
Abstract
Bovine respiratory disease (BRD), as one of the most common and costly diseases in the beef cattle industry, has significant adverse impacts on global food security and the economic stability of the industry. The bovine respiratory microbiome is strongly associated with health and disease and may provide insights for alternative therapy when treating BRD. The niche-specific microbiome communities that colonize the inter-surface of the upper and the lower respiratory tract consist of a dynamic and complex ecological system. The correlation between the disequilibrium in the respiratory ecosystem and BRD has become a hot research topic. Hence, we summarize the pathogenesis and clinical signs of BRD and the alteration of the respiratory microbiota. Current research techniques and the biogeography of the microbiome in the healthy respiratory tract are also reviewed. We discuss the process of resident microbiota and pathogen colonization as well as the host immune response. Although associations between the microbiota and BRD have been revealed to some extent, interpreting the development of BRD in relation to respiratory microbial dysbiosis will likely be the direction for upcoming studies, which will allow us to better understand the importance of the airway microbiome and its contributions to animal health and performance.
Similar content being viewed by others


1 Introduction to the bovine respiratory microbiome
Bovine respiratory disease (BRD), a leading cause of morbidity, mortality and economic cost, is one of the largest health challenges facing the modern-day beef cattle industry [1]. In the US, over 90% of large feedlots reported BRD as the most frequent disease [2]. Not only does this disease result in increased medication costs and death, but morbid beef cattle also grow slower, develop less efficient feed conversion ratios, and tend to need additional feed time to reach similar carcass quality of clinically healthy calves [3]. The wide use of vaccines and antimicrobials to prevent and treat BRD is a common approach worldwide [2]. However, the desired effects of vaccines to protect against BRD have not been reached, and mass administration of antimicrobials should be critically evaluated due to increased concerns over antibiotic resistance [4,5,6,7,8]. Alternative therapies, such as probiotics [9], are becoming increasingly investigated to treat BRD and improve management. For example, intranasal bacterial therapeutics developed from the bovine nasopharyngeal Lactobacillus spp. could reduce the colonization by pathogen Mannheimia haemolytica in dairy calves [10].
In past decades, next-generation sequencing (NGS) technology has contributed to the progressive understanding of the roles of the resident microbiota [11]. The microbiome, including both the community of the microbiota (microorganisms containing bacteria, archaea, fungi, protists and algae) and their “theatre of activity” (structural elements, metabolites/signal molecules, and the surrounding environmental conditions) in a specific environment (e.g., gut, lung), are important for animal health and disease [12, 13]. The contribution of the respiratory microbiota to maintaining health and its association with disease has attracted more attention [14,15,16]. It is well known that, in humans, the airway microbial system may cooperate with host immunity and metabolize products to generate key defenses against infections produced by opportunistic pathogens [17, 18]. Moreover, airway microbial composition and heightened respiratory pathogen incidence are associated with pneumonia in beef cattle [19, 20]. The respiratory ecosystem contains the upper (URT) and the lower (LRT) respiratory tract at the anatomical and physiological perspectives. Regarding the specific environments of each niche in the respiratory tract, niche-associated microbiota inhabit and differentiate within each specific environment [11]. Despite the remarkable progress made in recent studies using NGS, current research relevant to the bovine respiratory microbiome is only at an initial stage. The most commonly implicated bacterial pathogens in BRD cases, including Mycoplasma bovis, Histophilus somni, Mannheimia haemolytica and Pasteurella multocida, have traditionally been identified using culture-dependent approaches [1, 21]. However, these bacteria have been classified from both clinically healthy controls and morbid cattle. Although some studies have attempted to investigate the URT and lung microbiomes using NGS [1, 20, 22,23,24,25], the microbial movements within the respiratory tract are still unknown. Based on the results in previous studies, the notion that the respiratory microbiome is significantly important to cattle health has been confirmed [15, 20, 22, 26]. Since BRD results in massive economic losses and beef cattle are one of the key food sources of human society, additional research that elucidates the role of the respiratory microbiota in BRD pathophysiology is needed and may help us identify potential alternative therapies.
In this review, we summarize BRD clinical signs and pathogenesis, techniques applied in the respiratory microbiome analysis, the biogeography of the microbiota in the respiratory system, and the association between BRD and the microbiome, which provide implications of the respiratory microbiome in health, disease and animal production for further studies. Although there are many debates regarding the respiratory microbiome, the speculation and authors’ own opinions are presented. Notably, as the host-microbiota’s interactions and microbial drifts within the airway are contemporary hot topics and will continue to be highly relevant to future studies, it is deeply analyzed in this discussion.
2 The pathogenesis of bovine respiratory disease
BRD, also known as “shipping fever”, is the most frequent and costly disease of the modern beef cattle industry, especially for newly feedlot calves [6]. Recently weaned and transported beef cattle are at an even greater risk for developing BRD, which contributes to roughly 70–80% of feedlot total morbidity and roughly 10–50% of feedlot mortalities and additionally results in the subsequent loss of performance and health [27]. Moreover, BRD diagnosis is usually dependent on trained feedlot personnel and is commonly based on observed clinical signs (e.g., cattle depression, nasal discharge, ocular discharge, coughing, gaunt appearance, or inappetence). Additionally, treatment often consists of administering antimicrobials which may lead to the increase of antibiotic-resistance determinants [7, 28].
BRD is usually observed in cattle within four weeks of transportation to a feedlot [29]. There are multiple clinical signs of BRD, which vary greatly, depending on the phase and extent of the disease process. The general signs can include depression, inappetence, dullness and fever. Additionally, several respiratory signs, including ocular and nasal discharge, coughing, excessive salivation, and abnormal respiratory rate and rhythm, have been observed in BRD-diagnosed cattle [30]. However, this assessment has limited sensitivity and specificity which may result in unnecessary treatment and delayed or negative detection of BRD in truly sick animals [31]. White and Renter [30] found that the sensitivity for BRD detection based on clinical signs observed by trained personnel was only 62%, indicating many BRD cases go undetected, or are not detected until the advanced disease stage when successful treatment is less likely [32]. Moreover, although the extent of lung lesions (e.g., pleural adhesions, collapse/consolidation, parenchymal fibrosis, abscesses, or emphysema) resulting from BRD is associated with the risk of mortality and retreatment, the lesions are frequently found at slaughter, often in calves in which BRD has never been detected [33, 34]. Overall, current methods for the early detection, prognosis and diagnosis of BRD still have low accuracy and therefore additional researches exploring BRD diagnostics are needed.
The currently accepted theory regarding BRD pathogenesis is the complex synergistic interaction of bacteria and viruses under the influence of various stressors (i.e., weaning, comingling, transportation, and dietary changes) in addition to changes within the host and environment [35, 36] (Figure 1). A harmonious interaction between the host, properties of microbiome colonization and the local environment within the airways exists in healthy cattle. In contrast, a disequilibrium related to microbial dysbiosis, mucosal dysfunction as well as acute or chronic inflammation consequently generates an opportunity for the development of BRD [37]. So far, we know bacterial pathogen invasion produces the acute syndrome of BRD after the bovine respiratory system has been disturbed by factors such as viral infections, environmental changes and/or stress [38, 39]. There are multiple viral agents that can contribute to the development of BRD, including bovine viral diarrhea virus (BVDV), bovine respiratory syncytial virus (BRSV), bovine herpes virus 1 (BHV-1), and parainfluenza 3 virus (PI3V) [40]. Those viruses with stressors can lead to enhanced colonization and replication of bacterial pathogens and infect the lung subsequently. However, the incidences and abundances of these bacteria identified as BRD pathogens, which may be commensal organisms in healthy animals, do not correlate well with the occurrence of clinical BRD. For example, a high abundance of Mycoplasma bovis has been observed both in healthy steers and those diagnosed with BRD [41, 42]. Since knowledge gaps remain regarding BRD pathogenesis, a review of current research on community structure and composition of the microbiota of the bovine respiratory tract would allow for a better understanding of the pathobiology of BRD and emphasize an important direction for future research.

Triggers affecting the healthy respiratory ecosystem and leading to the onset of bovine respiratory disease (BRD) in newly weaned beef cattle. The bovine respiratory ecosystem has an increased risk of disequilibrium and subsequent BRD signs when the host is affected by pathogens, changes in the environment and managements (e.g., weaning, commingling, transportation, and dietary changes etc.).
3 Techniques used in the studies of the respiratory microbiome
Most BRD microbiology studies have been conducted using the nasopharyngeal swab (NPS) sampling approach [22]. Thus, sampling other niches within the respiratory tract is needed to better understand the cattle respiratory microbiome and BRD pathogenesis. This manuscript will describe the methods of sample collection in relation to the specific anatomical structures of the airway. The key points focus on subsequent measurements and data analysis of sequencing since most of the studies analyzed used the 16S rRNA sequencing technique.
3.1 Sampling techniques for the bovine respiratory microbiome
For sample collection, the literature reports various methods when comparing the URT and the LRT. Typically, a sterile swabbing approach is common for the collection of microbiome samples from the URT. There are many types of swabs such as those with cotton or polyester ends with various transport medium available depending on the subsequent diagnostics employed. In several previous cattle studies, researchers used short cotton swabs (17 cm length) for nasal sampling, and longer double-guarded polystyrene cotton swabs (84 cm length) for nasopharyngeal samples [1, 22, 43]. Common approaches for the collection of LRT samples are mainly using trans-tracheal wash (TTW) and bronchoalveolar lavage (BAL) techniques [1, 43, 44]. TTW utilizes the insertion of a catheter into the trachea of subjects to collect fluids samples from the LRT to provide molecular evidence about the LRT for cytologic and culture analysis. BAL, as a minimally invasive medical procedure, employs a broncho-alveolar lavage tube passed through the nares into the lungs for sampling of the lower airways [45]. It is not well documented whether TTW or BAL is the best approach to investigate the LRT microbiome in cattle. In previous studies, TTW and BAL were able to determine the bacterial pathogens including Mcoplasma and Mannheimia in the lungs of calves acutely affected with BRD [1, 43], and Mycoplasma spp. and other bacteria have been isolated from BAL samples from pneumonia calves [46, 47]. Although some concerns remain regarding the risk of nasal and nasopharyngeal contamination, BAL has become more widely used for bacterial diagnosis in pneumonia due to its advantages of being minimally invasive and useful for cytology [45, 48, 49]. In human studies, the contamination of the BAL microbiome from the URT is considered minimal and could be ignored when using guarded BAL [12, 50, 51]. In addition to the common URT and LRT sampling methods described, some researchers have collected lung tissue post-slaughter for microbiome analysis [52]. However, the value of these samples is limited due to the cost of euthanasia, lack of slaughter facilities appropriate for the sampling of lung tissue and lung traceability. Overall, the most useful approach to sample the airway will be dependent on research goals, the diagnostics utilized, the species being studied, and the category and the severity of the disease course [53].
3.2 Sequencing and bioinformatics for the bovine respiratory microbiome
Previous studies have analyzed the respiratory microbiota or pathogens using culture-dependent techniques [54,55,56]. However, these techniques only enable the detection of a small fraction of the microbiota. Also, various molecular techniques have been used to quantify specific microbes within the respiratory microbial community, including immunohistochemistry [57] and real-time PCR [58]. The improved availability of NGS techniques has allowed us to more broadly and specifically study the microbiome. Due to the low biomass of specimens in the LRT, the lungs have traditionally been considered a minimal source of bacteria when using culture-dependent or molecular techniques in the past [12, 15]. However, recent studies using NGS techniques have shown the complex microbial composition and found the common bacterial pathogens for BRD in the lungs of both healthy and sick cattle [1, 16, 23, 43, 52]. With the improvement of techniques, nanopore sequencing and multiple omics [i.e., metagenomics (the study of a collection of genomes and genes from the members of a microbiota by shotgun sequencing), metaproteomics, metatranscriptomics and metabolomics] are becoming more effective to investigate the composition and functions of the respiratory microbiome [59, 60]. In cattle, the bacterial community and BRD-associated pathogens were found when using metagenomics to measure all the genes of all the microbes in an environment [20, 25, 61,62,63], but still no reports using other omics were found. However, due to many factors such as cost, technique error, and time of analysis etc., NGS is still the most popular approach [64].
Big data analysis post-NGS is another key factor in respiratory microbiome analysis. After obtaining sequencing data from Illumina MiSeq /HiSeq or 454 pyrosequencing platforms [65,66,67,68], the quality control steps are carried out to remove low-quality reads with sequencing errors while the remaining high-quality sequences are classified to the genus- or species- level based on available databases such as RDP [69], Greengenes [70], NCBI [71] or BRD niche specific database [72]. Of note, it is critical to include negative (i.e., blanks during DNA extraction and PCR reactions) and positive controls (i.e., mock communities with known bacterial taxa) to rule out any environmental contaminations, especially when analyzing low biomass lung samples. The software, such as quantitative insights into microbial ecology (QIIME) and mothur, have been commonly used for 16S rRNA sequencing data analysis, including quality control, bacterial classification, and downstream analysis [73,74,75]. Then, basic analyses, such as community measures of alpha diversity (the diversity within a particular ecosystem, including richness and evenness) and beta diversity (comparison of diversity or the extent of changes between ecosystems) for overall community structure as well as major microbiota composition, are performed and reported.
3.3 Statistics for the bovine respiratory microbiome
The interactions among microbes within a community are essential to study the bovine respiratory microbiome and its association with BRD. Network theory can investigate and display the complex interactions of microbiota in a single network. A wide range of methods has been used to build ecological networks regarding microbiome data [76]. Those methods vary in their accuracy, efficiency, speed and computational requirements, as well as the span from simple measures of pairwise Spearman or Pearson correlations, to more complex multiple regression and even Gaussian graphical models. A study using co-occurrence analysis by calculating all coefficients of Spearman’s rank correlation found “core” community structures formed between bacteria in both healthy and diseased human airways [77]. Only one study using network analysis related to BRD was found based on our knowledge and this research distinguished the bacteria associated with BRD using network analysis [78]. Network analysis should be used broadly in BRD researches, which could allow us to understand the microbial interplays and find the potential probiotics.
Identification of bacterial taxa related to BRD is of particular interest to many scientists. Many machine learning techniques that allow algorithm to be more accurate at predicting outcomes could be used to identify specific bacteria deferentially represented between healthy versus BRD calves, including Random Forest [79], Support Vector Machine (SVM) [80], Linear discriminant analysis Effect Size (LEfSe) [81], etc. For example, Random Forest has been used as a robust machine learning technique to identify microbial biomarkers related to different human and animal diseases [82, 83]. It can deal with binary, categorical and continuous variables, and works well with unbalanced data sets. Although machine learning needs more computational power and resource, it is not a big problem with the development of computer science. Area-under-ROC curve (AUC) of the Random Forest (AUCRF), that has been previously published [84], has higher accuracy for feature prediction. A recent study used machine learning to predict viral-induced BRD with high accuracy based on public datasets [85]. Moreover, regression-based Random Forest models could be developed to identify bacterial taxa associated with continuous variables such as body weight and temperature using an updated method. This exciting area of research is continually developing as new techniques and modified techniques are being explored and will ultimately be key to improving our recognition of bacterial pathogens.
Another important ecological question involves the spatial dynamics (from the upper to the lower airway) of the respiratory microbiome in the context of BRD. In humans, the URT microbiota disperses and colonizes the lung via respiration and microaspiration [86], and the island model has been used to detect microbial movement/dispersion from the source to sink environment community [87]. The island model considers source ecological communities (URT) as dynamic assemblages of microbes whose existence, deficiency and relative abundances in the sink (LRT) environment are affected by random dispersion, speciation, elimination, and stochastic birth and death events [88]. Some alternate models, such as the neutral model and source tracker, are useful to characterize the spatial movement of the respiratory microbiota as well. Source tracker can determine the contribution of microbiota from one or more sources to a particular sink using the Bayesian algorithm [89]. The neutral model assesses the microbial drift from one source to one sink [86]. Due to the complex ecosystems of bovine respiratory tracts, these models may not perfectly fit the dispersals of bovine respiratory microbiota. However, the outputs of these models should clarify the activities of BRD pathogens somehow. Unfortunately, there are limited BRD studies to measure the spatial dynamics of bovine respiratory microbiota [90]. In future BRD studies, researchers need to determine the microbial movements from the URT to the lung in both healthy and sick calves using these developing models, which may be helpful for the understanding of BRD pathogenesis.
Several statistical models have been developed and could be applied to examine the associations between the bovine respiratory microbiome, host phenotypes (e.g., body weight, BRD) and genotypes, diets and environment. For example, permutational multivariate analysis of variance (PERMANOVA) was used to screen for the factors influencing bovine respiratory microbiota [90]. Correspondingly, a study used bovine respiratory pathogens to predict the clinical outcome using a univariate logistic regression model [21]. Additionally, some analyses, such as Procrustes and multiple co-inertia, are approaches to integrate multi-omics datasets [91], and could be potentially applied to study bacterial-host interactions in BRD cases. These algorithms could allow us to explore the mechanisms of BRD pathogenesis and better identify the factors leading to BRD if they will be used in future studies of the bovine respiratory microbiome.
4 Biogeography of the bovine respiratory tract and microbiota
The URT includes the nostrils, the tonsils, and the nasopharynx, whereas the LRT includes the larynx, trachea, tracheal bronchus, tracheal bifurcation, bronchi, bronchioles, alveolar ducts, alveolar sacs, and alveoli. Considering cattle, the tracheal bronchus specifically arises cranial to the tracheal bifurcation. The primary physiological function of the respiratory tract is to conduct gas exchange (inhalation of oxygen for the exchange and exhalation of carbon dioxide) from the blood. To achieve this, the respiratory tract must warm, filter, and humidify inhaled air and, in doing so, prevent the formation of poisonous or infectious agents that may have access to the respiratory system and threaten function and health. Thus, the pH and temperature gradually increase along the respiratory tract, while the partial pressures of airway oxygen (pO2) and carbon dioxide (pCO2) have opposing gradients that are regulated by environmental air circumstances and the ability of gas exchange at the surface of the lungs (Figure 2). The respiratory microbiota colonizes along the URT and further disperses into the LRT due to the respiratory tract’s anatomical connection with the external environment and direct dispersal along mucosal surfaces, where the microbiota lives. However, the niche-specific microbial communities along the respiratory tract are selectively grown due to the niche physiological parameters (e.g., temperature, pH, ventilation, etc.) that are present within the bovine respiratory tract [11]. In one study, the existence of M. haemolytica in the cattle nasal cavity affected its prevalence in the trachea, although their abundances in both sites were not well correlated [92]. Therefore, understanding the biogeography of the respiratory microbiome provides insights into the complexity of the respiratory ecosystem and BRD.

The harmonious interaction of the respiratory ecosystem in healthy cattle. The complex respiratory ecosystem in healthy cattle is harmonious and contains niche specific environmental properties, microbiota immigrations and host-microbiota interactions, which could resist pathogen colonization to some extent. The gradients of physiological features are along the respiratory tract and move from the nasal cavity to the nasopharynx to the trachea and terminate in the lungs. Lower temperature is reported in the nostrils while the lungs reach body temperature. The partial pressures of airway oxygen (pO2) and carbon dioxide (pCO2) have opposing gradients that are regulated by ventilation and gas exchange at the epithelial surface of the airway [11]. Concerning respiration and microaspiration, micro-particles from the external environment enter the upper respiratory tract (URT) and move to the lungs. Equilibrium is achieved when the host and the respiratory microbiota maintain harmonious interaction [15, 37, 108, 109].
The URT contains several diverse anatomical zones that have different physiological conditions and a greater prevalence to generate contact with the external environment (e.g., diet, water, feces, urine) and other cattle. The nasal cavity, most rostral to the external environment, contains a skin-like, keratinized squamous epithelium. The genera associated with common BRD pathogens such as Mycoplasma, Mannheimia, and Pasteurella are observed in the nostrils of both healthy and BRD affected cattle [1]. Other dominant genera in the bovine nasal cavity, including Psychrobacter, Aggregatibacter, Sphingomonas, Corynebacterium and Coprococcus, are also reported [1, 41]. The microbial community of the nasopharynx, the region near the caudal aspect of the nose, has been widely investigated. The four genera associated with BRD pathogens, including Mycoplasma, Mannheimia, Histophilus and Pasteurella, have also been observed in nasopharyngeal samples from both healthy and BRD-affected animals [19, 22, 24]. Other dominant genera include Pseudomonas, Psychrobacter, Actinobacillus, Clostridium, Acinetobacter, Bacillus, Proteus, Bifidobacterium, Rathayibacter, Cellulomonadaceae, Corynebacterium, Jeotgalicoccus, and Planomicrobium [22, 23, 25, 93, 94]. Temporal changes of the nasopharyngeal microbiota have also been reported. A previous study in cattle confirmed that the nasopharyngeal microbiota changed significantly within several days of arrival to the feedlot, resulting in greater microbial diversity and richness [93]. Pasteurella haemolytica was identified in the tonsils of calves [95]. In addition, the microbiome in the oral cavity and oropharynx is also of interest since Pasteurellaceae associated with the commonly isolated BRD pathogens was detected in the oral cavity of calves [96]. Considering cattle often lick their noses and can actually reach farther into their nostrils than other species, it is not surprising that there would be similarities between the microbes found in the oral cavity and oral pharynx and the URT. However, studies are currently limited regarding the characterization of the oral microbiome in bovines. The first study to investigate the oral microbiomes of cattle with bovine periodontitis found that the most prevalent bacterial microbiota in cattle considered healthy were Pseudomonas, Burkholderia and Actinobacteria, whereas Prevotella, Fusobacterium and Porphyromonas were significantly reported in diseased subjects [97]. A recent study found Streptococcus was the predominant bacteria on the mouth floor, while Streptococcus, Bibersteinia and Mycoplasma were found in the oropharyngeal community in healthy calves [16]. Currently, no published studies of the microbiota found in the oropharynx of BRD-affected cattle exist. The oral and oropharyngeal microbiome may be a new direction for BRD microbiome research, since overlaps of Mycoplasma between the oropharynx and the lungs in cattle have been reported [16].
The LRT is comprised of trachea, tracheal bronchi, bronchioles, and alveoli. Passageways entering the lungs distal to the tracheal bronchus and bifurcation contain bronchi (primary, secondary, and tertiary), bronchioles, alveolar ducts and sacs along the respiratory tree [98, 99]. Until now, little to no bovine research has been conducted that separates the LRT into the trachea and lung to investigate the LRT microbiome, likely due to the supported evidence showing their similarity in the identification of BRD pathogens [44]. However, the abundances of non-dominant abundance bacteria between the trachea and the lung were different [16], and they may have functions in BRD pathogenesis that we may not yet understand. There are some studies that have started to investigate the LRT microbial composition and structure in cattle with different sampling techniques. In the clinically healthy bovine LRT, the genera Mycoplasma, Moraxella, Pasteurella, Mannheimia, Bacteroides and Clostridium were observed in both TTA and BAL samples [1, 20, 43]. Other genera such as Bibersteinia and Prevotella were also observed in the bovine lung [43].
Microbial movement or dispersion within the respiratory tract is another new research direction since it could potentially explain the contribution of the URT microbiota to the lung microbiota. One study concluded that nasopharyngeal microbiota may serve as the primary source for the lungs in healthy calves since the nasopharyngeal region shared similar bacterial composition with the lungs compared to other sampling niches [16]. Similarly, bacterial overlaps between the URT and LRT in cattle have also been described in additional studies [1, 23, 43]. However, in cattle, no studies have yet evaluated the dispersion of the respiratory microbiota with effective statistical models. In healthy subjects, microbiota enter the lungs through an active and continuous process by inhalation of air, direct mucosal dispersal and microaspiration from the URT (Figure 2) [50]. In healthy humans, the adapted island model hypothesizes that the lung microbiome and its growth rate are more affected by microbial immigration and elimination processes than by the effects of the local or lung growth environments [100]. The composition of a healthy lung’s microbiome fit a neutral model when using oral microbiota as a source, which meant the oral microbiota is one of the major sources contributing to the human lung microbiome [51, 86]. Moreover, Venkataraman et al. [86] found that 75% of the oral OTUs were neutrally distributed bacteria from the upper gastrointestinal tract in humans. Shared microbiotas between the oropharynx and lung, such as Mycoplasma and Moraxella, have been found in healthy cattle [16]. Therefore, especially for ruminants, investigation of microbial movement within the airway affected by external sources is complex yet relevant. Since the specific ruminating activity of cattle causes rumen content and rumen microbiota entry to the oral cavity and oropharynx frequently, it is expected that this physiological activity would influence lung microbiota [101], perhaps even more so than it might in monogastric. The shifts in rumen microbiota affected by dietary changes and age may also contribute to the structure of the respiratory microbiome. A previous report found that weaned calves which consumed selenium-biofortified alfalfa hay for nine weeks resulted in favorably reformed microbial communities in the nostrils [102]. Simultaneously, the communication between cattle and the environment, as well as with each other, could also serve as a significant contributor to the URT microbiota and, subsequently, the lung microbiota. Cattle, in particular young calves and dairy breeds (like Holsteins and Jerseys), tend to investigate their environment with their mouths. They lick, suckle, and mouth things in the environment (and each other) frequently, which suggests the environment could serve as an important source for both the oral and URT microbiota. Also, social grooming through licking the head and neck of each other is an important behavioral feature that may additionally influence oral and URT microbiota. Another potential source to consider is the direct inhalation of air in different weather conditions as survivability of microorganisms in the air can be influenced by temperature, humidity, UV light, etc. [11, 103]. These dynamic movements of microbiota are relevant for our understanding of pathogenesis in pulmonary health and disease [104]. For example, altered respiratory rate and effort in a diseased animal may alter the impact that direct inhalation of air might have on the microbiome. Additionally, diseased animals often have altered eating habits, such as inappetence, are sometimes offered diets higher in roughages such as hay, and may also receive antimicrobials in their diet, which may also impact the respiratory microbiome. Moreover, there are multiple factors that affect the URT microbiota and eventual microbial shifts in newly weaned calves, including antibiotics, external environments, stress, and host immunity [11, 15, 105, 106]. Recently, a study confirmed that a single injection of antibiotic including oxytetracycline and tulathromycin led to changes in the nasopharyngeal and fecal microbiota, and increased the relative abundance of several antibiotic resistance genes in these two communities later [28]. Therefore, investigation of microbiota dispersion through the URT or the oral cavity or oropharyngeal region needs to consider all these potential factors that could influence the bovine respiratory ecosystem.
5 Association between the respiratory microbiome and BRD
Disease is one of the main factors influencing the respiratory microbiome. Many studies have confirmed that the alteration of the respiratory microbiota can be observed in calves with clinical BRD. For example, the nasopharyngeal microbiota in BRD-affected feedlot calves was distinct from pen-matched healthy controls [22, 103], a distinct longitudinal shift of microbial composition of the nasopharynx from feedlot arrival to when BRD was diagnosed was observed in another study [90, 107], and bacterial families associated with BRD, including Mycoplasma and Pasteurellaceae, had greater abundances and frequencies in lung tissue samples from calves with BRD signs [52]. Although these reports support the hypothesis that microbial dysbiosis is associated with BRD, we still do not know the accuracy of the association between the microbiome and BRD despite the microbial changes found. A key unknown factor is whether the pathogens and the rest of the microbiota are the inducers of inflammation and onset of BRD, or whether host inflammation or other changes in the respiratory immune system leads to the alteration of microbial structure by selectively overgrowing pathogenic microbes thriving in a more inflammatory milieu [37]. Therefore, integration of the mechanism of microbial dysbiosis or host immunity influenced by causal agents is important for clarity regarding the association of the respiratory microbiome and BRD.
5.1 The microbial ecosystems in healthy bovine respiratory tracts
Understanding the physiological functions of the healthy airway and resident microbial colonization help us further elucidate BRD pathogenesis. In healthy cattle, a mucosal layer covers the respiratory tract and provides immune and physical protection for maintaining homeostasis under the interactions of the host, microbiotas and the external environments [15, 37, 108, 109] (Figure 2). Mucus consists of a complex array of antimicrobial peptides, immunoglobulins, glycoproteins, mucins, polysaccharides, ions, cells, and bacteria which work to maintain harmonizability [15, 109]. In addition, mucus in the URT offers protective barriers against pathogens and toxins from the external environment [110]. Dynamic mucociliary escalator transport works to shift mucus and the small particles it traps (dust, infectious agents, bacteria, etc.) toward the nasopharynx or oropharynx to be swallowed, which helps prevent foreign material from entering the lungs during breathing [111]. The nostrils, nasopharynx and trachea are lined with respiratory epithelium encompassing goblet cells containing pseudostratified columnar epithelial cells for mucous production [112]. Epithelium in the bronchioles shifts gradually toward a cuboidal epithelium with some cilia and club cells which generate glycosaminoglycans and secretory proteins to maintain normal lung physiology and host defense. Epithelial cell surfaces of the alveoli consist of two types: type I cells that regulate gas exchange processes and barrier function, and type II alveolar cells producing lipid-rich surfactants with the ability to prevent bacterial growth. These two epithelial cell types provide the mechanical defenses based on antimicrobial peptides whose secretion increases during the process of inflammation due to their activation by dendritic cells and macrophages [113]. They might also yield cytokines and chemokines that employ and trigger immune cells in infected or damaged areas of the respiratory tract [114]. Surfactant proteins A and D in type II and club cells (formerly named clara cells) potentially have antimicrobial and immunomodulatory roles by binding and inactivating microbial agents [109]. Moreover, the airway microbiota and their metabolites can cooperate with the host to protect against pathogen invasion/overproduction. Steed et al. [115] reported a microbially produced metabolite in humans (desaminotyrosine) that can protect the host by amplifying type I IFN signaling. Other factors, such as the secretion of the ion chloride as well as sodium uptake, are essential to maintaining normal regulation of mucus production [116]. Altogether, the microbiome adapts to a state of microbial symbiosis and homeostasis with the host mucosal surface and immune system. This complex interaction of factors requires additional researches in order to fully understand how different factors (environmental, host, pathogen) can impact this delicate balance.
5.2 The microbial ecosystems in the respiratory tracts of BRD calves
Newly weaned beef calves experience numerous stressors that can cause dysbiosis of the respiratory ecosystem and result in subsequent BRD infection [22, 26, 102, 117, 118]. At feedlot arrival, the homeostasis of microbial communities in healthy cattle prevents pathogens from establishing infection on mucosal surfaces through the consumption of all presented nutrients, the adjustment of the local niche environments and microbial composition, the occupancy of receptor sites, the clustering of antimicrobial molecule construction, and the regulation of mucosal inflammation [11, 15, 109]. A previous study confirmed that the URT microbiota in healthy feedlot cattle rapidly changed from weaning day to arrival at the feedlot (a period of 2 days including 10 h shipping and overnight comingling with other calves) and within 40 days after feedlot arrival [119], indicating that the respiratory ecosystem responded to new challenges in the feedlot. In a previous study, the serotype 2 of Mannheimia haemolytica was more abundant in the nasal cavity before stress (before weaning and shipping to feedlot), while serotype 1, commonly considered to be more pathogenic, was more frequently isolated after feedlot arrival [120]. Likewise, viral and bacterial pathogen invasion through susceptible host defenses leads to microbial dysbiosis, damage of airway tissue and the subsequent development of BRD after feedlot arrival [15]. The proliferation and movement of BRD pathogens within the bovine respiratory tract were still unclear. A previous hypothesis stated that initiation of dysbiosis is directed by the stress and colonization of pathogens into the URT, subsequent shifting of the URT microbiome structure and then proliferation and ultimately infection of the lungs [121]. However, the mechanism of infections in BRD lungs caused by bacterial pathogens is not understood since pathogens are found in healthy lungs in calves. The spatial dynamics of pathogens and their subsequent influences on the bovine respiratory microbiome are necessary to be investigated in future studies.
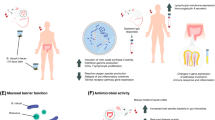
While limited information for the colonization, dispersion and infection of BRD pathogens have been reported, our efforts in preventing and predicting BRD would be improved by increasing our understanding of the initial colonization of pathogens in the URT. The pattern-recognition receptors in the URT that are expressed by the mucosal epithelium and immune cells (e.g., dendritic cells, macrophages, and neutrophils) can recognize pathogens or other noxious substances and then provide signals to regulate acquired immune responses [110]. Increasing pathogen loads generate the URT immune responses, alteration of the niche environment, and subsequent disequilibrium. Under the initial stage of dysbiosis representing an increase in pathogens and host inflammation, the host is considered to be at the “pre-BRD” state in which microbial communities are unstable and easily breach susceptible host defenses. The mucosal barrier function responds to respiratory ecosystem dysbiosis and pathogen invasion by secreting signaling molecules (e.g., inflammatory cytokines and chemokines) in mucus production, and the stimulation of an immune response in local niches [15]. This pre-BRD state can be considered a reversible phase. To resist pathogen invasion, protect the host, and reestablish barrier function, inflammatory events (including IgA production, immune cell recruitment) are activated until risk signals vanish, and reestablishment of the damaged area can begin to occur [37]. Alternatively, the onset of BRD occurs when cattle in the unstable pre-BRD state are subjected to the continuous increase of pathogens. Then, detectable mucosal damage accompanied by a deficiency of the mucosal barrier is usually found in the respiratory tracts of BRD-affected calves (Figure 3).

Respiratory pathogen invasion and the host immune response. Dysbiosis is developed by increased colonization of pathogens into the upper respiratory tract (URT), shifting the structure of the URT microbiome and then proliferating and infecting the lungs. In the pre-BRD state, the mucosal barrier’s function responds to dysbiosis of the bacterial community and the reproduction of pathogens across the airway epithelium by releasing chemokines and cytokines in mucus production and activates local immune cells [37]. The onset of BRD occurs when the unbalanced pre-BRD state suffers a decline into the clinical exacerbation state. Then, detectable damaged epithelium accompanied by a functional deficiency of the mucosal barrier is commonly found in the respiratory tracts of BRD calves.
Citing M. haemolytica as an example, it is a commensal resident in the URT of healthy calves. However, a sudden explosive proliferation associated with stress and viral infection occurs in the URT of susceptible animals [122]. One specific serotype of M. haemolytica (serotype 1) adheres to and colonizes bovine bronchial epithelial cells, and subsequently forms foci of infection through damaging tight-junction integrity, transcytosis and rapidly replicating intracellularly [123]. M. haemolytica produces leukotoxin and lipopolysaccharide which are two important virulence factors that contribute to M. haemolytica’s pathogenicity within the respiratory tract [124]. During the invasion process, M. haemolytica stimulates host epithelial cells producing proinflammatory mediators including cytokines (TNF-α, IL-6 and IL-1β) as well as chemokines (CXCL8). The generation of proinflammatory mediators and the release of lipopolysaccharide and leukotoxin together affect neutrophil movement into the lungs, as they are the primary cause of damaged respiratory tissue associated with BRD [125,126,127]. Then, the BRD pathogens, such as M. haemolytica, could attack differentiated bovine bronchial epithelial cells through cytopempsis, and replicate quickly in cells prior to BRD triggering subsequent widespread cellular injury and lung lesions [123, 125].
5.3 The remained questions for the association between BRD and microbiota
Until recently, the actual mechanism of epithelial damage in BRD cases has not been clear due to the complex interaction of multiple viral and opportunistic bacterial pathogens. Although the process of bovine respiratory viruses cooperating with bacterial pathogens is not clear, in humans, the summarizations of how respiratory viruses interacted with bacteria generating damage peripheral to the bronchi and bronchioles are reported [128]. The presence of viruses in the bovine respiratory tract may also alter the microenvironment of mucosal surfaces and bacterial structure, leading to reduced mucociliary function and cilia damage. Additionally, viral infection could reduce the concentration of antimicrobial peptides, bacterial adherence and invasion by adjusting the host immune system [129]. Segal et al. [130] observed that enhanced expression of inflammatory cytokines (Th17 immune activation) is associated with bacteria in the human LRT, which indicates that the respiratory microbiota could regulate the inflammatory condition at the mucosal surface in humans. Host defense and clearance of viral infections are generally facilitated by an equilibrium of the humoral immune response to neutralize antibodies and the T cell-mediated immune response. Undeniably, viruses in the bovine airway may injure ciliated cells and even the epithelial layer by disturbing cellular tasks and killing infected epithelial cells, decreasing mucociliary transport and consequently exposing the respiratory tracts basement membrane [15, 19]. Thus, an altered niche environment could lead to secondary infections by opportunistic bacterial pathogens following a host immune response by facilitating adhesion and colonization of bacterial pathogens [131, 132].
There are additional knowledge gaps regarding how the microbiota, including commensal bacterial and pathogens, move within the airway of both healthy and BRD calves. Achievements in understanding microbial shifts within respiratory tracts in other species provide alternative statistical techniques to study microbial drift associated with BRD. In humans, the neutral model can estimate microbial drifts from the URT to the lungs successfully in healthy individuals, but fails in patients with cystic fibrosis [51, 86]. In future studies, the microbial drifts in the bovine respiratory tract need to be investigated using similar statistical approaches by considering the specific factors influencing the lung microbiome in cattle, to provide more insights into the pathogenesis of BRD.
6 Conclusions and future perspectives
BRD is a multi-factorial disease and the pathogenesis of BRD is complex and not fully understood. Although the rapid development of sequencing techniques and machine learning or data science help us to better characterize the biogeography of microbial communities in the respiratory tracts of healthy cattle and those affected by BRD, there remain many knowledge gaps. The complexity of the respiratory ecological system limits our understanding of mechanisms such as microbial colonization and host-microbiota interactions, and drift between biogeographical locations. Suitable techniques for sampling niches and data analyses should be considered and optimized based on research goals. Many studies have analyzed the differences of the respiratory microbial composition in healthy or BRD affected cattle in different niche locations.
However, little is known about the dynamic microbial movements within the airway [90]. Moreover, characterizing the function of the respiratory microbiome is one of the next steps and will be an important component in advancing our understanding of pathogenesis. Additionally, advanced multi-omics techniques (i.e., metagenomics, metaproteomics) will provide key insights into the respiratory microbiota and its interaction with epithelial cells for maintenance of homeostasis and dysbiosis associated with BRD development. Future studies concerning BRD and the bovine respiratory microbiome should also consider greater exploration of the interactions between viral and bacterial communities, and the association of microbial dispersion and BRD severity, to determine which factors (especially stress) contribute to pathogen colonization and proliferation. Simultaneously, the bovine respiratory microbiome data should be integrated with host phenotypic data to elucidate the impact on BRD pathogenesis.
Future studies investigating the temporal and spatial dynamics of the bovine respiratory microbiome using multi-omics approaches not only improve our understanding of the pathogenicity of BRD but also provide novel biomarkers for the accurate prognosis and diagnosis of this disease. For example, a panel of biomarkers including bacterial taxa, metabolites, and gene transcripts generated from nasal swabs collected from healthy calves and those with BRD could be developed by machine learning techniques such as Random Forest to predict and diagnose BRD if the nasal microbiome is representative of the bovine respiratory microbiome (at least the upper respiratory system). Advanced tools in bioinformatics should be developed to integrate the high dimensional datasets generated from multi-omics studies using different platforms. Network analysis of the lower respiratory microbiome can reveal bacterial interactions between the opportunistic pathogens and other bacteria. The bacterial taxa that had antagonistic interactions with these pathogens could be further investigated and used as potential probiotics to kill these pathogens. Overall, the deep and continued investigation of the bovine respiratory microbiome opens new possibilities for therapeutics and rapid detection approaches for BRD or other respiratory diseases.
Abbreviations
- BRD:
-
bovine respiratory disease
- NGS:
-
next-generation sequencing
- URT:
-
upper respiratory tract
- LRT:
-
lower respiratory tract
- BVDV:
-
bovine viral diarrhea virus
- BRSV:
-
bovine respiratory syncytial virus
- BHV-1:
-
bovine herpes virus 1
- PI3V:
-
parainfluenza 3 virus
- NPS:
-
nasopharyngeal swab
- TTW:
-
trans-tracheal wash
- BAL:
-
bronchoalveolar lavage
- QIIME:
-
quantitative insights into microbial ecology
- SVM:
-
Support vector machine
- LEfSe:
-
Linear discriminant analysis Effect Size
- AUCRF:
-
Area-under-ROC curve (AUC) of the Random Forest
- PERMANOVA:
-
permutational multivariate analysis of variance
References
Nicola I, Cerutti F, Grego E, Bertone I, Gianella P, D’Angelo A, Peletto S, Bellino C (2017) Characterization of the upper and lower respiratory tract microbiota in piedmontese calves. Microbiome 5:152
USDA (2013) Feedlot. Part iv: health and health management on us feedlots with a capacity of 1,000 or more head, NAHMS Feedlot Studies. Fort Collins: National Animal Health Monitoring System
Holland B, Burciaga-Robles L, VanOverbeke D, Shook J, Step D, Richards C, Krehbiel C (2010) Effect of bovine respiratory disease during preconditioning on subsequent feedlot performance, carcass characteristics, and beef attributes. J Anim Sci 88:2486–2499
Cusack PMV (2004) Effect of mass medication with antibiotics at feedlot entry on the health and growth rate of cattle destined for the Australian domestic market. Aust Vet J 82:154–156
Baptiste KE, Kyvsgaard NC (2017) Do antimicrobial mass medications work? A systematic review and meta-analysis of randomised clinical trials investigating antimicrobial prophylaxis or metaphylaxis against naturally occurring bovine respiratory disease. Pathog Dis 75:ftx083
Peel DS (2020) The effect of market forces on bovine respiratory disease. Vet Clin North Am Food Anim Pract 36:497–508
Woolums AR, Karisch BB, Frye JG, Epperson W, Smith DR, Blanton J Jr, Austin F, Kaplan R, Hiott L, Woodley T, Gupta SK, Jackson CR, McClelland M (2018) Multidrug resistant Mannheimia haemolytica isolated from high-risk beef stocker cattle after antimicrobial metaphylaxis and treatment for bovine respiratory disease. Vet Microbiol 221:143–152
O’Connor AM, Hu D, Totton SC, Scott N, Winder CB, Wang B, Wang C, Glanville J, Wood H, White B, Larson R, Waldner C, Sargeant JM (2019) A systematic review and network meta-analysis of injectable antibiotic options for the control of bovine respiratory disease in the first 45 days post arrival at the feedlot. Anim Health Res Rev 20:163–181
Timsit E, McMullen C, Amat S, Alexander TW (2020) Respiratory bacterial microbiota in cattle: from development to modulation to enhance respiratory health. Vet Clin North Am Food Anim Pract 36:297–320
Amat S, Alexander TW, Holman DB, Schwinghamer T, Timsit E (2020) Intranasal bacterial therapeutics reduce colonization by the respiratory pathogen Mannheimia haemolytica in dairy calves. mSystems 5:e00629-19
Man WH, de Steenhuijsen Piters WA, Bogaert D (2017) The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat Rev Microbiol 15:259–270
Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB (2016) The microbiome and the respiratory tract. Annu Rev Physiol 78:481–504
Berg G, Rybakova D, Fischer D, Cernava T, Verges MC, Charles T, Chen X, Cocolin L, Eversole K, Corral GH, Kazou M, Kinkel L, Lange L, Lima N, Loy A, Macklin JA, Maguin E, Mauchline T, McClure R, Mitter B, Ryan M, Sarand I, Smidt H, Schelkle B, Roume H, Kiran GS, Selvin J, Souza RSC, van Overbeek L, Singh BK, Wagner M, Walsh A, Sessitsch A, Schloter M (2020) Microbiome definition re-visited: old concepts and new challenges. Microbiome 8:103
Lloyd-Price J, Abu-Ali G, Huttenhower C (2016) The healthy human microbiome. Genome Med 8:51
Zeineldin M, Lowe J, Aldridge B (2019) Contribution of the mucosal microbiota to bovine respiratory health. Trends Microbiol 27:753–770
McMullen C, Alexander TW, Leguillette R, Workentine M, Timsit E (2020) Topography of the respiratory tract bacterial microbiota in cattle. Microbiome 8:91
Shukla SD, Budden KF, Neal R, Hansbro PM (2017) Microbiome effects on immunity, health and disease in the lung. Clin Transl Immunol 6:e133
Gao Z, Kang Y, Yu J, Ren L (2014) Human pharyngeal microbiome may play a protective role in respiratory tract infections. Genomics Proteomics Bioinformatics 12:144–150
Lima SF, Teixeira AG, Higgins CH, Lima FS, Bicalho RC (2016) The upper respiratory tract microbiome and its potential role in bovine respiratory disease and otitis media. Sci Rep 6:29050
Klima CL, Holman DB, Ralston BJ, Stanford K, Zaheer R, Alexander TW, McAllister TA (2019) Lower respiratory tract microbiome and resistome of bovine respiratory disease mortalities. Microb Ecol 78:446–456
Cirone F, Padalino B, Tullio D, Capozza P, Losurdo M, Lanave G, Pratelli A (2019) Prevalence of pathogens related to bovine respiratory disease before and after transportation in beef steers: preliminary results. Animals 9:1093
Zeineldin M, Lowe J, de Godoy M, Maradiaga N, Ramirez C, Ghanem M, Abd El-Raof Y, Aldridge B (2017) Disparity in the nasopharyngeal microbiota between healthy cattle on feed, at entry processing and with respiratory disease. Vet Microbiol 208:30–37
Timsit E, Workentine M, van der Meer F, Alexander T (2018) Distinct bacterial metacommunities inhabit the upper and lower respiratory tracts of healthy feedlot cattle and those diagnosed with bronchopneumonia. Vet Microbiol 221:105–113
Holman DB, McAllister TA, Topp E, Wright A-DG, Alexander TMJJVm (2015) The nasopharyngeal microbiota of feedlot cattle that develop bovine respiratory disease. Vet Microbiol 180:90–95
Gaeta NC, Lima SF, Teixeira AG, Ganda EK, Oikonomou G, Gregory L, Bicalho RC (2017) Deciphering upper respiratory tract microbiota complexity in healthy calves and calves that develop respiratory disease using shotgun metagenomics. J Dairy Sci 100:1445–1458
Timsit E, Holman DB, Hallewell J, Alexander TW (2016) The nasopharyngeal microbiota in feedlot cattle and its role in respiratory health. Anim Front 6:44–50
Sanderson MW, Dargatz DA, Wagner BA (2008) Risk factors for initial respiratory disease in United States’ feedlots based on producer-collected daily morbidity counts. Can Vet J 49:373–378
Holman DB, Yang W, Alexander TW (2019) Antibiotic treatment in feedlot cattle: a longitudinal study of the effect of oxytetracycline and tulathromycin on the fecal and nasopharyngeal microbiota. Microbiome 7:7–86
Currin JF, Whittier WD (2005) Recognition and treatment of bovine respiratory disease complex. Vet Med 8:1–3
White BJ, Renter DG (2009) Bayesian estimation of the performance of using clinical observations and harvest lung lesions for diagnosing bovine respiratory disease in post-weaned beef calves. J Vet Diagn Invest 21:446–453
Abutarbush SM, Pollock CM, Wildman BK, Perrett T, Schunicht OC, Fenton RK, Hannon SJ, Vogstad AR, Jim GK, Booker CW (2012) Evaluation of the diagnostic and prognostic utility of ultrasonography at first diagnosis of presumptive bovine respiratory disease. Can J Vet Res 76:23–32
Kayser WC, Carstens GE, Jackson KS, Pinchak WE, Banerjee A, Fu Y (2019) Evaluation of statistical process control procedures to monitor feeding behavior patterns and detect onset of bovine respiratory disease in growing bulls. J Anim Sci 97:1158–1170
Schneider MJ, Tait RG, Busby WD, Reecy JM (2009) An evaluation of bovine respiratory disease complex in feedlot cattle: impact on performance and carcass traits using treatment records and lung lesion scores. J Anim Sci 87:1821–1827
Wittum TE, Woollen NE, Perino LJ, Littledike ET (1996) Relationships among treatment for respiratory tract disease, pulmonary lesions evident at slaughter, and rate of weight gain in feedlot cattle. J Am Vet Med Assoc 209:814–818
Taylor JD, Fulton RW, Lehenbauer TW, Step DL, Confer AW (2010) The epidemiology of bovine respiratory disease: what is the evidence for predisposing factors? Can Vet J 51:1095–1102
Snowder GD, Van Vleck LD, Cundiff LV, Bennett GL (2006) Bovine respiratory disease in feedlot cattle: environmental, genetic, and economic factors. J Anim Sci 84:1999–2008
Hakansson AP, Orihuela CJ, Bogaert D (2018) Bacterial-host interactions: physiology and pathophysiology of respiratory infection. Physiol Rev 98:781–811
Fulton RW, Purdy CW, Confer AW, Saliki JT, Loan RW, Briggs RE, Burge LJ (2000) Bovine viral diarrhea viral infections in feeder calves with respiratory disease: interactions with Pasteurella spp., parainfluenza-3 virus, and bovine respiratory syncytial virus. Can J Vet Res 64:151–159
Thompson PN, Stone A, Schultheiss WA (2006) Use of treatment records and lung lesion scoring to estimate the effect of respiratory disease on growth during early and late finishing periods in South African feedlot cattle. J Anim Sci 84:488–498
Klima CL, Zaheer R, Cook SR, Booker CW, Hendrick S, Alexander TW, McAllister TA (2014) Pathogens of bovine respiratory disease in North American feedlots conferring multidrug resistance via integrative conjugative elements. J Clin Microbiol 52:438–448
McDaneld TG, Kuehn LA, Keele JW (2018) Evaluating the microbiome of two sampling locations in the nasal cavity of cattle with bovine respiratory disease complex (BRDC). J Anim Sci 96:1281–1287
McMullen C, Orsel K, Alexander TW, van der Meer F, Plastow G, Timsit E (2019) Comparison of the nasopharyngeal bacterial microbiota of beef calves raised without the use of antimicrobials between healthy calves and those diagnosed with bovine respiratory disease. Vet Microbiol 231:56–62
Zeineldin MM, Lowe JF, Grimmer ED, de Godoy MRC, Ghanem MM, Abd El-Raof YM, Aldridge BM (2017) Relationship between nasopharyngeal and bronchoalveolar microbial communities in clinically healthy feedlot cattle. BMC Microbiol 17:138
Doyle D, Credille B, Lehenbauer TW, Berghaus R, Aly SS, Champagne J, Blanchard P, Crossley B, Berghaus L, Cochran S, Woolums A (2017) Agreement among 4 sampling methods to identify respiratory pathogens in dairy calves with acute bovine respiratory disease. J Vet Intern Med 31:954–959
Pardon B, Buczinski S (2020) Bovine respiratory disease diagnosis: what progress has been made in infectious diagnosis? Vet Clin North Am Food Anim Pract 36:425–444
Araghi Soure A, Mokhber Dezfuli MR, Zahrayi Salehi T, Rezakhani A, Afshari GR, Nadealian MG, Bahonar AR (2007) Bronchoalveolar lavage and bacteriological findings in calf pneumonia. J Vet Res 62:87–92
Thomas A, Dizier I, Trolin A, Mainil J, Linden A (2002) Comparison of sampling procedures for isolating pulmonary mycoplasmas in cattle. Vet Res Commun 26:333–339
Lavigne MC (2017) Nonbronchoscopic methods [nonbronchoscopic bronchoalveolar lavage (BAL), mini-BAL, blinded bronchial sampling, blinded protected specimen brush] to investigate for pulmonary infections, inflammation, and cellular and molecular markers: a narrative review. Clin Pulm Med 24:13–25
Van Driessche L, Bokma J, Deprez P, Haesebrouck F, Boyen F, Pardon B (2019) Rapid identification of respiratory bacterial pathogens from bronchoalveolar lavage fluid in cattle by MALDI-TOF MS. Sci Rep 9:18381
Dickson RP, Martinez FJ, Huffnagle GB (2014) The role of the microbiome in exacerbations of chronic lung diseases. Lancet 384:691–702
Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, Beck JM, Curtis JL, Huffnagle GB (2015) Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. Mbio 6:e00037
Johnston D, Earley B, Cormican P, Murray G, Kenny DA, Waters SM, McGee M, Kelly AK, McCabe MS (2017) Illumina MiSeq 16S amplicon sequence analysis of bovine respiratory disease associated bacteria in lung and mediastinal lymph node tissue. BMC Vet Res 13:118
Rossi H, Virtala A-M, Raekallio M, Rahkonen E, Rajamäki MM, Mykkänen A (2018) Comparison of tracheal wash and bronchoalveolar lavage cytology in 154 horses with and without respiratory signs in a referral hospital over 2009–2015. Front Vet Sci 5:61
Love WJ, Lehenbauer TW, Van Eenennaam AL, Drake CM, Kass PH, Farver TB, Aly SS (2016) Sensitivity and specificity of on-farm scoring systems and nasal culture to detect bovine respiratory disease complex in preweaned dairy calves. J Vet Diagn Invest 28:119–128
Timsit E, Hallewell J, Booker C, Tison N, Amat S, Alexander TW (2017) Prevalence and antimicrobial susceptibility of Mannheimia haemolytica, Pasteurella multocida, and Histophilus somni isolated from the lower respiratory tract of healthy feedlot cattle and those diagnosed with bovine respiratory disease. Vet Microbiol 208:118–125
Capik SF, White BJ, Lubbers BV, Apley MD, DeDonder KD, Larson RL, Harhay GP, Chitko-McKown CG, Harhay DM, Kalbfleisch TS, Schuller G, Clawson ML (2017) Comparison of the diagnostic performance of bacterial culture of nasopharyngeal swab and bronchoalveolar lavage fluid samples obtained from calves with bovine respiratory disease. Am J Vet Res 78:350–358
Sachse K, Pfützner H, Hotzel H, Demuth B, Heller M, Berthold E (1993) Comparison of various diagnostic methods for the detection of Mycoplasma bovis. Rev Sci Tech 12:571–571
Sachse K, Salam HSH, Diller R, Schubert E, Hoffmann B, Hotzel H (2010) Use of a novel real-time PCR technique to monitor and quantitate Mycoplasma bovis infection in cattle herds with mastitis and respiratory disease. Vet J 186:299–303
Bokma J, Vereecke N, Pas ML, Chantillon L, Vahl M, Weesendorp E, Deurenberg RH, Nauwynck H, Haesebrouck F, Theuns S, Boyen F, Pardon B (2021) Evaluation of nanopore sequencing as a diagnostic tool for the rapid identification of Mycoplasma bovis from individual and pooled respiratory tract samples. J Clin Microbiol 59:e0111021
Cui Y, Qi J, Cai D, Fang J, Xie Y, Guo H, Chen S, Ma X, Gou L, Cui H, Geng Y, Ye G, Zhong Z, Ren Z, Hu Y, Wang Y, Deng J, Yu S, Cao S, Zou H, Wang Z, Zuo Z (2021) Metagenomics reveals that proper placement after long-distance transportation significantly affects calf nasopharyngeal microbiota and is critical for the prevention of respiratory diseases. Front Microbiol 12:700704
Holman DB, Klima CL, Ralston BJ, Niu YD, Stanford K, Alexander TW, McAllister TA (2017) Metagenomic sequencing of bronchoalveolar lavage samples from feedlot cattle mortalities associated with bovine respiratory disease. Genome Announc 5:e01045-17
Zhang MD, Hill JE, Fernando C, Alexander TW, Timsit E, van der Meer F, Huang YY (2019) Respiratory viruses identified in western canadian beef cattle by metagenomic sequencing and their association with bovine respiratory disease. Transbound Emerg Dis 66:1379–1386
Zhang M, Hill JE, Godson DL, Ngeleka M, Fernando C, Huang Y (2020) The pulmonary virome, bacteriological and histopathological findings in bovine respiratory disease from western Canada. Transbound Emerg Dis 67:924–934
Boers SA, Jansen R, Hays JP (2019) Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory. Eur J Clin Microbiol 38:1059–1070
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624
Illumina, HiSeq™ sequencing systems. https://www.illumina.com/documents/products/datasheets/datasheet_hiseq_systems.pdf. Accessed 26 Oct 2010
Ravi RK, Walton K, Khosroheidari M (2018) Miseq: a next generation sequencing platform for genomic analysis. Methods Mol Biol 1706:223–232
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM (2013) Ribosomal database project: data and tools for high throughput rrna analysis. Nucleic Acids Res 42:D633–D642
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P (2012) An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618
Federhen S (2011) The ncbi taxonomy database. Nucleic Acids Res 40:D136–D143
Myer PR, McDaneld TG, Kuehn LA, Dedonder KD, Apley MD, Capik SF, Lubbers BV, Harhay GP, Harhay DM, Keele JW, Henniger MT, Clemmons BA, Smith TPL (2020) Classification of 16S rRNA reads is improved using a niche-specific database constructed by near-full length sequencing. PLoS One 15:e0235498
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq illumina sequencing platform. Appl Environ Microbiol 79:5112–5120
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Tumbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857
Layeghifard M, Hwang DM, Guttman DS (2018) Constructing and analyzing microbiome networks in R. Methods Mol Biol 1849:243–266
Einarsson GG, Zhao J, LiPuma JJ, Downey DG, Tunney MM, Elborn JS (2019) Community analysis and co-occurrence patterns in airway microbial communities during health and disease. ERJ Open Res 5:00128–02017
Zeineldin M, Elolimy AA, Barakat R (2020) Meta-analysis of bovine respiratory microbiota: link between respiratory microbiota and bovine respiratory health. Fems Microbiol Ecol 96:127
Breiman L (2001) Random forests. Mach Learn 45:5–32
Cortes C, Vapnik V (1995) Support-vector networks. Mach Learn 20:273–297
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12:R60
Knights D, Parfrey LW, Zaneveld J, Lozupone C, Knight R (2011) Human-associated microbial signatures: examining their predictive value. Cell Host Microbe 10:292–296
Ma T, Villot C, Renaud D, Skidmore A, Chevaux E, Steele M, Guan LL (2020) Linking perturbations to temporal changes in diversity, stability, and compositions of neonatal calf gut microbiota: prediction of diarrhea. ISME J 14:2223–2235
Calle ML, Urrea V, Boulesteix AL, Malats N (2011) AUC-RF: a new strategy for genomic profiling with random forest. Hum Hered 72:121–132
Scott MA, Woolums AR, Swiderski CE, Perkins AD, Nanduri B (2021) Genes and regulatory mechanisms associated with experimentally-induced bovine respiratory disease identified using supervised machine learning methodology. Sci Rep 11:22916
Venkataraman A, Bassis CM, Beck JM, Young VB, Curtis JL, Huffnagle GB, Schmidt TM (2015) Application of a neutral community model to assess structuring of the human lung microbiome. MBio 6:e02284-14
Dickson RP, Erb-Downward JR, Huffnagle GB (2014) Towards an ecology of the lung: new conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir Med 2:238–246
Hubbell SP (2001) The unified neutral theory of biodiversity and biogeography (MPB-32). Princeton University Press, Princeton
Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST (2011) Bayesian community-wide culture-independent microbial source tracking. Nat Methods 8:761–763
McMullen C, Alexander TW, Orsel K, Timsit E (2020) Progression of nasopharyngeal and tracheal bacterial microbiotas of feedlot cattle during development of bovine respiratory disease. Vet Microbiol 248:108826
Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, Gonzalez A, Kosciolek T, McCall LI, McDonald D, Melnik AV, Morton JT, Navas J, Quinn RA, Sanders JG, Swafford AD, Thompson LR, Tripathi A, Xu ZZ, Zaneveld JR, Zhu Q, Caporaso JG, Dorrestein PC (2018) Best practices for analysing microbiomes. Nat Rev Microbiol 16:410–422
Grey CL, Thomson RG (1971) Pasteurella haemolytica in the tracheal air of calves. Can J Comp Med 35:121–128
Holman DB, Timsit E, Amat S, Abbott W, Buret AG, Alexander TW (2017) The nasopharyngeal microbiota of beef cattle before and after transport to a feedlot. BMC Microbiol 17:70. https://doi.org/10.1186/s12866-017-0978-6
Amat S, Holman DB, Timsit E, Schwinghamer T, Alexander TW (2019) Evaluation of the nasopharyngeal microbiota in beef cattle transported to a feedlot, with a focus on lactic acid-producing bacteria. Front Microbiol 10:1988
Frank GH, Briggs RE (1992) Colonization of the tonsils of calves with Pasteurella haemolytica. Am J Vet Res 53:481–484
Barden M, Richards-Rios P, Ganda E, Lenzi L, Eccles R, Neary J, Oultram J, Oikonomou G (2020) Maternal influences on oral and faecal microbiota maturation in neonatal calves in beef and dairy production systems. Anim Microbiome 2:31
Borsanelli AC, Lappin DF, Viora L, Bennett D, Dutra IS, Brandt BW, Riggio MP (2018) Microbiomes associated with bovine periodontitis and oral health. Vet Microbiol 218:1–6
Pierson RE, Kainer RA (1980) Clinical classification of pneumonias in cattle. Bov Pract 3:73–79
Prohl A, Ostermann C, Lohr M, Reinhold P (2014) The bovine lung in biomedical research: visually guided bronchoscopy, intrabronchial inoculation and in vivo sampling techniques. J Vis Exp 3:51557
Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle GB, Curtis JL (2015) Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc 12:821–830
Glendinning L, Collie D, Wright S, Rutherford KMD, McLachlan G (2017) Comparing microbiotas in the upper aerodigestive and lower respiratory tracts of lambs. Microbiome 5:145
Hall JA, Isaiah A, Estill CT, Pirelli GJ, Suchodolski JS (2017) Weaned beef calves fed selenium-biofortified alfalfa hay have an enriched nasal microbiota compared with healthy controls. PLos One 12:e0179215
Padalino B, Cirone F, Zappaterra M, Tullio D, Ficco G, Giustino A, Ndiana LA, Pratelli A (2021) Factors affecting the development of bovine respiratory disease: a cross-sectional study in beef steers shipped from france to Italy. Front Vet Sci 8:627894
Segal LN, Blaser MJ (2014) A brave new world: the lung microbiota in an era of change. Ann Am Thorac Soc 11:S21-27
Zeineldin M, Lowe J, Aldridge B (2020) Effects of tilmicosin treatment on the nasopharyngeal microbiota of feedlot cattle with respiratory disease during the first week of clinical recovery. Front Vet Sci 7:115
Malmuthuge N, Howell A, Arsic N, Prysliak T, Perez-Casal J, Griebel P (2021) Effect of maternal separation and transportation stress on the bovine upper respiratory tract microbiome and the immune response to resident opportunistic pathogens. Anim Microbiome 3:62
McMullen C, Orsel K, Alexander TW, van der Meer F, Plastow G, Timsit E (2018) Evolution of the nasopharyngeal bacterial microbiota of beef calves from spring processing to 40 days after feedlot arrival. Vet Microbiol 225:139–148
Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, Alexander SN, Bellinghausen LK, Song AS, Petrova YM, Tuvim MJ, Adachi R, Romo I, Bordt AS, Bowden MG, Sisson JH, Woodruff PG, Thornton DJ, Rousseau K, De la Garza MM, Moghaddam SJ, Karmouty-Quintana H, Blackburn MR, Drouin SM, Davis CW, Terrell KA, Grubb BR, O’Neal WK, Flores SC, Cota-Gomez A, Lozupone CA, Donnelly JM, Watson AM, Hennessy CE, Keith RC, Yang IV, Barthel L, Henson PM, Janssen WJ, Schwartz DA, Boucher RC, Dickey BF, Evans CM (2014) Muc5b is required for airway defence. Nature 505:412–416
Ackermann MR, Derscheid R, Roth JA (2010) Innate immunology of bovine respiratory disease. Vet Clin North Am Food Anim Pract 26:215–228
Osman R, Malmuthuge N, Gonzalez-Cano P, Griebel P (2018) Development and function of the mucosal immune system in the upper respiratory tract of neonatal calves. Annu Rev Anim Biosci 6:141–155
Erickson HH, Goff JP, Uemura EE (2015) Dukes’ physiology of domestic animals. Wiley, Hoboken
Cozens D, Grahame E, Sutherland E, Taylor G, Berry CC, Davies RL (2018) Development and optimization of a differentiated airway epithelial cell model of the bovine respiratory tract. Sci Rep 8:853
Lee DF, Salguero FJ, Grainger D, Francis RJ, MacLellan-Gibson K, Chambers MA (2018) Isolation and characterisation of alveolar type ii pneumocytes from adult bovine lung. Sci Rep 8:11927
Su F, Liu X, Liu GH, Yu Y, Wang YS, Jin YP, Hu GD, Hua S, Zhang Y (2013) Establishment and evaluation of a stable cattle type ii alveolar epithelial cell line. PLos One 8:e76036
Steed AL, Christophi GP, Kaiko GE, Sun L, Goodwin VM, Jain U, Esaulova E, Artyomov MN, Morales DJ, Holtzman MJ, Boon ACM, Lenschow DJ, Stappenbeck TS (2017) The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science 357:498–502
Bartoszewski R, Matalon S, Collawn JF (2017) Ion channels of the lung and their role in disease pathogenesis. Am J Physiol Lung Cell Mol Physiol 313:L859–L872
Timsit E, Workentine M, Crepieux T, Miller C, Regev-Shoshani G, Schaefer A, Alexander T (2017) Effects of nasal instillation of a nitric oxide-releasing solution or parenteral administration of tilmicosin on the nasopharyngeal microbiota of beef feedlot cattle at high-risk of developing respiratory tract disease. Res Vet Sci 115:117–124
Pratelli A, Cirone F, Capozza P, Trotta A, Corrente M, Balestrieri A, Buonavoglia C (2021) Bovine respiratory disease in beef calves supported long transport stress: an epidemiological study and strategies for control and prevention. Res Vet Sci 135:450–455
Timsit E, Workentine M, Schryvers AB, Holman DB, van der Meer F, Alexander TW (2016) Evolution of the nasopharyngeal microbiota of beef cattle from weaning to 40 days after arrival at a feedlot. Vet Microbiol 187:75–81
Frank GH, Smith PC (1983) Prevalence of Pasteurella haemolytica in transported calves. Am J Vet Res 44:981–985
Caswell JL (2014) Failure of respiratory defenses in the pathogenesis of bacterial pneumonia of cattle. Vet Pathol 51:393–409
Singh K, Ritchey JW, Confer AW (2011) Mannheimia haemolytica: bacterial-host interactions in bovine pneumonia. Vet Pathol 48:338–348
Cozens D, Sutherland E, Lauder M, Taylor G, Berry CC, Davies RL (2019) Pathogenic Mannheimia haemolytica invades differentiated bovine airway epithelial cells. Infect Immun 87:e00078-19
McClenahan D, Hellenbrand K, Atapattu D, Aulik N, Carlton D, Kapur A, Czuprynski C (2008) Effects of lipopolysaccharide and Mannheimia haemolytica leukotoxin on bovine lung microvascular endothelial cells and alveolar epithelial cells. Clin Vaccine Immunol 15:338–347
Kiser JN, Lawrence TE, Neupane M, Seabury CM, Taylor JF, Womack JE, Neibergs HL (2017) Rapid communication: subclinical bovine respiratory disease - loci and pathogens associated with lung lesions in feedlot cattle. J Anim Sci 95:2726–2731
Zecchinon L, Fett T, Desmecht D (2005) How Mannheimia haemolytica defeats host defence through a kiss of death mechanism. Vet Res 36:133–156
Aulik NA, Hellenbrand KM, Klos H, Czuprynski CJ (2010) Mannheimia haemolytica and its leukotoxin cause neutrophil extracellular trap formation by bovine neutrophils. Infect Immun 78:4454–4466
Griffiths C, Drews SJ, Marchant DJ (2017) Respiratory syncytial virus: Infection, detection, and new options for prevention and treatment. Clin Microbiol Rev 30:277–319
Sudaryatma PE, Saito A, Mekata H, Kubo M, Fahkrajang W, Mazimpaka E, Okabayashi T (2020) Bovine respiratory syncytial virus enhances the adherence of Pasteurella multocida to bovine lower respiratory tract epithelial cells by upregulating the platelet-activating factor receptor. Front Microbiol 11:1676
Segal LN, Clemente JC, Tsay JCJ, Koralov SB, Keller BC, Wu BG, Li YH, Shen N, Ghedin E, Morris A, Diaz P, Huang L, Wikoff WR, Ubeda C, Artacho A, Rom WN, Sterman DH, Collman RG, Blaser MJ, Weiden MD (2016) Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol 1:16031
Czuprynski CJ, Leite F, Sylte M, Kuckleburg C, Schultz R, Inzana T, Behling-Kelly E, Corbeil L (2004) Complexities of the pathogenesis of Mannheimia haemolytica and Haemophilus somnus infections: challenges and potential opportunities for prevention? Anim Health Res Rev 5:277–282
Bosch AATM, Biesbroek G, Trzcinski K, Sanders EAM, Bogaert D (2013) Viral and bacterial interactions in the upper respiratory tract. Plos Pathog 9:e1003057
Acknowledgements
We thank Hunter Usdrowski and Samantha Howe, Department of Animal Science, University of Arkansas, for their help with copyediting.
Funding
This project was supported by Agriculture and Food Research Initiative Competitive Grant no. 20196701629869 from the USDA National Institute of Food and Agriculture.
Author information
Authors and Affiliations
Contributions
JC and JZ conceived the manuscript, collected data and interpreted the relevant literature. JC and JZ wrote the manuscript and generated figures. SC, BK, JR, JP and JZ assisted in revision and proofing. JZ acquired and managed the funding. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chai, J., Capik, S.F., Kegley, B. et al. Bovine respiratory microbiota of feedlot cattle and its association with disease. Vet Res 53, 4 (2022). https://doi.org/10.1186/s13567-021-01020-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-021-01020-x