravindran R, Khan N, Nakaya HI, Li S, Loebbermann J, Maddur MS, Park Y, Jones DP, Chappert P, Davoust J, Weiss DS, Virgin HW, Ron D, Pulendran B (2014) Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science 343:313–317. doi:10.1126/science.1246829
Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, Reis e Sousa C (2010) RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397–408. doi:10.1016/j.cell.2010.01.020 S0092-8674(10)00021-8 [PII]
Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y, Bose S (2009) Activation of innate immune antiviral responses by Nod2. Nat Immunol 10:1073–1080. doi:10.1038/ni.1782
Sadler AJ, Williams BR (2008) Interferon-inducible antiviral effectors. Nat Rev Immunol 8:559–568. doi:10.1038/nri2314
Sakabe S, Iwatsuki-Horimoto K, Takano R, Nidom CA, Le M, Nagamura-Inoue T, Horimoto T, Yamashita N, Kawaoka Y (2011) Cytokine production by primary human macrophages infected with highly pathogenic H5N1 or pandemic H1N1 2009 influenza viruses. J Gen Virol 92:1428–1434. doi:10.1099/vir.0.030346-0
Salentin R, Gemsa D, Sprenger H, Kaufmann A (2003) Chemokine receptor expression and chemotactic responsiveness of human monocytes after influenza A virus infection. J Leukoc Biol 74:252–259
Sancho D, Reis e Sousa C (2012) Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu Rev Immunol 30:491–529. doi:10.1146/annurev-immunol-031210-101352
Sasaki S, Jaimes MC, Holmes TH, Dekker CL, Mahmood K, Kemble GW, Arvin AM, Greenberg HB (2007) Comparison of the influenza virus-specific effector and memory B-cell responses to immunization of children and adults with live attenuated or inactivated influenza virus vaccines. J Virol 81:215–228. doi:10.1128/JVI.01957-06
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832. doi:10.1016/j.cell.2010.01.040 S0092-8674(10)00075-9 [PII]
Segura E, Valladeau-Guilemond J, Donnadieu MH, Sastre-Garau X, Soumelis V, Amigorena S (2012) Characterization of resident and migratory dendritic cells in human lymph nodes. J Exp Med 209:653–660. doi:10.1084/jem.20111457
Seo SU, Kwon HJ, Ko HJ, Byun YH, Seong BL, Uematsu S, Akira S, Kweon MN (2011) Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog 7:e1001304. doi:10.1371/journal.ppat.1001304
Seo SU, Kwon HJ, Song JH, Byun YH, Seong BL, Kawai T, Akira S, Kweon MN (2010) MyD88 signaling is indispensable for primary influenza A virus infection but dispensable for secondary infection. J Virol 84:12713–12722. doi:10.1128/JVI.01675-10
Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y (2006) Avian flu: influenza virus receptors in the human airway. Nature 440:435–436. doi:10.1038/440435a
Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, McAlees J, Gioannini TL, Weiss J, Chen WH, Ernst RK, Rossignol DP, Gusovsky F, Blanco JC, Vogel SN (2013) The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 497:498–502. doi:10.1038/nature12118
Short KR, Brooks AG, Reading PC, Londrigan SL (2012) The fate of influenza A virus after infection of human macrophages and dendritic cells. J Gen Virol 93:2315–2325. doi:10.1099/vir.0.045021-0
Short KR, Kroeze EJ, Fouchier RA, Kuiken T (2014) Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis 14:57–69. doi:10.1016/S1473-3099(13)70286-X S1473-3099(13)70286-X [PII]
Skehel JJ, Wiley DC (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Ann Rev Biochem 69:531–569. doi:10.1146/annurev.biochem.69.1.531
Smed-Sorensen A, Chalouni C, Chatterjee B, Cohn L, Blattmann P, Nakamura N, Delamarre L, Mellman I (2012) Influenza A virus infection of human primary dendritic cells impairs their ability to cross-present antigen to CD8 T cells. PLoS Pathog 8:e1002572. doi:10.1371/journal.ppat.1002572 PPATHOGENS-D-11-01789
Song JY, Cheong HJ, Hwang IS, Choi WS, Jo YM, Park DW, Cho GJ, Hwang TG, Kim WJ (2010) Long-term immunogenicity of influenza vaccine among the elderly: risk factors for poor immune response and persistence. Vaccine 28:3929–3935. doi:10.1016/j.vaccine.2010.03.067
Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E (2013) Innate lymphoid cells—a proposal for uniform nomenclature. Nat Rev Immunol 13:145–149. doi:10.1038/nri3365
Steinman RM, Banchereau J (2007) Taking dendritic cells into medicine. Nature 449:419–426. doi:10.1038/nature06175
Takeuchi O, Akira S (2009) Innate immunity to virus infection. Immunol Rev 227:75–86. doi:10.1111/j.1600-065X.2008.00737.x IMR737 [PII]
Tam VC, Quehenberger O, Oshansky CM, Suen R, Armando AM, Treuting PM, Thomas PG, Dennis EA, Aderem A (2013) Lipidomic profiling of influenza infection identifies mediators that induce and resolve inflammation. Cell 154:213–227. doi:10.1016/j.cell.2013.05.052 S0092-8674(13)00695-8 [PII]
Tate MD, Brooks AG, Reading PC (2008) The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir Res 9:57. doi:10.1186/1465-9921-9-57
Tate MD, Brooks AG, Reading PC, Mintern JD (2012) Neutrophils sustain effective CD8(+) T-cell responses in the respiratory tract following influenza infection. Immunol Cell Biol 90:197–205. doi:10.1038/icb.2011.26
Tate MD, Deng YM, Jones JE, Anderson GP, Brooks AG, Reading PC (2009) Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J Immunol 183:7441–7450. doi:10.4049/jimmunol.0902497
Tate MD, Ioannidis LJ, Croker B, Brown LE, Brooks AG, Reading PC (2011) The role of neutrophils during mild and severe influenza virus infections of mice. PLoS ONE 6:e17618. doi:10.1371/journal.pone.0017618
Tate MD, Pickett DL, van Rooijen N, Brooks AG, Reading PC (2010) Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J Virol 84:7569–7580. doi:10.1128/JVI.00291-10
Taubenberger JK, Morens DM (2008) The pathology of influenza virus infections. Ann Rev Pathol 3:499–522. doi:10.1146/annurev.pathmechdis.3.121806.154316
Thitithanyanont A, Engering A, Ekchariyawat P, Wiboon-ut S, Limsalakpetch A, Yongvanitchit K, Kum-Arb U, Kanchongkittiphon W, Utaisincharoen P, Sirisinha S, Puthavathana P, Fukuda MM, Pichyangkul S (2007) High susceptibility of human dendritic cells to avian influenza H5N1 virus infection and protection by IFN-alpha and TLR ligands. J Immunol 179:5220–5227 179/8/5220 [PII] doi:10.4049/jimmunol.179.8.5220
Thomas PG, Dash P, Aldridge JR Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD (2009) The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30:566–575. doi:10.1016/j.immuni.2009.02.006
Thompson CI, Barclay WS, Zambon MC, Pickles RJ (2006) Infection of human airway epithelium by human and avian strains of influenza a virus. J Virol 80:8060–8068. doi:10.1128/JVI.00384-06
Tripathi S, White MR, Hartshorn KL (2013) The amazing innate immune response to influenza A virus infection. Innate Immun. doi:10.1177/1753425913508992
Tsai SY, Segovia JA, Chang TH, Morris IR, Berton MT, Tessier PA, Tardif MR, Cesaro A, Bose S (2014) DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog 10:e1003848. doi:10.1371/journal.ppat.1003848 PPATHOGENS-D-13-00524 [PII]
Tsang JS, Schwartzberg PL, Kotliarov Y, Biancotto A, Xie Z, Germain RN, Wang E, Olnes MJ, Narayanan M, Golding H, Moir S, Dickler HB, Perl S, Cheung F (2014) Global analyses of human immune variation reveal baseline predictors of postvaccination responses. Cell 157:499–513. doi:10.1016/j.cell.2014.03.031
Tscherne DM, Garcia-Sastre A (2011) Virulence determinants of pandemic influenza viruses. J Clin Invest 121:6–13. doi:10.1172/JCI44947
Tse H, To KK, Wen X, Chen H, Chan KH, Tsoi HW, Li IW, Yuen KY (2011) Clinical and virological factors associated with viremia in pandemic influenza A/H1N1/2009 virus infection. PLoS ONE 6:e22534. doi:10.1371/journal.pone.0022534
Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S, Solorzano A, van Rooijen N, Katz JM, Basler CF (2005) Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 79:14933–14944. doi:10.1128/JVI.79.23.14933-14944.2005
Unkel B, Hoegner K, Clausen BE, Lewe-Schlosser P, Bodner J, Gattenloehner S, Janssen H, Seeger W, Lohmeyer J, Herold S (2012) Alveolar epithelial cells orchestrate DC function in murine viral pneumonia. J Clin Invest 122:3652–3664. doi:10.1172/JCI62139
Valkenburg SA, Rutigliano JA, Ellebedy AH, Doherty PC, Thomas PG, Kedzierska K (2011) Immunity to seasonal and pandemic influenza A viruses. Microbes Infect 13:489–501. doi:10.1016/j.micinf.2011.01.007 S1286-4579(11)00037-2 [PII]
van Riel D, Leijten LM, van der Eerden M, Hoogsteden HC, Boven LA, Lambrecht BN, Osterhaus AD, Kuiken T (2011) Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF-alpha induction. PLoS Pathog 7:e1002099. doi:10.1371/journal.ppat.1002099 PPATHOGENS-D-10-00224 [PII]
Vareille M, Kieninger E, Edwards MR, Regamey N (2011) The airway epithelium: soldier in the fight against respiratory viruses. Clin Microbiol Rev 24:210–229. doi:10.1128/CMR.00014-10
Verbist KC, Rose DL, Cole CJ, Field MB, Klonowski KD (2012) IL-15 participates in the respiratory innate immune response to influenza virus infection. PLoS ONE 7:e37539. doi:10.1371/journal.pone.0037539
Videira PA, Amado IF, Crespo HJ, Alguero MC, Dall’Olio F, Cabral MG, Trindade H (2008) Surface alpha 2-3- and alpha 2-6-sialylation of human monocytes and derived dendritic cells and its influence on endocytosis. Glycoconj J 25:259–268. doi:10.1007/s10719-007-9092-6
von der Malsburg A, Abutbul-Ionita I, Haller O, Kochs G, Danino D (2011) Stalk domain of the dynamin-like MxA GTPase protein mediates membrane binding and liposome tubulation via the unstructured L4 loop. J Biol Chem 286:37858–37865. doi:10.1074/jbc.M111.249037
Waithman J, Zanker D, Xiao K, Oveissi S, Wylie B, Ng R, Togel L, Chen W (2013) Resident CD8(+) and migratory CD103(+) dendritic cells control CD8 T cell immunity during acute influenza infection. PLoS ONE 8:e66136. doi:10.1371/journal.pone.0066136 PONE-D-13-05069
Wang J, Nikrad MP, Phang T, Gao B, Alford T, Ito Y, Edeen K, Travanty EA, Kosmider B, Hartshorn K, Mason RJ (2011) Innate immune response to influenza A virus in differentiated human alveolar type II cells. Am J Respir Cell Mol Biol 45:582–591. doi:10.1165/rcmb.2010-0108OC
Wang J, Nikrad MP, Travanty EA, Zhou B, Phang T, Gao B, Alford T, Ito Y, Nahreini P, Hartshorn K, Wentworth D, Dinarello CA, Mason RJ (2012) Innate immune response of human alveolar macrophages during influenza A infection. PLoS ONE 7:e29879. doi:10.1371/journal.pone.0029879 PONE-D-11-13862 [PII]
Wang J, Oberley-Deegan R, Wang S, Nikrad M, Funk CJ, Hartshorn KL, Mason RJ (2009) Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza A infection. J Immunol 182:1296–1304. doi:10.4049/jimmunol.182.3.1296
Wang SF, Huang JC, Lee YM, Liu SJ, Chan YJ, Chau YP, Chong P, Chen YM (2008) DC-SIGN mediates avian H5N1 influenza virus infection in cis and in trans. Biochem Biophys Res Commun 373:561–566. doi:10.1016/j.bbrc.2008.06.078
Wang X, Hinson ER, Cresswell P (2007) The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2:96–105. doi:10.1016/j.chom.2007.06.009 S1931-3128(07)00135-7 [PII]
Watanabe Y, Hashimoto Y, Shiratsuchi A, Takizawa T, Nakanishi Y (2005) Augmentation of fatality of influenza in mice by inhibition of phagocytosis. Biochem Biophys Res Commun 337:881–886. doi:10.1016/j.bbrc.2005.09.133 S0006-291X(05)02132-7 (PII)
Welliver TP, Garofalo RP, Hosakote Y, Hintz KH, Avendano L, Sanchez K, Velozo L, Jafri H, Chavez-Bueno S, Ogra PL, McKinney L, Reed JL, Welliver RC Sr (2007) Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J Infect Dis 195:1126–1136. doi:10.1086/512615
White MR, Tecle T, Crouch EC, Hartshorn KL (2007) Impact of neutrophils on antiviral activity of human bronchoalveolar lavage fluid. Am J Physiol Lung Cell Mol Physiol 293:L1293–L1299. doi:10.1152/ajplung.00266.2007
Wilson IA, Cox NJ (1990) Structural basis of immune recognition of influenza virus hemagglutinin. Ann Rev Immunol 8:737–771. doi:10.1146/annurev.iy.08.040190.003513
Wisskirchen C, Ludersdorfer TH, Muller DA, Moritz E, Pavlovic J (2011) The cellular RNA helicase UAP56 is required for prevention of double-stranded RNA formation during influenza A virus infection. J Virol 85:8646–8655. doi:10.1128/JVI.02559-10
Wong JP, Christopher ME, Viswanathan S, Karpoff N, Dai X, Das D, Sun LQ, Wang M, Salazar AM (2009) Activation of toll-like receptor signaling pathway for protection against influenza virus infection. Vaccine 27:3481–3483. doi:10.1016/j.vaccine.2009.01.048 S0264-410X(09)00126-1 [PII]
Woods CW, McClain MT, Chen M, Zaas AK, Nicholson BP, Varkey J, Veldman T, Kingsmore SF, Huang Y, Lambkin-Williams R, Gilbert AG, Hero AO 3rd, Ramsburg E, Glickman S, Lucas JE, Carin L, Ginsburg GS (2013) A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLoS ONE 8:e52198. doi:10.1371/journal.pone.0052198
Wright PF, Neumann G, Kawaoka Y (2013) Orthomyxoviruses. In: Knipe DM, Howley PM (eds) Fields virology, 6th edn. Wolters Kluwer Health: Lippincott Willams and Wilkins, Philadelphia, PA pp 1691–1740
Yan N, Chen ZJ (2012) Intrinsic antiviral immunity. Nat Immunol 13:214–222. doi:10.1038/ni.2229
Yu CI, Becker C, Wang Y, Marches F, Helft J, Leboeuf M, Anguiano E, Pourpe S, Goller K, Pascual V, Banchereau J, Merad M, Palucka K (2013) Human CD1c+ dendritic cells drive the differentiation of CD103+ CD8+ mucosal effector T cells via the cytokine TGF-beta. Immunity 38:818–830. doi:10.1016/j.immuni.2013.03.004 S1074-7613(13)00139-8 [PII]
Yu HB, Finlay BB (2008) The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe 4:198–208. doi:10.1016/j.chom.2008.08.007
Yu WC, Chan RW, Wang J, Travanty EA, Nicholls JM, Peiris JS, Mason RJ, Chan MC (2011) Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses. J Virol 85:6844–6855. doi:10.1128/JVI.02200-10
Zaslavsky E, Nudelman G, Marquez S, Hershberg U, Hartmann BM, Thakar J, Sealfon SC, Kleinstein SH (2013) Reconstruction of regulatory networks through temporal enrichment profiling and its application to H1N1 influenza viral infection. BMC Bioinformatics 14(Suppl 6):S1. doi:10.1186/1471-2105-14-S6-S1
Zeng H, Goldsmith C, Thawatsupha P, Chittaganpitch M, Waicharoen S, Zaki S, Tumpey TM, Katz JM (2007) Highly pathogenic avian influenza H5N1 viruses elicit an attenuated type i interferon response in polarized human bronchial epithelial cells. J Virol 81:12439–12449. doi:10.1128/JVI.01134-07
Zhou J, Law HK, Cheung CY, Ng IH, Peiris JS, Lau YL (2006) Differential expression of chemokines and their receptors in adult and neonatal macrophages infected with human or avian influenza viruses. J Infect Dis 194:61–70. doi:10.1086/504690 JID36098 [PII]
Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, MacPherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB (2010) Nomenclature of monocytes and dendritic cells in blood. Blood 116:e74–e80. doi:10.1182/blood-2010-02-258558
Zimmermann P, Manz B, Haller O, Schwemmle M, Kochs G (2011) The viral nucleoprotein determines Mx sensitivity of influenza A viruses. J Virol 85:8133–8140. doi:10.1128/JVI.00712-11
Acknowledgments
This work was supported by grants from the U.S. National Institutes of Health (grants HHSN272201400004C, N01 AI50019, and N01 AI50025, R37 DK057665, R37 AI048638, U19 AI090023, U19 AI057266, U54 AI057157, U54 AI057160).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Pulendran, B., Maddur, M.S. (2014). Innate Immune Sensing and Response to Influenza. In: Oldstone, M., Compans, R. (eds) Influenza Pathogenesis and Control - Volume II. Current Topics in Microbiology and Immunology, vol 386. Springer, Cham. https://doi.org/10.1007/82_2014_405
Download citation
DOI: https://doi.org/10.1007/82_2014_405
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-11157-5
Online ISBN: 978-3-319-11158-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)