
Abstract
Background
Cardiac tumors are a very rare entity. Leiomyosarcoma represents less than 1% of cases.
Case presentation
a 51-year-old woman diagnosed with primary left atrium leiomyosarcoma. She was treated by optimal surgery and adjuvant chemotherapy. She is still alive after a follow-up of 24 months without evidence of local or distant recurrence.
Conclusions
Cardiac leiomyosarcoma is a rare tumor with a dismal prognosis. Surgery is the mainstay of treatment. Adjuvant treatment is still controversial.
Similar content being viewed by others


Background
Cardiac tumors are a very rare entity with an incidence of 0.02%. Only 25% are malignant with a prevalence ranging from 0.001% to 0.28 [1]. Primary cardiac cancers are scarcer than cardiac metastases [2]. Most frequent histologic types are Angiosarcomas followed by rhabdomyosarcomas, mesotheliomas and fibrosarcomas. Leiomyosarcoma represents less than 1% of cases [3]. In spite of improvement of multidisciplinary treatment including surgery, chemotherapy and radiotherapy, the prognosis remains (Fig. 1) poor with a median survival of 6 months [3].
This case aimed to describe the clinical, histological, therapeutic and prognostic features of this rare pathology.
Case report
We present the case of a 51-year-old woman, with past medical history of hypertension, who consulted in September 2014 for bilateral lower limb pain and lower extremity edema since 6 months. The diagnosis of hypertrophic osteoarthropathy was suspected. In this context, a CT scan revealed a 6 cm defect, involving the right atrium and the right inferior pulmonary vein, which appears markedly enlarged. This aspect evoked a large intracavitary thrombus. A transesophageal cardiac ultrasound showed a 30 × 26 mm, with little mobility, lobed tumor, in the left atrium, connected to the atrial septum. The tumor wasn’t obstructive. Cardiac chambers dimensions and pulmonary pressure were normal. There was no systolic or diastolic left ventricular dysfunction. The patient was referred to a cardiovascular surgeon with a suspected diagnosis of left atrium myxoma. Surgery was performed through median sternotomy. The patient had a cardiopulmonary bypass (CPB) with aortic and bi-caval cannulation. The left atrium was dissected, revealing a voluminous, septal based tumor involving the right inferior pulmonary vein and the posterior wall of the left atrium, suggestive of malignancy. We did a wide en bloc excision of the tumor, extended from the posterior wall of the left atrium Fig. 2 and inferior pulmonary vein to pericardial reflection. Reconstruction of the left atrium with two pericardial patches (an anterior septal and posterior parietal) was achieved according to Sutureless de lacourt-Gayettechnique. Macroscopic examination showed a friable whitish mass measuring 40 × 30 × 30 mm.
Microscopic examination revealed the presence of conjunctival tumoral proliferation made of spindle cells with a fascicular organization infiltrating myocardial fibers. A high mitotic activity was noticed. Surgical margins were clear. Immunohistochemical staining showed an intense and diffuse positivity of alpha-smooth-muscle actin and caldesmon and the negativity of PS100, desmine and myogenin. Based on these findings, diagnosis of primary cardiac leiomyosarcoma grade 3 according to FNNCLCC was confirmed.
Post-operative CT scan revealed no metastases. According to a multidisciplinary staff, an adjuvant chemotherapy consisting on six cycles of Doxorubicin and Ifosfamide was prescribed.
The patient is regularly followed. After a follow-up of 24 months, she still has no clinical or radiological evidence of recurrence.
Discussion
We presented a rare case of cardiac leiomyosarcoma treated by surgery followed by an adjuvant chemotherapy. The patient is still alive after a follow-up of 24 months.
Until December 2015, we found only 32 cases of primary cardiac leiomyosarcoma with available data. In most studies, it was defined as tumors originating only from cardiac chambers, excluding those located in the pericardium and great vessels [4]. Epidemiological, clinical, therapeutic features and outcomes were listed in the Table 1.
Median age at diagnosis was 48 with a female predominance [4].
This tumor has a poor prognosis due essentially to advanced stages at presentation. It usually remains asymptomatic until advanced stages. Even when it becomes symptomatic, presentation is atypical and non specific. Symptoms of obstruction especially dyspnea is found in 78.1% of cases [4]. Physical examination isn’t helpful no more. In our case, the patient was asymptomatic and the diagnosis was suspected fortuitously on the findings of a CT performed for another aim.
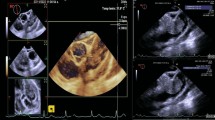
Echocardiography, especially the trans-esophageal route, is habitually the initial imaging modality. It may show the tumor, its extent and its hemodynamic consequences (Fig. 1). CT scan and cardiac MRI provide further information about morphology, location and extent of the mass (Fig. 2). Cardiac MRI is more efficient to evaluate myocardial involvement. CT scan is useful to assess extracardiac extent and metastasis [5].

Transoesophageal echocardiography showing mid esophageal 20° view. Left atrium tumor. RA right atrium, LA left atrium

Computed tomography; Transverse section: 6 cm defect involving the right atrium and the right inferior pulmonary vein, which appears markedly enlarged
Cardiac leiomyosarcomas have a high rate of local and distant recurrence, occurring even after an optimal resection of the primary tumor.
The left atrium is the most frequent location of cardiac leiomyosarcomas (51%). That joins operative and radiological findings, in our case.
Biopsy is the gold standard for histological confirmation but this step can sometimes be overtaken and the diagnosis is then made on the examination of resected mass.
Complete surgical resection, when it’s possible, is the mainstay of treatment.
Since soft tissue sarcoma is a heterogeneous group, benefits of adjuvant chemotherapy isn’t clearly established. Several studies defined subgroups associated with a high risk of local and distant relapse. Risk varies depending on factors like size >5 cm, high grade, depth and chemosensitive histologies with a metastatic potential. Leiomyosarcoma belongs to the high risk group. Major drugs used are Doxorubicin, Ifosfamide and Dacarbazine [6]. It’s what leaded us to propose six cycles of Doxorubicin and Ifosfamide in adjuvant setting to our patient, after a multidisciplinary team consultation.
Indications of radiation therapy are mostly restrained to palliative setting. It’s proposed when margins of resection are positive or for aggressive localized disease or for recurrences. Since the lack of evidence concerning the efficacy of radiotherapy in management of cardiac leiomyosarcomas and the poor tolerance, its use stills equivocal and unusual.
According to the reported cases of leiomyosarcomas with cardiac involvement, the mean survival time of patients who underwent surgery and chemotherapy was about 12 months [6]. Several factors are suspected to enworse the prognosis particularly the high grade, a high mitotic index, positive surgical margins and metastasis [7].
In the case above, our patient had a grade 3 tumor with a high mitotic activity. She underwent a carcinologic surgery and adjuvant chemotherapy. Currently, she is still alive and there is no evidence of local recurrence or metastasis, after a follow-up of 24 months.
Conclusions
Cardiac leiomyosarcoma is a rare tumor with a dismal prognosis. Wide margin resection is the mainstay of treatment. For soft tissue sarcoma, adjuvant chemotherapy is still controversial. Nevertheless, leiomyosarcoma belongs to the high risk group of soft tissue sarcoma associated with a metastatic potential. In this group, there is a trend to outcomes improvement with adjuvant chemotherapy. Drugs used in this context are Doxorubicin, Ifosfamide and Dacarbazine.
Regarding the scarcity of this disease, it’s important to report all cases with a longer follow-up to refine indications and modalities of adjuvant treatment and prognostic factors.
References
Smith S, Grange S, Wilson P. Leiomyosarcoma of the left atrium. A case study. Radiography. 2012;18(3):225–8.
Isambert N, Ray-Coquard I, Italiano A, Rios M, Kerbrat P, Gauthier M, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group 1990. Eur J Cancer Oxf Engl. 2014;50(1):128–36.
Andersen RE, Kristensen BW, Gill S. Cardiac leiomyosarcoma, a case report. Int J Clin Exp Pathol. 2013;6(6):1197–9.
Wang J-G, Cui L, Jiang T, Li Y-J, Wei Z-M. Primary cardiac leiomyosarcoma: an analysis of clinical characteristics and outcome patterns. Asian Cardiovasc Thorac Ann. 2015;23(5):623–30.
Galeone A, Validire P, Debrosse D, Folliguet T, Laborde F. Leiomyosarcoma of the right inferior pulmonary vein: 2 years survival with multimodality therapy. Gen Thorac Cardiovasc Surg. 2013;61(9):534–7.
Steen S, Stephenson G. Current treatment of soft tissue sarcoma. Proc Bayl Univ Med Cent. 2008;21(4):392–6.
Lv Y, Pang X, Zhang Q, Jia D. Cardial leiomyosarcoma with multiple lesions involved: a case report. Int J Clin Exp Pathol. 2015;8(11):15412–6.
Kornberg A, Wildhirt SM, Kreuzer E, Reichart B. Asymptomatic right atrial leiomyosarcoma with tricuspid valve obstruction in a young female patient. Eur J Cardio Thorac Surg Off J Eur Assoc Cardio Thorac Surg. 1998;14:635–8.
Takamizawa S, Sugimoto K, Tanaka H, Sakai O, Arai T, Saitoh A. A case of primary leiomyosarcoma of the heart. Intern Med Tokyo Jpn. 1992;31:265–8.
Fox JP, Freitas E, McGiffin DC, Firouz-Abadi AA, West MJ. Primary leiomyosarcoma of the heart: a rare cause of obstruction of the left ventricular outflow tract. Aust N Z J Med. 1991;21:881–3.
Hattori Y, Iriyama T, Watanabe K, Negi K, Takeda I, Sugimura S. Rapidly growing primary cardiac leiomyosarcoma: report of a case. Surg Today. 2000;30:838–40. doi:10.1007/s005950070069.
Minakata K, Konishi Y, Matsumoto M, Nonaka M, Yamada N. Primary leiomyosarcoma of the left atrium. Jpn Circ J. 1999;63:414–5.
Pins MR, Ferrell MA, Madsen JC, Piubello Q, Dickersin GR, Fletcher CD. Epithelioid and spindle-celled leiomyosarcoma of the heart. Report of 2 cases and review of the literature. Arch Pathol Lab Med. 1999;123:782–8. doi:10.1043/0003-9985(1999)123<0782:EASCLO>2.0.CO;2.
Minardi G, Pulignano G, Sentinelli S, Narducci C, Giovannini M. Left atrial leiomyosarcoma: double occurrence and double recurrence—report of one case. J Am Soc Echocardiogr Off Publ Am Soc Echocardiogr. 1998;11:1171–6.
Burnett RA. Primary cardiac leiomyosarcoma with pulmonary metastases: a diagnostic problem. Scott Med J. 1975;20:125–8.
Andersen RE, Kristensen BW, Gill S. Cardiac leiomyosarcoma, a case report. Int J Clin Exp Pathol. 2013;6:1197–9.
Ogimoto A, Hamada M, Ohtsuka T, Hara Y, Shigematsu Y, Yokoyama A, et al. Rapid progression of primary cardiac leiomyosarcoma with obstruction of the left ventricular outflow tract and mitral stenosis. Intern Med Tokyo Jpn. 2003;42:827–30.
Willaert W, Claessens P, Shoja A, Heremans A, Deferm H, Roelandts J, et al. Ventricular outflow tract obstruction secondary to leiomyosarcoma of the right ventricle. Jpn Heart J. 2001;42:377–86.
Strina C, Zannoni M, Parolin V, Cetto GL, Zuliani S. Bone metastases from primary cardiac sarcoma: case report. Tumori. 2009;95:251–3.
Lee SH, Kim WH, Choi JB, Lee SR, Rhee KS, Chae JK, et al. Huge primary pleomorphic leiomyosarcoma in the right ventricle with impending obstruction of both inflow and outflow tracts. Circ J Off J Jpn Circ Soc. 2009;73:779–82.
Malyshev M, Safuanov A, Gladyshev I, Trushyna V, Abramovskaya L, Malyshev A. Primary left atrial leiomyosarcoma: literature review and lessons of a case. Asian Cardiovasc Thorac Ann. 2006;14:435–40. doi:10.1177/021849230601400520.
Smith S, Grange S, Wilson P. Leiomyosarcoma of the left atrium. A case study. Radiography. 2012;18:225–8. doi:10.1016/j.radi.2012.01.002.
Antunes MJ, Vanderdonck KM, Andrade CM, Rebelo LS. Primary cardiac leiomyosarcomas. Ann Thorac Surg. 1991;51:999–1001. doi:10.1016/0003-4975(91)91031-P.
Rastan AJ, Walther T, Mohr FW, Kostelka M. Leiomyosarcoma-an unusual cause of right ventricular outflow tract obstruction. Thorac Cardiovasc Surg. 2004;52:376–7. doi:10.1055/s-2004-821276.
Astarcioglu MA. Multimodality imaging of a recurrent case of right-sided cardiac leiomyosarcoma with an unusual clinical course. Turk Kardiyol Dernegi Arsivi Arch Turk Soc Cardiol. 2016. doi:10.5543/tkda.2015.09125.
Glaoui M, Benbrahim Z, Belbaraka R, Naciri S, Errihani H, Lescene A. An uncommon long-term survival case of primary cardiac leiomyosarcoma. World J Surg Oncol. 2014;12:338. doi:10.1186/1477-7819-12-338.
Nakanishi H, Furukawa K, Noguchi R, Furutachi A, Itoh M, Kamohara K, et al. Primary cardiac leiomyosarcoma originating from the left atrium. Kyobu Geka. 2012;65:1057–61.
Wilbring M, Kappert U, Daubner D, Matschke K, Tugtekin SM. Metastasizing primary atrial leiomyosarcoma causing a functional high-grade mitral stenosis. Heart Surg Forum. 2012;15:E108–10. doi:10.1532/HSF98.20111147.
Parissis H, Akbar MT, Young V. Primary leiomyosarcoma of the right atrium: a case report and literature update. J Cardiothorac Surg. 2010;5:80. doi:10.1186/1749-8090-5-80.
Mazzola A, Spano J-P, Valente M, Gregorini R, Villani C, Eusanio MD, et al. Leiomyosarcoma of the left atrium mimicking a left atrial myxoma. J Thorac Cardiovasc Surg. 2006;131:224–6. doi:10.1016/j.jtcvs.2005.07.061.
Esaki M, Kagawa K, Noda T, Nishigaki K, Gotoh K, Fujiwara H, et al. Primary cardiac leiomyosarcoma growing rapidly and causing right ventricular outflow obstruction. Intern Med Tokyo Jpn. 1998;37:370–5.
Guschmann M, Hofmeister J. Primary leiomyosarcoma in the left atrium—a rarity. Case report and literature review. J Pathol. 1997;18:474–9.
Davis GK, Jones EL, Bonser RS, Roberts DH. Coronary arteriographic and pathological findings in a case of primary leiomyosarcoma of the heart. Int J Cardiol. 1997;59:313–6. doi:10.1016/S0167-5273(97)02967-7.
Panday VRN, Cramer MJM, Elbers HRJ, Riviere AB, Ernst SMPG, Plokker HWT. Primary leiomyosarcoma of the heart presenting as obstruction to the pulmonary trunk. Am Heart J. 1997;133:465–6. doi:10.1016/S0002-8703(97)70191-1.
Pessotto R, Silvestre G, Luciani GB, Anselmi M, Pasini F, Santini F, et al. Primary cardiac leiomyosarcoma: seven-year survival with combined surgical and adjuvant therapy. Int J Cardiol. 1997;60:91–4.
Lo FL, Chou YH, Tiu CM, Lan GY, Hwang JH, Chern MS, et al. Primary cardiac leiomyosarcoma: imaging with 2-D echocardiography, electron beam CT and 1.5-Tesla MR. Eur J Radiol. 1998;27:72–6.
Authors’ contributions
Conception and design: FK; YY. Administrative support: AM; HR; NC. Provision of study materials or patients: AM; KM. Collection and assembly of data: EM. Data analysis and interpretation: KB; MA. Manuscript writing: All authors. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
In tunisian law, patients consent is not mandatory for retrospective studies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Behi, K., Ayadi, M., Mezni, E. et al. Two years survival of primary cardiac leiomyosarcoma managed by surgical and adjuvant therapy. Clin Sarcoma Res 7, 5 (2017). https://doi.org/10.1186/s13569-017-0069-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13569-017-0069-3