
Abstract
Background
Aquaculture successfully meets global food demands for many fish species. However, aquaculture production of Atlantic cod (Gadus morhua) is just 2.5% of total market production. For cod farming to be a viable economic venture specific challenges on how to increase growth, health and farming productivity need to be addressed. Feed ingredients play a key role here. Macroalgae (seaweeds) have been suggested as a functional feed supplement with both health and economic benefits for terrestrial farmed animals and fish. The impact of such dietary supplements to cod gut integrity and microbiota, which contribute to overall fish robustness is unknown. The objective of this study was to supplement the diet of juvenile Atlantic cod with macroalgae and determine the impacts on fish condition and growth, gut morphology and hindgut microbiota composition (16S rRNA amplicon sequencing). Fish were fed one of three diets: control (no macroalgal inclusion), 10% inclusion of either egg wrack (Ascophyllum nodosum) or sea lettuce (Ulva rigida) macroalgae in a 12-week trial.
Results
The results demonstrated there was no significant difference in fish condition, gut morphology or hindgut microbiota between the U. rigida supplemented fish group and the control group at any time-point. This trend was not observed with the A. nodosum treatment. Fish within this group were further categorised as either ‘Normal’ or ‘Lower Growth’. ‘Lower Growth’ individuals found the diet unpalatable resulting in reduced weight and condition factor combined with an altered gut morphology and microbiome relative to the other treatments. Excluding this group, our results show that the hindgut microbiota was largely driven by temporal pressures with the microbial communities becoming more similar over time irrespective of dietary treatment. The core microbiome at the final time-point consisted of the orders Vibrionales (Vibrio and Photobacterium), Bacteroidales (Bacteroidetes and Macellibacteroides) and Clostridiales (Lachnoclostridium).
Conclusions
Our study indicates that U. rigida macroalgae can be supplemented at 10% inclusion levels in the diet of juvenile farmed Atlantic cod without any impact on fish condition or hindgut microbial community structure. We also conclude that 10% dietary inclusion of A. nodosum is not a suitable feed supplement in a farmed cod diet.
Similar content being viewed by others
Background
Sustainable food production is gaining increased attention from consumers, farmers, and policy-makers. This is spurred on by concerns on the impact of climate change, consumer awareness, and the growing global food demand [1,2,3]. Currently, 151 million tonnes of fish are produced annually for human consumption [4]. Wild fish stocks cannot sustain further increases in demand. Several wild fish populations have collapsed or are in danger of critical decline from over-fishing and habitat damage [5,6,7,8]. A significant depletion in 4 European cod stocks (Kattegat, North Sea, West of Scotland and Irish Sea) by the early 2000s led to wild stock enhancement initiatives, such as the European Union’s Cod Recovery Plan (EC No 1342/2008 [9], and ultimately included enforced catch quotas. However, despite their initial recovery, the International Council for the Exploration of the Sea (ICES) have advised that the North Sea, Celtic Sea, Irish Sea and West of Scotland cod stocks are in decline once more [10,11,12,13]. The cause of this decline is unclear, with climate change, destruction of nearshore cod nurseries by trawling, grey seal predation and fisheries management being implicated [14,15,16]. The precarious condition of these wild cod stocks means this is simply not a sustainable option to meet consumer demand for this fish.
Commercial aquaculture has successfully met the market demand for a range of fish species, including Atlantic salmon (Salmo salar), rainbow trout (Oncorhynchus mykiss), gilt-head seabream (Sparus aurata), and seabass (Dicentrarchus labrax), thereby alleviating demand on wild resources [4]. Despite the popularity of cod as a food fish, aquaculture production of farmed cod is a business that has hit both highs and lows since the 1970’s. Currently, only 2.5% of the total Atlantic cod produced for food is through aquaculture [3, 4]. The economic viability of a commercial cod farming operation presents a number of challenges including a consistent high-quality broodstock, reducing high mortality, disease susceptibility, market pricing and high costs associated with aquafeeds [17,18,19]. For this to be fully realised, a number of issues need to be addressed not least the challenge associated with fish meal (FM). Carnivorous fish (e.g. salmon, trout, cod) in farmed systems are fed a diet of fishmeal (FM) consisting of milled and dried fish parts from small pelagic fish caught in the wild. This practice is environmentally unsustainable as these small pelagic fish are a food source for wild fish stocks and there is a considerable ‘bycatch’ associated with FM fishing further reducing and adding pressure on wild stocks [20]. Developing novel feed ingredients and alternatives to marine by-products in formulated diets not only could improve the economic viability of commercial cod farming but could also have implications in the sustainability status of cod farming and consumer perception of farmed cod.
One potential solution is to supplement the diet of farmed cod with high-quality functional ingredients that provide health benefits, nutrition and are environmentally sustainable and low cost. The use of macroalgae (also known as seaweeds) as a potential feed supplement could further this sustainability agenda. The biomass can often be grown or sustainably harvested at quantities that would meet commercial aquafeed demand. Dietary supplementation of macroalgae has been trialled in many farmed fish species including Atlantic salmon (S. salar [21]), rainbow trout (O. mykiss [22]), European seabass (D. labrax, [23]), carp (C. carpio, [24]), and tilapia (Oreochromis. niloticus [25]). Some studies have advocated that macroalgae could partially replace protein constituents in formulated diets [26, 27]. However, the lower algal protein content (up to 27%) in comparison to highly proteinaceous fishmeal (e.g. LT94 fish meal, ~ 67%) and plant by-products (e.g. soy protein concentrates, ~ 76%) typically used in formulated diets, indicate it would be better to exploit algal meal as a functional feed supplement instead [28]. As a functional feed supplement macroalgae can be used to supply trace metals, carotenoids, long-chain omega-3 polyunsaturated fatty acids, and vitamins that potentially benefit the growing farmed fish [29]. In general, the inclusion of macroalgae into formulated fish diets without causing decreased growth performance is approximately 10% [28]. However, the dietary macroalgae inclusion level that fish can tolerate is dependent on the algal species and fish species, i.e. carnivorous versus omnivorous fish, brown versus green macroalgae or indeed morphological differences in the fish gastrointestinal tract [30].
There is a wealth of evidence outside of fish research showing that the gut microbiota is intrinsically linked to digestive function, metabolism, immune support and general health as shown in dogs [31] and rats [32] for example. For fish gut microbiota these links are less clearly defined, though this is the focus of intensive research efforts in Atlantic salmon (S. salar) for example [33, 34]. We know that the digestibility and metabolism of a substrate is impacted by the gut microbiome [33, 35]. Fish gut microbiota produce exogenous digestive enzymes (e.g. lipases, amylases and proteases), which can significantly influence nutrient digestibility and uptake and renewal of the gut epithelial layer [35]. The hindgut (distal gut) in particular in cod (G. morhua) is a primary area of fermentation with a stable microbial community [36, 37]. A comprehensive review on the role of the gut microbiome in aquaculture is described in [38]. Research has indicated that dietary amendment can cause changes in the gut microbiome which may or may not be associated with physiological changes to the fish [39,40,41]. Algae can possess unique taxon specific polysaccharides known as phycocolloids (e.g. alginic acid and fucoidan), which have been shown using in vitro studies to exhibit antiviral, immunostimulatory, anticoagulant, and antioxidant bioactivity [42, 43]. In addition, complex components of dietary macroalgae such as cellulose, hemicellulose, and lignin, could alter the gut microbiome in the fish by providing a substrate source to hydrolytic bacteria [44]. It, therefore, follows that any manipulation of farmed cod diets with macroalgae could also impact the gut microbiome, which in turn may have yet to be understood implications for fish welfare and growth. Indeed, Gupta et al. [45] observed with prebiotic supplements containing alginate (extracted from Laminaria sp. of brown seaweed) corresponded to distinct microbiome changes in Atlantic salmon. Their study, however, did not comment on the associated health or growth impacts. Thus, a complementary approach combining fish growth, internal physiology, and the gut microbiome is more appropriate as microbiome changes alone are not indicative of condition or response to new feed supplements.
To further develop Atlantic cod aquaculture and to address the food sustainability agenda, this study aimed to evaluate if the diet of juvenile Atlantic cod could be supplemented with macroalgae and to determine the effects on fish growth, condition, gut morphology and gut microbiome structure. We focused on the hindgut as a primary site of fermentation and to maximise the potential for obtaining digesta. Two species of individual macroalgae dietary supplements were compared - egg wrack Ascophyllum nodosum (brown) and sea lettuce Ulva rigida (green), alongside a non-amended fishmeal diet. A. nodosum is a common algae species in northern temperate Atlantic waters and is harvested from the wild for commercial alginate (phycocolloid) production and farm animal feeds. U. rigida is ubiquitous and can be a nuisance species, proliferating in large quantities resulting in green tides. It is this significant algal biomass availability in the wild that would provide the upscale requirement in commercial aquafeed production [46]. Our hypothesis was that Atlantic cod growth and condition would not be negatively impacted by macroalgae inclusion, but that the addition of the macroalgae would lead to changes in the gut microbiome compared to the control diet. To the author’s knowledge, this is the first study to examine the impact of dietary macroalgae supplementation on the gut integrity and hindgut microbiome in juvenile Atlantic cod.
Results
Fish growth, condition and survival
The mean weight of Atlantic cod juveniles increased from 123 g at the start of the feeding trial to 236 ± 62 g (Control – CTRL), 243 ± 72 g (10% Ulva rigida – ULVA) and 190 ± 84 g (10% Ascophyllum nodosum – ASCO) at the end of the trial. An almost two-fold increase in weight was measured in the fish fed the ULVA and CTRL diets. The analysis showed there were significant (P = 0.024 0.05, F = 73.53, df = 2) differences between the dietary groups, with a Tukey test revealing ASCO fish weighed significantly less than CTRL and ULVA groups (P < 0.05). The ASCO treatment was not split into ASCO Normal (ASCO_N) and ASCO Lower Growth (ASCO_LG) for tank population data (Materials and Methods; Supplementary Information Figure S1). All tank populations had individual fish with condition factor of < 0.85. The prevalence of fish with condition factor < 0.85 in the ASCO treatment was 33%, as compared to just 3% in the CTRL and 8% in the ULVA treatment at Week 12. Correspondingly, the ASCO treatment had a greater percentage mortality rate at 21%, while the CTRL and ULVA treatments had values of 19 and 16%, respectively.
Within the fish sampled for microbiology and histology, statistically significant differences (Kruskal-Wallis; P < 0.05) were observed in total weight (g) and condition factor (K) of the ASCO_LG group as compared to the remaining treatments (Supplementary Figure S2a and b). At Week 8 within the microbiology samples, the ASCO_LG group had a condition factor of 0.77 ± 0.02 (SD), compared to 1.16 ± 0.14 (SD) in the ASCO_N group. In addition, there were no significant differences (P > 0.05) found between the weight or condition factors for the CTRL, ULVA or ASCO_N treatments (229–256 g fish− 1 and condition factor of 1.16–1.21). This trend continued in Week 12 with a further decrease in condition factor in the ASCO_LG group observed.
Intestinal morphology
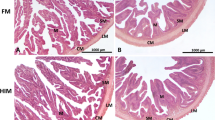
In the ULVA and CTRL groups, the microvilli were in a densely packed arrangement of uniform shape (Fig. 1). In contrast, both ASCO groupings (Normal and Lower Growth) had a more heterogeneous structural arrangement. Analysis of Week 8 data indicated that the hindgut of the ASCO_LG grouping had significantly reduced mucosal fold height (116 ± 42 μm, P < 0.0005) and significantly increased lamina propria width (106 ± 44 μm P < 0.01) compared to 220 ± 60 μm and 69 ± 28 μm in the CTRL for height and width, respectively (Table 2). It was noted upon dissection even after the starvation period that undigested material was contained in the digestive tract of fish within this group. There was no significant difference between the lamina propria width and mucosal fold height of the ULVA group compared to the CTRL group (P > 0.05 and P > 0.05, respectively). A similar trend was observed at Week 12, with ULVA and CTRL groups not being significantly different in terms of lamina propria width and mucosal fold height. Interestingly, the ULVA diet had increased microvilli density (AU) when compared to the CTRL, ASCO_N and ASCO_LG groupings (Fig. 1, Table 1). However, this relationship was only significant between the ASCO_N at Week 8 (P < 0.01) and ASCO_LG groups at both Week 8 (P < 0.01) and Week 12 (P < 0.05).

Scanning electron microscopy images of the hindgut of juvenile Atlantic cod (Gadus morhua) from each treatment (CTRL, ULVA, ASCO) at Week 8 and Week 12. Scale bar represents 1 μm
Hindgut microbial community composition
The hindgut of Atlantic cod (Gadus morhua) was characterised using amplicon sequencing of the 16S rRNA V4 region. The data presented here are based on the sequences from 67 samples obtained following quality filtering as described in the Methods section. A total of 12,655,273 reads were generated, resulting in 411,123 quality-filtered paired-end reads once singletons were removed, corresponded to 3612 unique OTUs at the 97% similarity level. A rarefaction curve is provided in Supplementary Figure S3. The taxonomically assigned data (> 97% similarity) showed that members of the Proteobacteria and Firmicutes phyla were dominant across all samples comprising as much as 98 and 90% relative abundance respectively (Fig. 2a). The phylum Bacteroidetes increased over time to as much as 80% relative abundance in the CTRL fish at Week 12. The most frequently observed OTUs (Fig. 2b) were OTU_307 and OTU_372, both Photobacterium spp. Over time OTUs identified as Bacteroidetes increased - OTU_9 [Bacteroides sp.] and OTU_20 [Alistipes sp.] appeared in the Top 25 phyla. The ASCO_LG samples at Week 12 (ASCO_LG 12) showed a high abundance of OTU_1968 [Psychromonas sp.]. A large proportion of the OTU-level taxonomically assigned data was assigned to the category ‘Others’, i.e. less abundant genera, as much as 99% in some Week 0 samples (Fig. 2b).

Horizontal bar-plot showing the relative abundance of the top 25 most abundant, a phyla, and b Operational taxonomic units (OTUs) in the hindgut of juvenile Atlantic cod ordered by the dietary groups over time. “Others” represents the remaining lesser abundant taxa
Diversity of the hindgut microbial communities
The alpha-diversity measures used in this study included the following a) species evenness - how similar in numbers each species is, b) species richness - the number of different species, and c) Shannon index - a measure of entropy i.e. the degree of uncertainty as a proxy for diversity. A significant decrease in all these tested measures was observed from Week 0 to Week 12, irrespective of dietary treatment (P < 0.05, Fig. 3a and Supplementary Information Figure S4). No significant difference was observed between Week 8 and Week 12 samples (excluding ASCO_LG samples). The ASCO_LG fish at Week 12 were less rich than those from the CTRL (P < 0.05), ULVA (P < 0.05) and ASCO_N (P < 0.001) individuals from the same time point (Fig. 3a).

a Alpha diversity measure: Species Richness with the corresponding colour legend for the treatment groups. Pair-wise ANOVA P-values are displayed P < 0.05*, P < 0.01**, P < 0.001***. b Non-metric distance scaling (NMDS) plot based on Unifrac phylogenetic distance with colour coded circles representing the differing treatment groups over time (R2 = 0.27182, P = 0.001)
We observed clear differentiation in beta-diversity between Week 0 samples and the CTRL and macroalgae dietary supplement groups at Week 8 and Week 12 (Fig. 3b). The microbial communities across all three treatments (CTRL, ULVA, ASCO_N, ASCO_LG) became increasingly similar over time – forming overlapping clusters (irrespective of treatment) by Week 12 (Fig. 3b). Dissimilarity observed could partly be explained by variation in OTUs of low abundance as weighted Unifrac (phylogenetic distance weighted by abundance counts) analysis showed all treatments and time points within overlapping clusters (Supplementary Information Figure S4). Despite showing a pattern of convergence over time treatment groups (Week 0, CTRL, ULVA, ASCO_N, ASCO_LG at Week 8 and Week 12) were significantly different in all beta-diversity non-metric multi-dimensional scaling plots (P < 0.001).
Temporal changes in the hindgut microbial community
The data above indicates that the hindgut microbial communities were strongly influenced by time, as opposed to treatment. Over time, the number of unique OTUs reduced from 151 at Week 0 to only 12 that were unique at Week 12 (Supplementary Information Figure S5). To identify what OTUs were changing over time ‘differential’ analysis was used. Discriminating OTUs between Week 0 and treatments (excluding ASCO_LG) at Week 8 and Week 12 (CTRL, ULVA and ASCO_N) were identified using Sparse Partial Least Squares Discriminant Analysis (sPLS-DA) and displayed in a heatmap (Supplementary Information S6). The microbial profile of Week 0 fish was distinct from the subsequent time points and the discriminating OTUs from Week 0, Week 8 and Week 12 were used to create differential trees to visually demonstrate the taxonomic changes over time (Fig. 4). The differential OTUs at Week 0 and Week 8 contain many Xanthomonadales, Rhodobacteriales, Desulfovibrionales, Mollicutes, Gemmatimonodales, Clostridaceae, and Bacilli taxonomic nodes. While Week 12 taxonomic nodes consisted of Ruminococcaceae, Lachnospiraceae, Christensenellaceae, Erysipelotrichia, and Bacteroidetes members.

Differential taxa in the hindgut microbiota of juvenile Atlantic cod (Gadus morhua). These taxa were first determined using sPLS-DA with the discriminating operational taxonomic units (OTUs) at > 1% relative abundance at Week 0, Week 8 and Week 12 (excluding ASCO_LG fish). The taxonomic map is a legend to show the identity of the differentiating taxa. This legend is read to inform the differential heat trees. The differential heat trees show the microbiota shifts from Week 0, Week 8, and Week 12. The branches where they are upregulated are coloured according to their respective categories shown for each tree (Week 0, Week 8, and Week 12). The nodes legend can be read as follows: Left side ‘Log2 ratio’ of the median proportions of the taxa corresponding to the colour scale, Right side ‘Number of OTUs’ where the diameter reflects the number of OTUs corresponding to the nodes on the heat trees
Influence of macroalgae supplement on the hindgut microbiome
Changes in diversity across treatments
The microbial diversity in the hindgut of juvenile cod decreased over time and the microbial communities became increasingly similar over time. Physiologically, however, significant changes were occurring with respect to the fish condition and the ASCO group were categorised into ASCO_N and ASCO_LG groupings from Week 8. Variances in beta-diversity within the groups was tested at Week 8 and Week 12 (beta.disper/PERMDISP), with no significant differences in variance observed across all dissimilarity matrices. This was with the exception of the ASCO_N samples which had less variance tested with Unifrac phylogenetic distance at Week 12 (CTRL; P = 0.019, ULVA; P = 0.47, ASCO_LG; P = 0.002). To understand if the fish condition could be linked to microbial community composition patterns Unifrac phylogenetic distance of OTUs was used and compared using Principal Coordinates Analysis (PCoA) plots containing contour maps with the condition factor values (Fig. 5). The culminative explained variance was as follows; Week 8 - Dim1 = 20.55% and Dim2 = 8.77%, Week 12 - Dim1 = 17.9% and Dim2 8.81%). At Week 8, ASCO_LG fish clustered around contour values of ~ 1. A clear distinction was not observed between treatment groups at this time point. ASCO_LG fish condition decreased from Week 8 to Week 12, and this corresponded with a response in the microbial community structure as evidenced by ASCO_LG individuals at Week 12 clustered around condition factor contours of values < 0.75 while the remaining groups (CTRL, ULVA and ASCO_N) clustered together with values generally above 1.

Unifrac unweighted principal co-ordinate analysis of the gut microbiota of juvenile Atlantic cod (Gadus morhua) community composition based on the different dietary treatments (CTRL, ULVA, ASCO_N and ASCO_LG) at a) Week 8, and b) Week 12. The values given on each axis (Dim1 or Dim2) indicate the total percentage variation for the treatment groups. Contour lines represent condition factor (K) fit through generalised additive modelling. Deviance in ordination space at Week 8 is explained by 24.5%; R2 = 0.18; * P = 0.043. Deviance in ordination space at Week 12 is explained by 57.8%; R2 = 0.5; *** P = 0.0002
Differential OTUs across treatments
sPLS-DA was used to discriminate the specific OTUs which were influenced by the dietary treatments and therefore determine the influence of macroalgae supplement on the hindgut microbiome. This was performed with the Week 0 samples removed and Weeks 8 and 12 were assessed separately to exclude any artefacts with respect to temporal sampling. The macroalgal treatments were compared to the corresponding CTRL group and the information displayed in a heatmap. Week 8 sPLS-DA is contained within Supplementary Information Figure S7. At Week 12, the ASCO_LG group formed a separate branch (Fig. 6), while the remaining treatments (CTRL, ULVA, ASCO_N) clustered together. The differential OTUs between treatments formed three large phylogenetic branches. The bottom branch contained OTUs (Oscillibacter, Erysipelotrichaceae and Rikenellaceae spp.) primarily downregulated in the ASCO_LG fish compared to the other diet treatments. The middle branch contained OTUs (Psychrosomonas spp., Propionigenium, Clostridium sensu stricto, and Rhodospirillaceae) primarily upregulated in the ASCO_LG fish compared to the other diets. The microbial profiles of the CTRL, ULVA, and ASCO_N groups showed a high degree of similarity within this plot.

sPLS-DA heatmap of the discriminant operational taxonomic units (OTUs) > 3.5% relative abundance between the CRTL, ULVA, ASCO_N and ASCO_LG groups of juvenile Atlantic cod (Gadus morhua) at Week 12. Rows and columns are ordered using hierarchical (average linkage) clustering to identify blocks of genera of interest. The heatmap depicts TSS + CLR (Total Sum Scaling followed by Centralised Log Ratio) normalised abundances: high abundance (orange/yellow) and low abundance (dark purple). Fish weight (g) and condition factor (K) is shown per grouping. sPLS-DA was fine-tuned using centroids.dist and 3 tuning components
Core microbiome of control diet versus Ulva rigida supplement
The data highlighted above shows that the differences over time were a stronger influence than treatment. The core microbiome (found in 85% of individuals) across diets and treatments consisted of just 3 OTUs – OTU 2746 [Vibrio sp.], OTU 307 and 2746 [Photobacterium spp.]. Given the palatability issues observed with the A. nodosum supplement, it is unlikely that this would be used in its current form as a feed supplement. As such, in the following section, the core microbiome was compared for the CTRL and ULVA fish only at Week 12 (Supplementary Information Figure S8). The core microbiome of the CTRL group consisted of 8 OTUs listed in order of abundance and prevalence (OTU 9 [Bacteroidetes sp.], OTU 2746 [Vibrio], OTU 307 [Photobacterium sp.], OTU 2624 [Lachnoclostridium sp.], OTU 3265 and 3130 [Photobacterium spp.] and OTU 982 and OTU 2550 [Macellibacteroides spp.]). The fish fed on the ULVA diet shared all OTUs within the CTRL group, except for OTU 3265 and OTU 3130, which were identified as Photobacterium spp. In addition, the ULVA group contained OTUs not found in the CTRL group. These were broadly identified as Ruminococcaceae spp., Shewanella, Tyzerella, Rikenellaceae spp., Peptococcaceae, Bacteroides, and Fusobacterium.
Discussion
In this work we evaluated the feasibility of supplementing juvenile Atlantic cod (G. morhua) diet with 10% macroalgae with specific consideration to the health of the intestine. To the author’s knowledge, few publications have considered the impact of macroalgal dietary supplementation on the gut physiology and microbiome in cod. Our research study aimed to address the following knowledge gap, examining two different macroalgal species: U. rigida and A. nodosum on fish condition and hindgut microbiome.
The data presented in this study indicated that a diet fed to juvenile Atlantic cod could be supplemented with 10% U. rigida without negative impacts on growth performance, gut physiology, or microbiome shifts as compared to the control treatment. This is in contrast, with reports from rainbow trout (O. mykiss) supplemented with a different Ulva species (U. lactuca) at 10% inclusion level had poorer growth compared to those fed control diets [47]. Differences in the response to specific macroalgae ingredients across fish species is unsurprising given the variation in multiple factors including water habitat type, gut morphology, digestive enzymes, and fish genetics. This highlights that extensive research is needed for individual fish species to ascertain the suitability of specific ingredients. Our positive result correlates with studies on European sea bass (D. labrax [48];) and Senegalese sole (Solea senegalensis [49];), indicating that U. rigida can be supplemented in the diets of a variety of finfish species. U. rigida is ubiquitous and can be a nuisance species, proliferating in large quantities resulting in green tides. It is this significant algal biomass availability in the wild that would provide the upscale requirement in commercial aquafeed production [46].
Based on the current study, A. nodosum was determined not to be a suitable dietary supplement for juvenile Atlantic cod at 10% inclusion levels. In a subset of the population, growth, fish condition, intestinal physiology and the gut microbiome were all impacted by this dietary inclusion. Individuals that tolerated the A. nodosum supplement (ASCO_N) had similar condition factors to those in the CTRL and ULVA dietary groups. There appeared to be two reasons for fish not tolerating the A. nodosum supplement: primarily – palatability, and to a lesser extent -digestibility. The ASCO diet was unpalatable to many fish within this treatment, evidenced by fish rejecting pellets (‘spit outs’) during a feeding event. Palatability issues have been documented in many fish species, including Atlantic cod fed a microalgal supplement [50]. Taste experiments with the common periwinkle have shown a preference for Ulva species over A. nodosum, even during starvation [51]. Cod is a benthic species, living close to the seafloor, and uses an enhanced chemoreceptor system with a large number of external cutaneous taste buds on areas like the pelvic fin to locate prey [52, 53]. Thus, they may have heightened sensitivity to stimuli or antinutritional factors (ANF). It is unknown why certain individuals within the ASCO treatment found the diet palatable whilst others did not. Though genetic variability may have been a factor. The Atlantic cod used in this study were the broodstock of wild cod from the Celtic Sea and thus, variation in the response to dietary inclusion may be attributed to genetic variation within the population. To draw a parallel with mammals, genetic polymorphisms in taste receptor genes (TRs) are attributed to variations in taste perception [54, 55]. While fish are more primitive, authors have attempted to find homologs of mammal TRs within fish. Notably taste receptor type 2 genes (T2R), which are responsible for bitter taste have been found in teleost fish such as the common carp (Cyprinus carpio [56]; and rainbow trout (Oncorhynchus mykiss [57]. Moreover, ANFs (e.g. phenolics, polysaccharides, and tannins) in the supplement ingredients can have a bitter taste [58]. These ANFs are also known to influence substrate digestibility by impeding digestive function [59]. During dissection, it was noted that the fish in the ASCO treatment had undigested material at the rear of the digestive tract. This was particularly prominent in the ASCO_LG fish. The ASCO_LG fish not surprisingly had significantly reduced weight and condition factor compared to the ULVA, CTRL and ASCO_N groups. These results correlate with work by Yone et al. [60] where 10% A. nodosum supplementation decreased the final weight and feed efficiency in red sea bream.
Gut morphology plays a key role in the digestive function and overall health of a fish species [61,62,63]. Absorption of nutrients is mediated by the function and morphology of the gut. The microvilli sit on a single layer of epithelial cells (enterocytes) which facilitates the transport of nutrients through the gut wall barrier. Healthy microvilli (long and densely packed) are proposed to be associated with improved nutrient uptake due to increased surface area for absorption [61, 62]. It is well established that aquafeed ingredients can positively or negatively impact intestinal morphology [64, 65]. Negative changes can include shortening of the microvilli, thickening of the laminar propria and possible invasion of inflammatory cells [66,67,68]. This is typically termed ‘enteritis’. Thus, changes in the cod hindgut microvilli density or mucosal-fold length in response to a change in diet could reveal potential changes in nutrient absorption. In this study, the microvilli density, mucosal fold height and lamina propria width in the ULVA group had higher mean values than all groupings (CTRL, ASCO_N and ASCO_LG), though a significant difference was only observed in the case of the ASCO fish. A similar finding was reported by Moutinho et al. [49] for Senegalese sole (S. senegalensis) showing a trend towards higher mucosal fold height but this was not significant when compared to the control. The ASCO_LG fish had an altered hindgut morphology. Thesealterations included, fatter laminar propria, decreased mucosal-fold height and a modest reduction in microvilli density. Thickening of the lamina propria is suggested to be due to the infiltration of inflammatory cells [69]. This may have increased the susceptibility of these fish to potential disease and predation by larger juveniles [70]. Though we observed no clinical signs of disease in any of the dietary groups.
In some aquafeed studies, supplementing the diet with plant ingredients (soybean, pea and canola) has resulted in negative impacts on fish gut morphology and reduced growth with combined disruption to the gut microbiota as observed in rainbow trout [71]. While in contrast, in a replacer study (where the complete protein source is replaced) no change in the gut microbiota was observed despite substantial changes in gut morphology (e.g. Atlantic salmon soybean replacer study [72]). Recent research on a variety of fish species has indicated that gut microbiota can be linked to digestion [35], immunity [73, 74], and disease prevention. Therefore, this is an important parameter to consider when determining the impacts of diet supplementation. We showed that macroalgae supplementation did not impact the hindgut microbiome. Rather, time was a greater driver with hindgut microbial communities converging to be more similar over time irrespective of treatment. Indeed, before the start of the experiment, the fish were fed the same diet (commercial fish pellet) and showed the greatest difference in community composition among individuals. Over the course of the experiment, as the juvenile cod grew, the microbial diversity in all treatments decreased and despite differences in the feed the microbial community structure converged among treatments over the 12-week period. These changes were accompanied by the differential increase and simultaneous decrease of a set of key OTUs (25 OTUs) over the course of the experiment. In fact, over the 12-week period the number of unique OTUs decreased from 152 at Week 0 to just 12 at Week 12. In terms of composition, the early hindgut microbiome was dominated by Proteobacteria and Firmicutes. The dominance of these phyla is not surprising as these bacteria are ubiquitous. In the context of the fish gut microbiome, Proteobacteria and Firmicutes have been found in high abundances in the gut of many omnivorous and carnivorous fish species. These have ranged from diverse environments [75] and coastal seawater samples [76]. The reason for their dominance has yet to be fully understood. However, over time, the relative abundance of Bacteroidetes members increased across all treatments, except for the ASCO_LG fish group. By week 12, Bacteroidetes was the dominant phyla present in the hindgut of the majority of fish. Bacteroidetes have been found in the hindgut of carps and other species [77]. Within gut environments Bacteroidetes can outcompete other phyla due to their metabolic flexibility and tractability in response to host pressures, e.g. the host immune system and gut environment pressures such as low pH [78]. Interestingly our work aligns with the finding by Xia et al. [40] where 12 days of starvation led to a shift towards Bacteroidetes members in the gut microbiome of Asian seabass (Lates calcarifer) [32]. The 24 h starvation periods for sampling within the experimental design may have been sufficient to provide a competitive advantage to the more flexible Bacteroidetes species. However, without a non-starved ‘control’ group this cannot be determined.
At week 0 and 8, we identified the discriminating taxa as Xanthomonadales, Rhodobacterales, Desulfovibrionales, Mollicutes, Gemmatimonadetes, Clostridiaceae, and Bacilli, which were overrepresented in terms of differential abundance within the hindgut microbial community. While, the presence of Xanthomonadales are less frequently found in the fish gut microbiota, they have been identified as bacterial symbionts [79] aiding the breakdown of complex hydrocarbon sources [80]. The group Rhodobacterales were postulated as a key bacterial species responsible for sea cucumber growth linked to polyhydroxybutyrate (energy storage molecules) metabolism [81]. Desulfovibrionales species are sulfate-reducers and produce hydrogen sulfide. They are commonly found in anoxic environments, their exact role and impact on fish health is unknown but within the human gut, they are linked with gut inflammation in disorders such as ulcerative colitis [82]. Mollicutes are an interesting group of bacteria, lacking a cell wall, and include Mycoplasmas which are well-known in the fish microbiome. Some studies have proposed that Mycoplasmas are commensal gut or even intra-host dependent microorganisms in Atlantic salmon [83, 84]. Though others have described Mycoplasmas as parasites [85]. Gemmatimonadetes are largely uncharacterised though have been found previously in the gut of sea bream (S. aurata) and sea bass (D. labrax), at low abundances [86]. Clostridiaceae (Clostridium sensu stricto) are common gut bacteria and are metabolically flexible, capable of metabolising a range of carbohydrates to produce alcohols and acids [87]. Bacilli are also common fish gut bacterial species [88] that can produce a range of hydrolytic enzymes [89]. Indeed, Bacilli are perceived as part of a healthy gut microbiome. Bacilli-based probiotics have been used as dietary supplements to fish and shellfish species [90]. The microbial composition is noted with the caveat that amplicon sequencing of the V4 region in particular does not yield species or strain level taxonomic resolution [91].
By Week 12, Ruminococcaceae, Lachnospiraceae, Christensenellaceae, Erysipelotrichia and Bacteroidetes members were the differentially abundant species from the former time points. Ruminococcaceae and Lachnospiraceae are commonly found genera, which together are responsible for protein break down and fermentation [92]. Christensenellaceae species are fermentative species and common in gut microbiome studies [93]. Erysipelotrichia and Bacteroidetes are saccharolytic species [92]. These patterns tend to indicate a change in microbial functioning within the gut and interactions over time towards fermentative activity and likely not a case of functional redundancy. In contrast, the ASCO_LG fish did not follow this pattern of gut microbiome development across week 8 and week 12 had upregulated Psychromonas, Propionigenium, Clostridium sensu stricto and Rhodospirillaceae species as compared to the remaining treatments. Psychromonas have been proposed to provide nutritional compensation for an unbalanced diet in deep-sea leeches (Piscicolidae), although this link remains to be further elucidated [94]. While, Propionigenium is known to degrade cellulose and complex carbohydrates, and are also shown to produce anti-inflammatory products as end products that could play a beneficial role in gut health [62]. Future work to understand the ecological assembly processes underpinning the microbial community development in juvenile Atlantic cod could shed valuable insights into the mechanisms responsible for the gut microbiota patterns. As has been observed in methods employed in previous gut microbiome studies [95, 96].
It has been evidenced in European seabass (D. labrax) that fish across different dietary regimes have a core gut microbial community that is stable over a six-week time period. While our results indicate that a core microbial community in Atlantic cod over a 12-week period consisted of just three OTUs – identified as Photobacterium spp. and a general Vibrio sp. Interestingly, the most frequently observed OTUs in the present study were identified as Photobacterium spp. a gamma proteobacteria within the Vibrionales order, widely occurring in the marine environment. These species have been found associated with numerous fish studies (for a review see [97]), and range from proposed pathogens [98] to proposed mutualistic species for the digestion of complex polysaccharides such as chitin [35]. The ubiquitous presence of Photobacterium spp. as part of the core microbiome suggests it may play a role in the breakdown of food and fermentation in the hindgut of juvenile Atlantic cod. The core microbiome of the ULVA treatment at Week 12 shared the same core community. These largely consisted of members of the bacterial orders Vibrionales (Vibrio and Photobacterium), Bacteroidales (Bacteroidetes and Macellibacteroides), and Clostridiales (Lachnoclostridium). This indicates this macroalgae supplement did not alter the core microbial community in juvenile Atlantic cod.
The overall temporal trend of converging microbial communities indicated no effect of dietary supplement (at 10% inclusion levels) on the hindgut microbiota of juvenile farmed Atlantic cod. There are no studies that consider the gut microbiome development in farmed Atlantic cod from larvae to maturation. Our work provides valuable insight into the development of the hindgut microbiome of farmed Atlantic cod in the juvenile life cycle stage. It has previously been shown in other fish, that the gut microbiota is highly malleable during the first-feeding and initial lifecycle period (e.g. rainbow trout [99]), and stabilises over time as we observed. The microbial associations observed need to be further characterised. Indeed, a major challenge in gut microbiome research is linking the microbiota to specific physiological and metabolic responses. In fish this includes the challenge of gut section sampling and obtaining a representative community from fully digested material. Promising developments such as the fish-gut-on-chip and SalmoSim gut model may offer opportunities to test the response of the fish gut microbiota to environmental parameters while studying the microbial populations in vitro [100, 101]. The logical progression of this work would be to undertake a full metabolomics evaluation to provide a more robust interpretation of the significance of changes in the context of the functionality and whole animal response to variations in diet and the environment.
Conclusions
Limiting the environmental impact of food production is paramount in ensuring future food security. We conclude that Ulva rigida species of macroalgae represent a low-cost, nutritious and palatable supplement to the diet of juvenile Atlantic cod (Gadus morhua). U. rigida can be supplemented up to 10% inclusion levels without negative impacts on the growth, gut morphology, or microbiome. We did not see any apparent increase in growth performance, though it should be noted that functional feed supplements and additives rarely manifest directly as having significant effects on improving fish growth rates under optimum rearing conditions. In contrast, we also determined that 10% dietary inclusion of Ascophyllum nodosum was not a suitable supplement for cod. This was evidenced by poor fish growth performance and appeared to negatively impact gut microbiome development and microbiome diversity. In the quest for economic and environmentally sustainable marine ingredients for aquafeeds, we advocate the potential for macroalgae (U. rigida in particular) to play a prominent role and support fish health and welfare as a sustainable feed ingredient.
Methods
Feed formulation and production
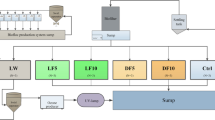
Macroalgae used in the feed trial were harvested from Muigh Inis, Co. Galway, Ireland (Ascophyllum nodosum) and Harbour View Bay, Co. Cork, Ireland (Ulva rigida, green tide, [46]). The biomass was washed with freshwater to remove debris and dried for 48 h at 40 °C in a dehumidifying cabinet. The resulting dried biomass was milled using a hammer mill (Timatic, Spello, Italy) and sieved to a particle size of < 0.8 mm. The proximate composition profile of the U. rigida biomass was 17.59 ± 0.09% protein, 0.48 ± 004% lipid, 22.53 ± 0.27% ash, and 10.08 ± 0.11 MJ kg− 1 energy. While, A. nodosum had 7.01 ± 0.10% protein, 0.94 ± 0.08% lipid, 24.83 ± 0.50% ash, and 11.77 ± 0.11 MJ kg− 1 energy. The inclusion of macroalgae into the test diets was predominately at the expense of fish meal and potato starch. Diets were formulated to be nutritionally balanced and were iso-nitrogenous (50%), iso-lipidic (20%) and iso-energetic (20 MJ kg− 1). Three diets (nominally CTRL, ULVA, ASCO, Table 2) were produced in-house through cold extrusion as described in Wan et al. [16]. Briefly, the formulations were passed through a single screw extruder using a 2 mm die and were subsequently dried at 40 °C. Proximate composition of the finished diets was analysed to confirm diet quality [102]. Quantification of protein was carried out by Kjeldahl procedure (DT220 and Kjeltec 8200, Foss A/S, Hillerød, Denmark) using × 6.25 conversion factor and lipid level was determined through Soxhlet extraction using petroleum ether (ST243, Foss A/S, Hillerød, Denmark). Ash content was measured through incineration of samples at 550 °C for 16 h. In addition, energy determination was carried out through bomb calorimetry (6200, Parr Instruments, Moline, Illinois, USA).
Experimental fish, design and fish physical condition
A total of 540 juvenile hatchery-reared Atlantic cod (G. morhua, the first-generation offspring of wild Celtic sea broodstock sourced in 2013) were employed in the feed trial (366 days post hatch). The stock fish of juvenile Atlantic cod (Gadus morhua) used were sampled prior to starting the experiment (Week 0). These fish had been fed a commercial fish pellet diet (Amber Neptun for marine fish, Skretting, Stavanger, Norway). Fish were hand-graded (123 ± 1 g) and randomly assigned to one of nine tanks (1200 L, n = 60 per tank, 3 replicate tanks per diet). The research tanks were fed with flow-through filtered ambient seawater (13.0 ± 1.5 °C) and were aerated to maintain a dissolved oxygen level of > 6 mg L− 1. A chiller problem was observed in Week’s 10 and 11 whereby the ambient tank temperature observed was 16 °C. A photoperiod of 8:16 light:dark was employed throughout the experiment.
Fish were acclimated for 1 week during which all tanks were fed a control diet (Table 1; CTRL) before starting the experimental diets. Following this acclimation period, each tank was randomly assigned to one of three diets: Control (CTRL), 10% supplement of Ulva rigida (ULVA) and 10% supplement of Ascophyllum nodosum (ASCO). Fish were hand-fed three times daily (evenly spaced throughout the day). The feeding rate was ~ 1% of the total tank body weight and was adjusted every 2 weeks to account for mortality, temperature and growth using standard cod growth models [103, 104]. Briefly, as is standard practice, fish were starved for 24 h before sampling, for total body weight (g) and total length (cm), of all individuals per experimental tank to calculate tank population growth rate. Fish were sampled for tank population data once monthly from the start of the acclimation period (i.e. from Week 0). Any mortalities were removed daily and recorded.
From Week 8 on, it was noted via visual distinction (Supplementary Information: Figure S1) that there were two subsets of fish within the A. nodosum supplemented diet: those that found the diet palatable and those that did not. This was confirmed by the presence of uneaten feed pellets in ASCO tanks following feeding events (not visible in tanks fed with CTRL or ULVA diets). Individuals that found the diet unpalatable had a markedly reduced size. These groups were referred to as ASCO_N (N – Normal) and ASCO_LG (LG – Lower Growth).
Sample collection
For both histology and microbiome analysis, sacrificial sampling was necessary. Fish were humanely euthanised using an overdose of tricaine methanesulphonate solution (MS222, Pharmaq, Overhalla, Norway), which was followed by the destruction of the brain to confirm death (EU Directive 2010/63/EU). The number of fish used for sacrificial sampling was kept to a minimum while remaining statistically sound. Separate individuals were necessary for histological and hindgut microbiome measurements as the former required a 24 h starvation period while the latter required gut content. For all fish sampled, the total weight (g) and total length (cm) of each fish were recorded.
Extra samples were taken from the ASCO treatment during the Week 8 and 12 sampling periods, with individuals classified as ‘ASCO_N’ or ‘ASCO_LG’ based initially on a visual assessment (i.e. slimmer body shape) and confirmed by calculation of condition factor. For histology analysis, three fish were taken per treatment (CTRL, ULVA, ASCO_N and ASCO_LG) at Week 8 and Week 12. For microbiome analysis, eight fish were taken at Week 0, 28 fish at Week 8 (6 CTRL, 9 ULVA, 8 ASCO_N, 5 ASCO_LG) and 31 fish at Week 12 (8 CTRL, 8 ULVA, 8 ASCO_N, 7 ASCO_LG). Variability in microbiome sample numbers was due to some fish not having sufficient gut content for analysis, resulting in additional fish being sampled.
Gut morphology analysis
Tissue samples for light microscopy (LM) and scanning electron microscopy (SEM) were taken in Weeks 8 and 12. Whole intestines (from the stomach to the anus) were dissected from each fish and the gastrointestinal (GI) tract unfolded and removed. The GI tract was subsequently flushed with phosphate buffer saline (PBS, pH 7.3) to remove gut digesta and mucosal layer. The total length of the intestine was measured and to standardise sampling, the hindgut was defined as the last 10–15% of the total gastrointestinal tract and was dissected. The hindgut was subsequently fixed in 10% neutral buffered marine formalin at room temperature for 24 h. Samples were subjected to serial dehydration steps in alcohol (100, 90, 70, 50, and 30%) before equilibration in xylene in a tissue processor. Samples were then embedded in paraffin wax according to standard histological procedures [105]. Transverse sections (6 μm) were sectioned and mounted on silane-covered (TESPA, 3-aminopropyl-triethoxysilane, Sigma-Aldrich, St. Louis, Missouri, USA) glass slides. Slides were stained with haematoxylin and eosin and mounted for light microscopy analysis. Slides were examined by light microscopy using an Olympus Vanox-T microscope and images were taken with a digital camera (Olympus camedia C-2020 Z).
For scanning electron microscopy analysis (SEM), hindgut samples were dipped in 1% 5-carboxymethyl-L-cysteine (Sigma-Aldrich, St. Louis, Missouri, USA) PBS solution for 30 s to remove surface mucus and digesta. Samples were fixed in 2.5% glutaraldehyde (pH 7.2) for 24 h and were dehydrated gradually in serial dilutions of 30, 50, 70, 90 and 100% ethanol for critical point drying (K850, Emitech Ltd., Ashford, Kent, UK). The resulting samples were mounted on stubs with the use of agar silver paint and gold sputter coated (K550, Emitech Ltd., Ashford, Kent, UK). Images of the microvilli density were taken using a low vacuum scanning electron microscope (5600, Jeol, Tokyo, Japan).
Gut microbiome analysis
Collection of hindgut content
The undersides of the fish were swabbed with ethanol before dissection and removal of the gut. The length of the digestive tract was recorded (cm). The hindgut was defined as the last 10–15% of the total gastrointestinal tract and was dissected. The gut contents of the hindgut were collected by squeezing the contents into a sterile tube and the weight of the material was recorded. The tubes were then placed on dry ice, transported and stored at − 80 °C for subsequent molecular analysis.
Sample DNA extraction and 16S rRNA amplicon sequencing
DNA extractions were carried out as described using a modified Griffiths et al procedure [106, 107]. Briefly, 250 μL TE buffer was added to the contents of the hindgut (digesta) while still frozen, mixed, and added to Lysing Matrix E tubes (MP Biomedical, Illkirch-Graffenstaden, France) containing 500 μL cetyl trimethylammonium bromide (CTAB) buffer and 250 μL of phenol-chloroform-isoamyl alcohol (25:24:1; pH 8). The mixture containing 0.25 g of digesta content was lysed by bead beating for 10 min at 3.2 K x g in a Vortex-Genie2™ (Scientific Industries Inc. Bohemia, New York, USA) and phase separation achieved by centrifugation at 13.3 K x g for 10 min. The remaining steps were followed as outlined in [107]. DNA plus a negative extraction control (nuclease-free water Qiagen, Venlo, The Netherlands) were sent to the Research Technology Support Facility at Michigan State University (Michigan, USA), for amplicon sequencing of the 16S rRNA gene targeting the V4 hypervariable region using the universal primer set (515f/806r [108];) with indexed fusion primers to generate amplicons compatible for multiplexed Illumina sequencing. This primer set was chosen in order to simultaneously detect bacterial and archaeal sequences. PCR products were normalised using an Invitrogen SequalPrep DNA normalisation plate and the normalised products per individual sample were then pooled to create an equimolar 16S V4 library. This library was quality checked using the Qubit dsDNA assay (Life Technologies, Darmstadt, Germany), Caliper LabChipGX (Caliper Life Sciences, Inc. Waltham, Massachusetts, USA) and Kapa Biosystems qPCR assay (Sigma-Aldrich, St. Louis, Missouri, USA). The library was then sequenced by Illumina Miseq at Michigan State University (Michigan, USA) using a standard flow cell and 500 cycle v2 reagent cartridge (Illumina Inc., Hayward, California, USA).
Sequencing analysis
Raw sequences were submitted to the SRA database under Bioproject Submission PRJNA636649. A total number of 12,655,273 reads were obtained from 68 samples. The subsequent paired-end reads were demultiplexed and converted to FastQ format sequence files for further analysis outlined below. The sequence reads were filtered using Sickle (v1.2 [109];) by applying a sliding window approach and trimming regions where the average base quality drops below 20. Pandaseq (v 2.4 [110];) was used with a minimum overlap of 20 bp to assemble the forward and reverse reads into a single sequence spanning the entire V4 region [110]. After obtaining the consensus sequences from each sample, UPARSE (v7.0.1001 [111];) was used for operational taxonomic unit (OTU) construction. The sequencing analysis protocol can be accessed as outlined in the data availability section. The approach was as follows: reads from different samples were pooled together and barcodes were added to keep an account of sample origin. Reads were then de-replicated (output - 3,264,891 reads) and sorted by decreasing abundance and singletons were discarded (output - 411,123 reads). Reads were then clustered based on 97% similarity, which followed by de novo chimera removal from most abundant sequences (output - 3894 reads). Additionally, a reference-based chimera filtering step using a gold database [112] was employed to remove chimeras that may have been missed in the previous step (output - 3612 reads). This left 3612 clean operational taxonomic units (OTUs). The assign_taxonomy.py script from the Qiime workflow [113] was used to taxonomically classify the representative OTUs against the SILVA SSU Ref NR database release (v123 [114];). These taxonomic assignments were then integrated with the abundance table using the make_otu_table.py function from the Qiime workflow to produce a biom file. To find the phylogenetic distances between OTUs, the OTUs were multisequence aligned against each other using mafft [115] and then FastTree (v2.1.7 [116]) was used on these alignments to generate an approximate maximum-likelihood phylogenetic tree in NEWICK format.
Data analyses
Calculation of fish condition
Fultons Condition Factor (K) growth parameter was calculated from the equations by recording fish length (cm) and fish body weight (g). This was carried out for a) tank populations, b) gut morphology sampled fish and c) gut microbiota sampled fish.
Microscopy analysis
ImageJ software (version 1.46, [117] was used to measure the following light microscopy gut parameters (μm): (1) mucosal-fold height (measured from the tip to the base of the mucosal fold) and (2) lamina propria width. The microvilli (MV) density measurements taken using SEM were processed by initially cropping images to a standard size and transforming to the 8-bit image. Using the threshold function, an arbitrary value (arbitrary units, AU) for the ratio of white (MV) to black (spaces between the MV) was calculated for each image [105].
Statistical analysis for growth and histology
Statistical analysis on the experimental trial growth data for tank populations was carried out using one-way ANOVA with a posthoc Tukey test to discern statistical differences. For the histology data (mucosal fold length, microvilli density and lamina propria width) was compared using the non-parametric Kruskal-Wallis test within Prism software (v6.03, GraphPad, San Diego, California, USA). This compared the mean rank of each treatment with the mean rank of every other treatment. A significance value (P) of 0.05 or under was used unless otherwise stated. This methodology was also used to determine the statistical differences in sampled fish condition (K), fish weight (g) and fish length (cm) for both gut morphology and gut microbiota sampled fish (tested separately – Figure S2).
Microbiome 16S rRNA sequencing statistical analysis
The microbial community of the hindgut of samples across treatments was characterised to assess whether treatment impacted the microbial composition of the gut. Samples were grouped into defining categories bases on treatment and/or time (Week 0, CTRL_8, ULVA_8, ASCO_N_8, ASCO_LG_8, CTRL_12, ULVA_12, ASCO_N_12, ASCO_LG_12). These groupings were then used to investigate changes in the microbiota with respect to treatment and time. Microbiome statistical analyses were carried out via the software R (v 3.4.2 [118]) with the OTU biom file, phylogenetic tree and the associated metadata files for the study. Spurious OTUs (not a bacterial or archaeal species) and OTUs found in the negative control were removed from all samples before statistical analyses.
Alpha and Beta-diversity statistical analyses
were carried out using R’s ‘Vegan’ package [119]. The following alpha diversity measures were then calculated - Pielou’s evenness (how equal in number the relative OTUs are); Richness (the number of unique OTUs) and Shannon’s index of entropy (the degree of uncertainty within a grouping assuming that a high degree of uncertainty corresponds to a highly diverse sample [120]). These were calculated on rarefied microbiome data (Supplementary Figure S3). Pair-wise ANOVA P-values were drawn on top of alpha diversity figures. Beta-diversity measures were visualised using non-metric multidimensional scaling (NMDS) plots using standard dissimilarity distance measures: Bray-Curtis and Unifrac (Unweighted and Weighted). Unifrac distances were calculated using the R package ‘Phyloseq’ [121]. Samples were grouped for different treatments as well as the mean ordination value and spread of points (ellipses were drawn using Vegan’s ordiellipse() function that represents the 95% confidence interval of the standard errors). R’s ‘Vegan’ package and ordisurf() function was used to determine if the main pattern in species composition described in principal co-ordinate analysis (PCOA) could be explained by the variable of fish condition factor (K). Ordisurf() internally fits a generalised additive model “gam” smoothing with formula K ~ s (PCoA1, PCoA2).
Characterising the microbial profiles across treatment groups and over time
In order to interpret this complex high-throughput dataset we used R’s mixOmics package [122] and Sparse Projection to Latent Structure – Discriminant Analysis (sPLS-DA) to select the most important and distinguishing OTUs between the treatments and over time. Analysis was performed at genus level with genera > 3.5% abundance and with TSS + CLR (Total Sum Scaling followed by Centralised Log Ratio) normalisation applied. OTU feature selection and multiple data integration was achieved by constructing artificial latent components (inferred variables) of the OTU table by first factorizing the abundance table and response variable (that contains categorical information of samples) matrices into scores and loading vectors in a new space such that the covariance between the scores of these matrices are maximised. In doing so, the algorithm also enforces a constraint on the loading vector associated with the abundance table (that has corresponding weight for each feature) such that some components of the loading vector go to zero and only significant features in terms of their discriminatory power remain. In this study, sPLS-DA was applied to determine the effect of ‘Treatment’ and ‘Time’. ‘Time’ contained Week 0 and all treatments except the ASCO_LG fish (i.e. CTRL, ULVA, ASCO_N) and longitudinal information (Week 0, Week 8, Week 12). ‘Treatment’ examines the independent treatments including all groups (CTRL, ULVA, ASCO_N and ASCO_LG) at either Week 8 or Week 12. The number of latent components and the number of discriminants were calculated. In finding the number of discriminants the model was fine-tuned using leave-one-out cross-validation and subsequent balanced error rates which accounts for sample number differences (fine-tuning parameters indicated in figure legends for Fig. 6 and Figure S5). The final output was a heatmap containing the differential genera over the number of tuned components which is discussed in the Results section. Further description of the implementation of sPLS-DA for microbial data-sets can be found in Gauchotte-Lindsay et al. [123]. The OTUs identified in sPLS-DA were subsequently used in differential tree analysis to show the taxonomic shifts in microbial community structure occurring over time. The complete methodology is outlined in [124] and uses the DESeqDataSetFromMatric() function from the DESeq2 package [125] Metacoder package to generate the tree visualisations [126] in R.
Finding a core microbiome and core microbiome per treatment
We followed a method for identifying the core microbiome as outlined McKenna et al. [124], and Shetty et al. [127] using R’s microbiome package [128] adjustable parameters for percentage relative abundance and percentage prevalence within samples. For the samples in this study, the core microbiome was defined as prevalent in 85% of samples [129].
Availability of data and materials
Raw sequences have been deposited in the NCBI sequence read archive (SRA) under bioproject number PRJNA636649. Sequencing analysis details are described in the Materials and Methods section and further scripts can be found at https://github.com/umerijaz/microbiomeSeq.
Abbreviations
- ICES:
-
International Council for the Exploration of the Sea
- CTRL:
-
Control diet treatment
- ULVA:
-
Ulva rigida supplement diet treatment
- ASCO:
-
Ascophyllum nodosum supplement diet treatment
- ASCO_N:
-
Ascophyllum nodosum supplement diet treatment – Normal growth
- ASCO_LG:
-
Ascophyllum nodosum supplement diet treatment – Lower growth
- LM:
-
Light Microscopy
- SEM:
-
Scanning electron microscopy (SEM)
- MV:
-
Microvilli
- AU:
-
Arbitrary units
- PBS:
-
Phosphate buffer saline
- CTAB:
-
Cetyl trimethylammonium bromide
- DNA:
-
Deoxyribonucleic acid
- RNA:
-
Ribonucleic Acid
- PCR:
-
Polymerase chain reaction
- OTU:
-
Operational Taxonomic Unit
- K:
-
Fulton’s condition factor
- ANOVA:
-
Analysis of Variance
- NMDS:
-
Non-metric multidimensional scaling
- PCoA:
-
Principal Coordinates Analysis
- sPLS-DA:
-
Sparse Projection to Latent Structure – Discriminant Analysis
- TSS + CLR:
-
Total Sum Scaling followed by Centralised Log Ratio
- ANF:
-
Antinutritional Factor
References
Allen M, Antwi-Agyei P, Aragon-Durand F, Babiker M, Bertoldi P, Bind M, et al. Technical Summary: Global warming of 1.5°C. An IPCC Special Report on the impacts of global warming of 1.5°C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty: Intergovernmental Panel on Climate Change; 2019.
Springmann M, Wiebe K, Mason-D’Croz D, Sulser TB, Rayner M, Scarborough P. Health and nutritional aspects of sustainable diet strategies and their association with environmental impacts: a global modelling analysis with country-level detail. Lancet Planet Health. 2018;2(10):e451–61.
FAO. The state of world fisheries and aquaculture 2020: In brief Sustainability in action Rome; 2020. https://doi.org/10.4060/ca9229en.
FAO. The State of World Fisheries and Aquaculture 2018 - Meeting the sustainable development goals. Rome: Licence: CC BY-NC-SA 3.0 IGO; 2018.
Edgar GJ, Ward TJ, Stuart-Smith RD. Rapid declines across Australian fishery stocks indicate global sustainability targets will not be achieved without an expanded network of “no-fishing” reserves. Aquat Conserv Mar Freshwat Ecosyst. 2018;28(6):1337–50 https://doi.org/10.1002/aqc.2934.
Kendall NW, Marston GW, Klungle MM. Declining patterns of Pacific northwest steelhead trout (Oncorhynchus mykiss) adult abundance and smolt survival in the ocean. Can J Fish Aquat Sci. 2017; https://doi.org/10.1139/cjfas-2016-0486.
Jaffa M. Concurrent Collapses of Demersal Fish and Sea Trout (Salmo trutta) on Scotland’s West Coast following the Removal of the “Three-Mile Fishing Limit”; 2019.
Palomares ML, Froese R, Derrick B, Meeuwig JJ, Nöel SL, Tsui G, Woroniak J, Zeller D, Pauly D. Fishery biomass trends of exploited fish populations in marine ecoregions, climatic zones and ocean basins. Estuar Coast Shelf Sci. 2020;243:106896 https://doi.org/10.1016/j.ecss.2020.106896.
Kraak SBM, Bailey N, Cardinale M, Darby C, De Oliveira JAA, Eero M, et al. Lessons for fisheries management from the EU cod recovery plan. Mar Policy. 2013;37:200–13.
ICES. Cod (Gadus morhua) in subarea 4, division 7.D, and subdivision 20 (North Sea, eastern English Channel, Skagerrak). In report of the ICES advisory committee, 2019. ICES advice 2019, cod.27.47d20. 2019a.
ICES. Cod (Gadus morhua) in divisions 7.E–k (western English Channel and southern Celtic seas) in report of the ICES advisory committee, 2019. ICES advice 2019, cod.27.7e–k. 2019b.
ICES. Cod (Gadus morhua) in division 7.A (Irish Sea). In report of the ICES advisory committee, 2019. ICES advice 2019, cod.27.7a. 2019c.
ICES. Cod (Gadus morhua) in division 6.A (west of Scotland). In report of the ICES advisory committee, 2019. ICES advice 2019, cod.27.6a. 2019d.
Cook RM, Holmes SJ, Fryer RJ. Grey seal predation impairs recovery of an over-exploited fish stock. J Appl Ecol. 2015;52(4):969–79.
Thurstan RH, Hawkins JP, Roberts CM. Origins of the bottom trawling controversy in the British Isles: 19th century witness testimonies reveal evidence of early fishery declines. Fish Fish. 2014;15(3):506–22.
Wright PJ, Pinnegar JK, Fox C. Impacts of climate change on fish, relevant to the coastal and marine environment around the UK. MCCIP Sci Rev. 2020;2020:354–81.
Kjesbu OS, Taranger GL, Trippel EA. Gadoid mariculture: development and future challenges. ICES J Mar Sci. 2006;63(2):187–91.
Korsøen ØJ, Dempster T, Fosseidengen JE, Karlsen Ø, Oppedal F, Stien LH, et al. Towards cod without spawning: artificial continuous light in submerged sea-cages maintains growth and delays sexual maturation for farmed Atlantic cod Gadus morhua. Aqua Environ Inter. 2013;3(3):245–55.
Lanes CFC, Bizuayehu TT, Bolla S, Martins C, de Oliveira Fernandes JM, Bianchini A, et al. Biochemical composition and performance of Atlantic cod (Gadus morhua L.) eggs and larvae obtained from farmed and wild broodstocks. Aquaculture. 2012;324–325:267–75.
Pauly D, Zeller D. Comments on FAOs state of world fisheries and aquaculture (SOFIA 2016). Mar Policy. 2017;77:176–81 https://doi.org/10.1016/j.marpol.2017.01.006.
Wan AHL, Soler-Vila A, O’Keeffe D, Casburn P, Fitzgerald R, Johnson MP. The inclusion of Palmaria palmata macroalgae in Atlantic salmon (Salmo salar) diets: effects on growth, haematology, immunity and liver function. J Appl Phycol. 2016;28(5):3091–100.
Soler-Vila A, Coughlan S, Guiry MD, Kraan S. The red alga Porphyra dioica as a fish-feed ingredient for rainbow trout (Oncorhynchus mykiss): effects on growth, feed efficiency, and carcass composition. J Appl Phycol. 2009;21(5):617–24.
Wassef EA, El-Sayed A-FM, Sakr EM. Pterocladia (Rhodophyta) and Ulva (Chlorophyta) as feed supplements for European seabass, Dicentrarchus labrax L., fry. J Appl Phycol. 2013;25(5):1369–76.
Diler I, Tekinay AA, Guroy D, Guroy BK, Soyuturk M. Effects of Ulva rigida on the growth, feed intake and body composition of common carp, Cyprinus carpio. J Biol Sci. 2007;7(2):305–8.
Azaza MS, Mensi F, Ksouri J, Dhraief MN, Brini B, Abdelmouleh A, et al. Growth of Nile tilapia (Oreochromis niloticus L.) fed with diets containing graded levels of green algae ulva meal (Ulva rigida) reared in geothermal waters of southern Tunisia. J Appl Ichthyol. 2008;24(2):202–7.
Pereira R, Valente LMP, Sousa-Pinto I, Rema P. Apparent nutrient digestibility of seaweeds by rainbow trout (Oncorhynchus mykiss) and Nile tilapia (Oreochromis niloticus). Algal Res. 2012;1(1):77–82.
Walker AB, Fournier HR, Neefus CD, Nardi GC, Berlinsky DL. Partial replacement of fish meal with laver Porphyra spp. in diets for Atlantic cod. N Am J Aquac. 2009;71(1):39–45.
Wan AHL, Davies SJ, Soler-Vila A, Fitzgerald R, Johnson MP. Macroalgae as a sustainable aquafeed ingredient. Rev Aquac. 2019;11(3):458–92.
Valente LMP, Rema P, Ferraro V, Pintado M, Sousa-Pinto I, Cunha LM, et al. Iodine enrichment of rainbow trout flesh by dietary supplementation with the red seaweed Gracilaria vermiculophylla. Aquaculture. 2015;446:132–9.
Wilson JM, Castro LFC. Morphological diversity of the gastrointestinal tract in fishes. In: Grosell M, Farrell AP, Brauner CJ, editors. Fish Physiology: The multifunctional gut of fish, vol. 30: Academic Press; 2010. p. 1–55.
Sandri M, Dal Monego S, Conte G, Sgorlon S, Stefanon B. Raw meat based diet influences faecal microbiome and end products of fermentation in healthy dogs. BMC Vet Res. 2016;13(1):65.
Lecomte V, Kaakoush NO, Maloney CA, Raipuria M, Huinao KD, Mitchell HM, Morris MJ. Changes in gut microbiota in rats fed a high fat diet correlate with obesity-associated metabolic parameters. PLoS One. 2015;10(5):e0126931.
Dvergedal H, Sandve SR, Angell IL, Klemetsdal G, Rudi K. Association of gut microbiota with metabolism in juvenile Atlantic Salmon. bioRxiv. 2020:2020.02.10.941245 https://doi.org/10.1101/2020.02.10.941245.
Webster TMU, Consuegra S, Hitchings M, de Leaniz CG. Interpopulation Variation in the Atlantic Salmon Microbiome Reflects Environmental and Genetic Diversity. Appl Environ Microbiol. 2018;84(16) https://doi.org/10.1128/AEM.00691-18.
Ray AK, Ghosh K, Ringø E. Enzyme-producing bacteria isolated from fish gut: a review. Aquac Nutr. 2012;18(5):465–92 https://doi.org/10.1111/j.1365-2095.2012.00943.x.
Seppola M, Olsen RE, Sandaker E, Kanapathippillai P, Holzapfel W, Ringø E. Random amplification of polymorphic DNA (RAPD) typing of carnobacteria isolated from hindgut chamber and large intestine of Atlantic cod (Gadus morhua l.). Syst Appl Microbiol. 2006;29(2):131–7 https://doi.org/10.1016/j.syapm.2005.07.006.
Zhang H, Jackson TA. Autochthonous bacterial flora indicated by PCR-DGGE of 16S rRNA gene fragments from the alimentary tract of Costelytra zealandica (Coleoptera: Scarabaeidae). J Appl Microbiol. 2008;105(5):1277–85 https://doi.org/10.1111/j.1365-2672.2008.03867.x.
Perry WB, Lindsay E, Payne CJ, Brodie C, Kazlauskaite R. The role of the gut microbiome in sustainable teleost aquaculture. Proc R Soc B Biol Sci. 2020;287(1926):20200184 https://doi.org/10.1098/rspb.2020.0184.
Li T, Long M, Ji C, Shen Z, Gatesoupe FJ, Zhang X, Zhang Q, Zhang L, Zhao Y, Liu X, Li A. Alterations of the gut microbiome of largemouth bronze gudgeon (Coreius guichenoti) suffering from furunculosis. Sci Rep. 2016;6(1):1–9.
Xia JH, Lin G, Fu GH, Wan ZY, Lee M, Wang L, Liu XJ, Yue GH. The intestinal microbiome of fish under starvation. BMC Genomics. 2014;15(1):266.
Egerton S, Wan A, Murphy K, Collins F, Ahern G, Sugrue I, Busca K, Egan F, Muller N, Whooley J, McGinnity P. Replacing fishmeal with plant protein in Atlantic salmon ( Salmo salar ) diets by supplementation with fish protein hydrolysate. Sci Rep. 2020;10(1):Art. no. 1 https://doi.org/10.1038/s41598-020-60325-7.S.
Hernández-Garibay E, Zertuche-González JA, Pacheco-Ruíz I. Isolation and chemical characterization of algal polysaccharides from the green seaweed Ulva clathrata (Roth) C. Agardh J Appl Phycol. 2011;23(3):537–42.
Sakthivel M, Deivasigamani B, Rajasekar T, Kumaran S, Alagappan KM. Immunostimulatory effects of polysaccharide compound from seaweed Kappaphycus alvarezii on Asian seabass (Lates calcarifer) and it’s resistance against Vibrio parahaemolyticus. J Mar Biol Oceanogr. 2015;4:2.
Liu H, Guo X, Gooneratne R, Lai R, Zeng C, Zhan F, et al. The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci Rep. 2016;6(1):24340.
Gupta S, Jep L, Abdelhafiz YA, Siriyappagouder P, Pierre R, Sørensen M, Fernandes JM, Kiron V. Macroalga-derived alginate oligosaccharide alters intestinal bacteria of Atlantic salmon. Front Microbiol. 2019;10:2037.
Wan AHL, Wilkes RJ, Heesch S, Bermejo R, Johnson MP, Morrison L. Assessment and characterisation of Ireland’s Green tides (Ulva species). PLoS One. 2017;12(1):e0169049.
Yıldırım Ö, Ergün S, Yaman S, Türker A. Effects of two seaweeds (Ulva lactuca and Enteromorpha linza) as a feed additive in diets on growth performance, feed utilization, and body composition of rainbow trout (Oncorhynchus mykiss). Kafkas Üniv Vet Fakült Dergisi. 2009;15(3):455–60.
Valente LMP, Gouveia A, Rema P, Matos J, Gomes EF, Pinto IS. Evaluation of three seaweeds Gracilaria bursa-pastoris, Ulva rigida and Gracilaria cornea as dietary ingredients in European sea bass (Dicentrarchus labrax) juveniles. Aquaculture. 2006;252(1):85–91.
Moutinho S, Linares F, Rodríguez JL, Sousa V, Valente LMP. Inclusion of 10% seaweed meal in diets for juvenile and on-growing life stages of Senegalese sole (Solea senegalensis). J Appl Phycol. 2018;30(6):3589–601.
Walker AB, Berlinsky DL. Effects of partial replacement of fish meal protein by microalgae on growth, feed intake, and body composition of Atlantic cod. N Am J Aquac. 2011;73(1):76–83.
Watson DC, Norton TA. Dietary preferences of the common periwinkle, Littorinalittorea (L.). J Exp Mar Biol Ecol. 1985;88(3):193–211.
Kasumyan AO, Doving KB. Taste preferences in fishes. Fish Fish. 2003;4(4):289–347.
Harvey R, Batty RS. Cutaneous taste buds in gadoid fishes. J Fish Biol. 2002;60(3):583–92.
Garneau NL, Nuessle TM, Sloan MM, Santorico SA, Coughlin BC, Hayes JE. Crowdsourcing taste research: genetic and phenotypic predictors of bitter taste perception as a model. Front Integr Neurosci. 2014;8:33 https://doi.org/10.3389/fnint.2014.00033.
Meyerhof W. In: Korsching S, editor. Chemosensory systems in mammals, fishes, and insects. Heidelberg: Springer; 2009.
Kong S, Dong C, Lv H, Chen L, Zhang J, Pu F, Li X, Xu P. Genome wide identification of taste receptor genes in common carp (Cyprinus carpio) and phylogenetic analysis in teleost. Gene. 2018;678:65–72 https://doi.org/10.1016/j.gene.2018.07.078.
Liu X, Yu Y, Qin D, Song Z, Huang Z, Meng K, Cao J, Xu F, Cheng G, Ji W, Xu Z. Expression analysis of taste receptor genes (T1R1, T1R3, and T2R4) in response to bacterial, viral and parasitic infection in rainbow trout, Oncorhynchus mykiss. Fish Shellfish Immunol. 2020; https://doi.org/10.1016/j.fsi.2020.03.055.
Sørensen M, Berge GM, Thomassen MS, Ruyter B, Hatlen B, Ytrestøyl T, Aas TS, Åsgård TE. Today’s and tomorrow’s feed ingredients in Norwegian aquaculture. Nofima Rapportserie. 2011.
Fleurence J. Seaweed proteins: biochemical, nutritional aspects and potential uses. Trends Food Sci Technol. 1999;10(1):25–8.
Yone Y, Hossain MA, Furuichi M, Kato F. Effect of fermented and fermented-Resteamed scrap meals on growth and feed efficiency of Red Sea bream. Nippon Suisan Gakkaishi. 1986;52(3):549–52.
Li X-Q, Zhang X-Q, Chowdhury MAK, Zhang Y, Leng X-J. Dietary phytase and protease improved growth and nutrient utilization in tilapia (Oreochromis niloticus × Oreochromis aureus) fed low phosphorus and fishmeal-free diets. Aquac Nutr. 2019;25(1):46–55.
Smith AA, Dumas A, Yossa R, Overturf KE, Bureau DP. Effects of soybean meal and high-protein sunflower meal on growth performance, feed utilization, gut health and gene expression in Arctic charr (Salvelinus alpinus) at the grow-out stage. Aquac Nutr. 2018;24(5):1540–52.
Raskovic B, Stankovic M, Markovic Z, Poleksic V. Histological methods in the assessment of different feed effects on liver and intestine of fish. J Agric Sci Belgrade. 2011;56(1):87–100 https://doi.org/10.2298/JAS1101087R.
Klurfeld DM. Nutritional regulation of gastrointestinal growth. Front Biosci. 1999;4(1–3):d299 https://doi.org/10.2741/klurfeld.
Adeoye AA, Yomla R, Jaramillo-Torres A, Rodiles A, Merrifield DL, Davies SJ. Combined effects of exogenous enzymes and probiotic on Nile tilapia (Oreochromis niloticus) growth, intestinal morphology and microbiome. Aquaculture. 2016;463:61–70 https://doi.org/10.1016/j.aquaculture.2016.05.028.
Baeverfjord G, Krogdahl A. Development and regression of soybean meal induced enteritis in Atlantic salmon, Salmo salar L., distal intestine: a comparison with the intestines of fasted fish. J Fish Dis. 1996;19(5):375–87 https://doi.org/10.1046/j.1365-2761.1996.d01-92.x.
Krogdahl Å, Kortner TM, Jaramillo-Torres A, Gamil AA, Chikwati E, Li Y, Schmidt M, Herman E, Hymowitz T, Teimouri S, Storebakken T. Removal of three proteinaceous antinutrients from soybean does not mitigate soybean-induced enteritis in Atlantic salmon (Salmo salar, L). Aquaculture. 2020;514:734495 https://doi.org/10.1016/j.aquaculture.2019.734495.
Tran-Ngoc KT, Schrama JW, Le MT, Nguyen TH, Roem AJ, Verreth JA. Salinity and diet composition affect digestibility and intestinal morphology in Nile tilapia (Oreochromis niloticus). Aquaculture. 2017;469:36–43 https://doi.org/10.1016/j.aquaculture.2016.11.037.
Stone DAJ, Bellgrove EJ, Forder REA, Howarth GS, Bansemer MS. Inducing subacute enteritis in yellowtail kingfish Seriola lalandi: the effect of dietary inclusion of soybean meal and grape seed extract on hindgut morphology and inflammation. N Am J Aquac. 2018;80(1):59–68.
Ames EP. Atlantic cod stock structure in the Gulf of Maine. Fisheries. 2004;29(1):10–28.
Desai AR, Links MG, Collins SA, Mansfield GS, Drew MD, Van Kessel AG, et al. Effects of plant-based diets on the distal gut microbiome of rainbow trout (Oncorhynchus mykiss). Aquaculture. 2012;350–353:134–42.
Booman M, Forster I, Vederas JC, Groman DB, Jones SRM. Soybean meal-induced enteritis in Atlantic salmon ( Salmo salar) and Chinook salmon ( Oncorhynchus tshawytscha ) but not in pink salmon ( O . gorbuscha ). Aquaculture. 2018;483:238–43.
Friberg IM, Taylor JD, Jackson JA. Diet in the driving seat: natural diet-immunity-microbiome interactions in wild fish. Front Immunol. 2019;10:243.
Xiong J-B, Nie L, Chen J. Current understanding on the roles of gut microbiota in fish disease and immunity. Zool Res. 2019;40(2):70–6.
Ghanbari M, Kneifel W, Domig KJ. A new view of the fish gut microbiome: advances from next-generation sequencing. Aquaculture. 2015;448:464–75.
King GM, Smith C, Tolar B, Hollibaugh JT. Analysis of composition and structure of coastal to mesopelagic bacterioplankton communities in the northern gulf of Mexico. Front Microbiol. 2013;3:438.
Mukherjee A, Rodiles A, Merrifield DL, Chandra G, Ghosh K. Exploring intestinal microbiome composition in three Indian major carps under polyculture system: a high-throughput sequencing based approach. Aquaculture. 2020;524:735206 https://doi.org/10.1016/j.aquaculture.2020.735206.
Flint HJ, Duncan SH. Bacteroides and Prevotella. In: Batt CA, Tortorello ML, editors. Encyclopedia of food microbiology. 2nd ed. Oxford: Academic Press; 2014. p. 203–8.
Kautz S, Rubin BER, Russell JA, Moreau CS. Surveying the microbiome of ants: comparing 454 pyrosequencing with traditional methods to uncover bacterial diversity. Appl Environ Microbiol. 2013;79(2):525–34.
Gutierrez T. Aerobic hydrocarbon-degrading Gammaproteobacteria: Xanthomonadales. Tax Genom Ecophysiol Hydrocarbon Degrading Microbes. 2019:191–205.
Yamazaki Y, Meirelles PM, Mino S, Suda W, Oshima K, Hattori M, Thompson FL, Sakai Y, Sawabe T, Sawabe T. Individual Apostichopus japonicus fecal microbiome reveals a link with polyhydroxybutyrate producers in host growth gaps. Sci Rep. 2016;6:21631.
Kushkevych I, Dordević D, Kollár P. Analysis of physiological parameters of Desulfovibrio strains from individuals with colitis. Open Life Sci. 2019;13(1):481–8.
Holben WE, Williams P, Gilbert MA, Saarinen M, Särkilahti LK, Apajalahti JHA. Phylogenetic analysis of intestinal microflora indicates a novel mycoplasma phylotype in farmed and wild salmon. Microb Ecol. 2002;44(2):175–85.
Llewellyn MS, McGinnity P, Dionne M, Letourneau J, Thonier F, Carvalho GR, et al. The biogeography of the Atlantic salmon ( Salmo salar ) gut microbiome. ISME J. 2016;10(5):1280–4.
Razin S, Yogev D, Naot Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev. 1998;62(4):1094–156.
Nikouli E, Meziti A, Antonopoulou E, Mente E, Kormas K. Gut bacterial communities in geographically distant populations of Farmed Sea bream (Sparus aurata) and sea bass (Dicentrarchus labrax). Microorganisms. 2018;6(3):92.
Alou MT, Ndongo S, Frégère L, Labas N, Andrieu C, Richez M, et al. Taxonogenomic description of four new Clostridium species isolated from human gut: ‘Clostridium amazonitimonense’ , ‘Clostridium merdae’ , ‘Clostridium massilidielmoense’ and ‘Clostridium nigeriense’. New Microbes New Infect. 2018;21:128–39.
Gajardo K, Rodiles A, Kortner TM, Krogdahl Å, Bakke AM, Merrifield DL, Sørum H. A high-resolution map of the gut microbiota in Atlantic salmon (Salmo salar): a basis for comparative gut microbial research. Sci Rep. 2016;6:30893.
Pohl S, Harwood CR. Heterologous protien section by bacillus species from the cradle to the grave. Adv Appl Microbiol. 2010;73:11–25. https://doi.org/10.1016/S0065-2164(10)73001-X.
Soltani M, Ghosh K, Hoseinifar SH, Kumar V, Lymbery AJ, Roy S, Ringø E. Genus bacillus, promising probiotics in aquaculture: aquatic animal origin, bio-active components, bioremediation and efficacy in fish and shellfish. Rev Fish Sci Aqua. 2019;27(3):331–79.
Johnson JS, Spakowicz DJ, Hong BY, Petersen LM, Demkowicz P, Chen L, Leopold SR, Hanson BM, Agresta HO, Gerstein M, Sodergren E. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10(1):Art no 1 https://doi.org/10.1038/s41467-019-13036-1.
Amaretti A, Gozzoli C, Simone M, Raimondi S, Righini L, Pérez-Brocal V, García-López R, Moya A, Rossi M. Profiling of protein degraders in cultures of human gut microbiota. Front Microbiol. 2019;10:2614.
Waters JL, Ley RE. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019;17(1):83.
Goffredi SK, Morella NM, Pulcrano ME. Affiliations between bacteria and marine fish leeches (Piscicolidae), with emphasis on a deep-sea species from Monterey canyon. CA Environ Microbiol. 2012;14(9):2429–44.
Darcy JL, Washburne AD, Robeson MS, Prest T, Schmidt SK, Lozupone CA. A phylogenetic model for the recruitment of species into microbial communities and application to studies of the human microbiome. ISME J. 2020;14(6):Art. no. 6 https://doi.org/10.1038/s41396-020-0613-7.
Verster AJ, Borenstein E. Competitive lottery-based assembly of selected clades in the human gut microbiome. Microbiome. 2018;6(1):186 https://doi.org/10.1186/s40168-018-0571-8.
Egerton S, Culloty S, Whooley J, Stanton C, Ross RP. The gut microbiota of marine fish. Front Microbiol. 2018;9:873.
Hassanzadeh Y, Bahador N, Baseri-Salehi M. First time isolation of Photobacterium damselae subsp. damselae from Caranx sexfasciatus in Persian Gulf, Iran. Iran J Microbiol. 2015;7(3):178.
Ingerslev HC, von Gersdorff JL, Strube ML, Larsen N, Dalsgaard I, Boye M, Madsen L. The development of the gut microbiota in rainbow trout (Oncorhynchus mykiss) is affected by first feeding and diet type. Aquaculture. 2014;424:24–34.
Drieschner C, Könemann S, Renaud P, Schirmer K. Fish-gut-on-chip: development of a microfluidic bioreactor to study the role of the fish intestine in vitro. Lab Chip. 2019;19(19):3268–76.
Kazlauskaite R, Cheaib B, Heys C, Ijaz UZ, Connelly S, Sloan WT, Russell J, Martinez-Rubio L, Sweetman J, Kitts A, McGinnity P. Development of a three-compartment in vitro simulator of the Atlantic Salmon GI tract and associated microbial communities: SalmoSim. bioRxiv. 2020; https://doi.org/10.1101/2020.10.06.327858.
AOAC. Official methods of analysis. 17th edition, The assosation of official analytical chemist. Gaithersburg, MD, USA. Methods. 2000:925. 10,65.17, 974.24,992.16.
Björnsson B, Steinarsson A, Árnason T. Growth model for Atlantic cod (Gadus morhua): effects of temperature and body weight on growth rate. Aquaculture. 2007;271(1–4):216–26.
Bolton-Warberg M, FitzGerald RD. Benchmarking growth of farmed Atlantic cod, Gadus morhua: a case study in Ireland: benchmarking growth of farmed Atlantic cod. Aquac Res. 2012;43(5):670–8.
Dimitroglou A, Merrifield DL, Moate R, Davies SJ, Spring P, Sweetman J, et al. Dietary mannan oligosaccharide supplementation modulates intestinal microbial ecology and improves gut morphology of rainbow trout, Oncorhynchus mykiss (Walbaum). J Anim Sci. 2009;87(10):3226–34.
Griffiths RI, Whiteley AS, O’Donnell AG, Bailey MJ. Rapid method for Coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl Environ Microbiol. 2000;66(12):5488–91.
Keating C, Chin JP, Hughes D, Manesiotis P, Cysneiros D, Mahony T, Smith CJ, McGrath JW, O’Flaherty V. Biological phosphorus removal during high-rate, low-temperature, anaerobic digestion of wastewater. Front Microbiol. 2016;7:226.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6(8):1621–4.
Joshi NA, Fass JN. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. (version 1.33) [software]. 2011. Available at https://github.com/najoshi/sickle.
Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13(1):31.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–8.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–200.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41(D1):D590–6.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80.
Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. https://doi.org/10.1371/journal.pone.0009490.
Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5.
Core TR. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2013. Available from https://www.r-project.org/.
Oksanen J. Vegan : community ecology package. R package version. 2007;1:8–5.
Daly A, Baetens J, De Baets B. Ecological diversity: measuring the Unmeasurable. Mathematics. 2018;6(7):119.
McMurdie PJ, Holmes S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One. 2013;8(4):e61217.
Rohart F, Gautier B, Singh A, Cao K-AL. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol. 2017;13(11):e1005752.
Gauchotte-Lindsay C, Aspray TJ, Knapp M, Ijaz UZ. Systems biology approach to elucidation of contaminant biodegradation in complex samples – integration of high-resolution analytical and molecular tools. Faraday Discuss. 2019;218:481–504.
McKenna A, Ijaz UZ, Kelly C, Linton M, Sloan WT, Green BD, Lavery U, Dorrell N, Wren BW, Richmond A, Corcionivoschi N, Gundogdu O. Impact of industrial production system parameters on chicken microbiomes: mechanisms to improve performance and reduce campylobacter. Microbiome. 2020;8(1):128 https://doi.org/10.1186/s40168-020-00908-8.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Foster ZSL, Sharpton TJ, Grünwald NJ. Metacoder: an R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput Biol. 2017;13(2):e1005404.
Shetty SA, Hugenholtz F, Lahti L, Smidt H, de Vos WM. Intestinal microbiome landscaping: insight in community assemblage and implications for microbial modulation strategies. FEMS Microbiol Rev. 2017;41(2):182–99.
Leo Lahti et al. (Bioconductor, 2017-2019). Tools for microbiome analysis in R. microbiome package version. Available from http://microbiome.github.io/microbiome.
Jalanka-Tuovinen J, Salonen A, Nikkilä J, Immonen O, Kekkonen R, Lahti L, Palva A, De Vos WM. Intestinal microbiota in healthy adults: temporal analysis reveals individual and common core and relation to intestinal symptoms. PLoS One. 2011;6(7):e23035.
Acknowledgements
We dedicate this paper to Richard Fitzgerald, a dear colleague and friend, 1957-2016. The authors also kindly acknowledge all staff at the Carna Research Station, Carna, Co. Galway.
Funding
This study was part funded by the EIRCOD (Cod Broodstock and Breeding) project, under the Sea Change Strategy with the support of the Marine Institute and the Marine Research Sub-programme of the Ireland’s National Development Plan 2007–2013 co-funded by the European Regional Development Fund, and also NutraMara programme (Grant-Aid Agreement No. MFFRI/07/01) under the Sea Change Strategy with the support of Ireland’s Marine Institute and the Department of Agriculture, Food and the Marine, funded under the National Development Plan 2007–2013, Ireland. C.S. was funded by Science Foundation Ireland & the Marie-Curie Action COFUND under Grant Number 11/SIRG/B2159 and a Royal Academy of Engineering-Scottish Water Research Chair Grant Number: RCSRF1718643. UZI is supported by NERC, UK, NE/L011956/1.
Author information
Authors and Affiliations
Contributions
CK dissected the fish for microbiome sampling, DNA extraction optimisation and extractions on the gut content, bioinformatics, and statistical analysis (excluding tank population data) and data interpretation. MBW managed the feeding trial, coordinated sampling, data management and statistical analysis of tank populations. JH ran the feeding trial, assisted with sampling for growth and carried out sampling for and analysis of data from light microscopy. RD ran the feeding trial, assisted with sampling for growth and carried out sampling for and analysis of data from SEM. SW ran the feeding trial, carried out sampling for growth and assisted with all dissections for microbiome, light microscopy, and SEM analysis. AHLW seaweed identification and collection designed the test diets and feed analysis. SJD designed the research study, supervised the growth and gut morphology data analysis and data interpretation. RF designed research study, EIRCOD Project Co-ordinator. UZI wrote the analysis scripts to generate the microbiota figures in this paper and performed the microbiota bioinformatic and statistical analysis with CK. CJS designed the research study and supervised the microbiome work. The study was designed by SJD, MBW, AHLW, CJS and RF. CK, MBW, AHLW, SJD, UZI and CJS wrote the manuscript, which was reviewed and edited by all authors. RF was EIRCOD Project Coordinator. All authors made a substantial contribution towards the research study and all authors (except RF) approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The feeding trial was carried out at the Carna Research Station, Carna, Co. Galway, Ireland, a Health Products Regulatory Authority (HPRA) licensed institution. All personnel involved in the feed trial work was Laboratory Animal Safety Trained Ireland (LAST-Ireland) with individual authorisation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Photographic images of individual juvenile Atlantic cod (Gadus morhua) showing the visual distinction between A) ASCO_N, and B) ASCO_LG groups at Week 8. Note the smaller body size in the ASCO_LG individual which is consistent with poor food uptake. Figure S2. Bar-plots of fish sampled for microbiome analysis (A) and fish sampled for histology B) with i) the recorded weights (g), the recorded lengths (cm) in ii) and iii) the calculated condition factor K over the course of the trial. The associated statistical measurements were calculated using Kruskal-wallis significance testing. Figure S3. Rarefaction curves showing the number of reads from the 16S rRNA gene in DNA from 67 samples from Atlantic cod (Gadus morhua) on the x-axis and the number of OTUs within a 97% percent sequence similarity threshold on the y-axis. Groups are indicated by colour coded lines. Figure S4. Microbial diversity and community structure according to variances in the 16S rRNA gene in DNA from 67 samples from Atlantic cod (Gadus morhua) microbiome. A) Alpha diversity box plots of Pielou’s evenness, Shannon Entropy and Simpson Diversity index. Pair-wise ANOVA P-values are displayed P < 0.05*, P < 0.01**, P < 0.001***. B) Beta diversity Non-Metric Multidimensional Scaling (NMDS) plots using Bray-Curtis dissimilarity (R2 = 0.24142, P = 0.001) and weighted UniFrac distances (R2 = 0.296, P = 0.001), where each point corresponds to the community structure of one sample, groups are indicated by colour coded circles, the ellipses are drawn at a 95% standard error. Figure S5. Venn diagram showing the number of shared operational taxonomic units (OTUs) and unique OTUs in the hindgut of juvenile Atlantic cod (Gadus morhua) from Week 0, Week 8 (CTRL, ULVA, ASCO_N and ASCO_LG and Week 12 (CTRL, ULVA, ASCO_N and ASCO_LG). Figure S6. sPLS-DA heatmap of the discriminant operational taxonomic units (OTUs) > 1% relative abundance in the hindgut microbiota between the Week 0, Week 8 (CRTL, ULVA, ASCO_N and ASCO_LG) and Week 12 (CRTL, ULVA, ASCO_N and ASCO_LG) groups of juvenile Atlantic cod (Gadus morhua). Rows and columns are ordered using hierarchical (average linkage) clustering to identify blocks of genera of interest. The heatmap depicts TSS + CLR (Total Sum Scaling followed by Centralised Log Ratio) normalised abundances: high abundance (orange/yellow) and low abundance (dark purple). Fish weight (g) and condition factor K is shown per grouping. sPLS-DA was fine-tuned using centroids.dist and 3 tuning components. Figure S7. sPLS-DA heatmap of the discriminant operational taxonomic units (OTUs) > 3.5% relative abundance between the CRTL, ULVA, ASCO_N and ASCO_LG groups of juvenile Atlantic cod (Gadus morhua) at Week 8. Rows and columns are ordered using hierarchical (average linkage) clustering to identify blocks of genera of interest. The heatmap depicts TSS + CLR (Total Sum Scaling followed by Centralised Log Ratio) normalised abundances: high abundance (orange/yellow) and low abundance (dark purple). Fish weight (g) and condition factor K is shown per grouping. sPLS-DA was fine-tuned using centroids.dist and 3 tuning components. Figure S8. The core microbiome consisting of the operational taxonomic units (OTUs) found in 85% of individuals in the A) CTRL and B) ULVA dietary treatment at Week 12. Green text indicates those that are shared between the two dietary treatments.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Keating, C., Bolton-Warberg, M., Hinchcliffe, J. et al. Temporal changes in the gut microbiota in farmed Atlantic cod (Gadus morhua) outweigh the response to diet supplementation with macroalgae. anim microbiome 3, 7 (2021). https://doi.org/10.1186/s42523-020-00065-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-020-00065-1