
Abstract
Background
Borrelia miyamotoi is a newly described relapsing fever spirochete transmitted by ixodid tick species. Little is known about the prevalence of B. miyamotoi infections in humans and ticks in Inner Mongolia, China. Therefore, we investigated the prevalence of B. miyamotoi in Ixodes persulcatus ticks, and we aimed to isolateB. miyamotoi from I. persulcatus from four regions of Greater Khingan, Inner Mongolia, China.
Methods
From May to June each year during the period 2016–2019, host-seeking adult I. persulcatus ticks were collected from vegetation. Genomic DNA was prepared from half of each tick body for PCR template, and the remaining half was used to cultivate B. miyamotoi in BSK-M medium. We employed quantitative real-time PCR (qPCR) to detect Borrelia DNA in the ticks and to calculate the prevalence of B. miyamotoi and infections with other borreliae. For characterization of the isolated B. miyamotoi, we performed draft genome sequencing and multilocus sequencing analysis (MLSA).
Results
A total of 2656 adult I. persulcatus ticks were collected. The overall prevalence of relapsing fever (RF) borreliae in ticks was 5.0% (134/2656) and that of Lyme disease (LD) borreliae was 43.8% (1164/2656). Co-infection with RF and LD borreliae was observed in 63 ticks (2.4%). Ticks that were positive for RF borreliae by qPCR were subjected to glycerophosphodiester diester phosphodiesterase gene (glpQ) PCR amplification and sequencing, through which we identified the RF borrelia specimens as B. miyamotoi. Furthermore, the B. miyamotoi strain Hetao-1 was isolated from I. persulcatus, and a draft genome sequence was obtained from the isolate. Sequencing determined the strain Hetao-1 genome to be approximately 906.1 kbp in length (28.9% average GC content), and MLSA identified the strain as ST633, which has previously been reported in Japan and Mongolia.
Conclusion
We detected B. miyamotoi from I. persulcatus ticks collected in Inner Mongolia, and successfully isolated a B. miyamotoi strain. To our knowledge, this is the first study to culture a B. miyamotoi isolate from China. The data on the prevalence of B. miyamotoi and other borreliae in I. persulcatus ticks will be fundamental for future epidemiological studies of B. miyamotoi disease in Inner Mongolia.

Similar content being viewed by others


Background
Borrelia miyamotoi and other genetically related relapsing fever (RF) borreliae are transmitted by Ixodes ticks, which are also vectors for the agents of Lyme disease [1, 2]. B. miyamotoi was first discovered from the tick I. persulcatus and the rodent Apodemus argenteus in Japan [3] and is considered an emerging pathogen in humans [4]. The spirochete B. miyamotoi has been shown to cause an infectious disease in humans now referred to as B. miyamotoi disease (BMD), and has been reported in Russia, the United States, several European countries, Japan, and China [4,5,6,7,8,9,10,11,12]. BMD manifests as a high fever (up to 40 °C), fatigue, headache, myalgia, chills, nausea, and arthralgia, and meningoencephalitis has been reported in immunocompromised patients [8, 13]. To date, B. miyamotoi has been found in I. scapularis and I. pacificus ticks in North America [14, 15], I. ricinus in Europe [16], and I. persulcatus, I. ovatus, and I. pavlovskyi in Asia [17, 18].
In China, cases of Lyme disease (LD) have been reported in Greater Khingan and Lesser Khingan in the northeast, where the principal LD vector, I. persulcatus, is abundant [19]. I. persulcatus has been confirmed to also carry B. miyamotoi in northeastern China, and BMD was reported in the region in 2018 [12]. However, there has been a dearth of regional surveys of B. miyamotoi infection in tick populations. Not only do the epidemiology and prevalence in China remain unclear, but also the genetic characteristics of the resident B. miyamotoi, due to difficulty in cultivating the bacteria. This basic information on the prevalence of B. miyamotoi infection in ticks, and the genetic characterization of the pathogen, are urgently required for risk assessment of BMD in northeastern China.
The Greater Khingan region in northeastern China offers favorable environmental conditions for the survival and proliferation of I. persulcatus. In this area, tick bites in people are common, and human ixodid tick-borne infections, including those caused by LD borreliae and tick-borne encephalitis virus (genus Flavivirus), are endemic and transmitted by the same tick species [20]. However, some febrile patients have a history of tick bite, and despite the possibility of tick-borne infection, laboratory diagnosis has not been able to identify the infectious agent. In this study, large-scale surveillance for B. miyamotoi was conducted in Greater Khingan to estimate the infection rate of host-seeking adult Ixodes ticks. The tick-derived isolates of B. miyamotoi discovered in this study were subjected to molecular analysis to characterize their genetic profile. The resultant field and laboratory data will serve as a baseline for research aiming to understand the epidemiology of B. miyamotoi in Inner Mongolia, China.
Methods
Study area
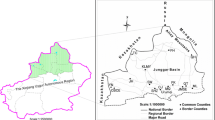
The tick samples in this study were collected in different forested areas throughout Greater Khingan in Hulun Buir City of Inner Mongolia, northeastern China (Fig. 1) [21]. The Greater Khingan forest region of Inner Mongolia is in the northernmost area of the Greater Khingan Mountains, accounting for 46% of the total area, with geographical coordinates of 119° 36′ 30″ to 125° 24′ 00″ E and 47° 03′ 40″ to 53° 20′ 00″ N. The main habitat is primeval forest at an altitude of 250–1745 m, an average annual temperature of −3.5 °C, and annual precipitation of 300–450 mm. In these areas, no specific permission was required for the collection of ticks, and this study did not involve endangered or protected species.

Tick collection areas in this study. Ticks were collected from the areas shown by red stars
Tick collection, DNA extraction, and borrelial cultivation
From May to June each year from 2016 through 2019, host-seeking adult ticks were collected by flagging from vegetation. The collected tick samples were placed in collection tubes, which were classified and numbered according to the sampling time and place. I. persulcatus ticks were identified by morphological characteristics [22]. Ticks were washed with 0.1% sodium hypochlorite and 75% ethanol containing povidone iodine for 5 min, washed again with 3% hydrogen peroxide for 5 min, and then rinsed with sterile water. A genomic DNA PCR template was prepared from half of each tick body according to Yamazaki-Matsune et al. [23]. The remaining half was used to cultivate B. miyamotoi in modified Barbour–Stoenner–Kelly medium (BSK-M: using minimal essential medium alpha [Bio West, Germany] as a substitute for CMRL-1066) under microaerophilic conditions [17, 24]. The tick samples that were positive for RF borreliae and negative for LD borreliae on qPCR were cultivated at 30 °C for 4 weeks, and the growth of spirochetes was examined by dark-field microscopy every 2 weeks.
Detection of borrelial DNA from ticks
Tick lysates were subjected to qPCR assay to detect borrelial infection. The assay was designed by Barbour et al. to specifically detect RF borreliae, including B. miyamotoi, and LD borreliae in tick lysates by multiplex qPCR targeting the 16S rRNA gene (16S rDNA) [25]. To enable the detection of most Borrelia spp., common primers targeted conserved sequences, and specific DNA probes conjugated to non-fluorescent quencher (NFQ) and minor groove-binder architectural protein (MGB) were designed. The two probes were labeled with either the fluorescence reporter group FAM or VIC, and the multi-qPCR reaction system was able to simultaneously detect RF and LD borreliae. The forward and reverse primers were 5′-GCTGTAAACGATGCACACTTGGT-3′ and 5′-GGCGGCACACTTAACACGTTAG-3′, respectively. The corresponding dye-labeled probes, FAM-TTCGGTACTAACTTTTAGTTAA-NFQ-MGB and VIC-CGGTACTAACCTTTCGATTA-NFQ-MGB, were purchased from Applied Biosystems (Foster City, CA). The qPCR was performed using Premix Ex Taq (Probe qPCR, Takara Bio Inc., Shiga, Japan) according to the manufacturer’s instructions and run on a Bio-Rad CFX96 system with 42 PCR cycles. Quality control in the nucleic acid amplification method in this study was performed as previously reported by Espy et al. [26]. A negative (blank) control was used in all qPCR runs. Plasmid DNA was used as a positive control for qPCR as previously reported by Takano et al. [17]. For conventional PCR, genomic DNA extracted from B. miyamotoi strain HT31 was used as a positive control.
Conventional PCR and phylogeny reconstruction using glpQ sequences
To confirm the qPCR results, we performed conventional PCR on the tick-derived isolates. Ticks that were found to be RF-DNA-positive by qPCR were subjected to glycerophosphodiester diester phosphodiesterase gene (glpQ) analysis with PCR-based DNA sequencing [27] using primers purchased from Nanjing GenScript Biological Technology Company: forward primer (glpQ-F), 5′-CATACGCTTATGCYTTRGGMGCTGA-3′, and reverse primer (glpQ-R), 5′-GCAACCTCTGYCATACCTTCTTSTG-3′. The amplicon was approximately 610 bp in length. The reaction conditions of the first PCR were 3 min at 94 °C, then 30 cycles of 30 s at 94 °C, 30 s annealing at 53 °C, 30 s at 72 °C, and finishing with 5 min at 72 °C. In the second PCR, the annealing temperature was changed to 55 °C. We employed the Blend Tag-Plus enzyme (TOYOBO, Osaka, Japan) in the PCR reactions, and the operation was conducted in accordance with the instructions. A negative control was used in each PCR amplification. After amplification, 5 μL of PCR product was separated on 1% agarose gel electrophoresis and visualized by ethidium bromide staining. PCR products containing the target fragment were sent to the Nanjing GenScript Biological Technology Company for bidirectional sequencing. We conducted phylogenetic analyses based on the nucleotide sequences of glpQ (555 bp) using the maximum likelihood method [28] in MEGA 6.0 [29]. We searched for homologous sequences with BLAST in NCBI and downloaded them. Clustal W software was used for sequence alignment analysis, and its reliability was tested with bootstrap analysis with 1000 replicates.
De novo sequencing and multilocus sequencing analysis based on draft genome data of cultured isolate
Genomic DNA was extracted from the B. miyamotoi strain Hetao-1 according to Lim et al. [30]. For genomic library construction, 1 μg of DNA was used for DNA sample preparation, and sequencing libraries were generated using the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, USA) following the manufacturer’s instructions. Briefly, the DNA sample was fragmented by sonication to approximately 350 bp, then DNA fragments were end-polished, A-tailed, and ligated with the full-length adaptor for Illumina sequencing with further PCR amplification. The PCR products were purified (AMPure XP system), and libraries were analyzed for size distribution on the Agilent 2100 Bioanalyzer and quantified using real-time PCR. The whole genome of B. miyamotoi strain Hetao-1 was sequenced using the Illumina NovaSeq PE150. For genome assembly, the raw data were independently assembled using SOAPdenovo v.1.0 [31], SPAdes [32], and ABySS v.2.0 [33]. The assembly results for the three software packages were integrated with CISA software [34], and the assembly result with the fewest scaffolds was selected. De novo sequencing and assembly were performed at Beijing Novogene Bioinformatics Technology Co. Ltd.
Multilocus sequencing analysis (MLSA) was performed on the draft genome sequence of strain Hetao-1 using the concatenated loci of eight genes (clpA, clpX, nifS, pepX, pyrG, recG, rplB, and uvrA) according to Margos et al. [35].
Results
Ticks infected with borreliae in Inner Mongolia
A total of 2656 adult I. persulcatus ticks were collected from the Daxingan mountains in Hulun Buir City of Inner Mongolia, China (Fig. 1). All the collected ticks were screened for borreliae DNA by qPCR targeting of 16S rDNA. As shown in Table 1, ticks harboring B. miyamotoi were found from four districts: Genhe, Yakeshi, Arong Banner, and Arxan. The overall prevalence of RF borreliae, including B. miyamotoi, in ticks of Hulun Buir was 5.0% (134/2656). Specifically, the percentages of ticks positive for RF borreliae were 8.6% in Genhe, 5.1% in Yakeshi, 2.6% in Arong Banner, and 14.0% in Arxan (Table 1). The overall prevalence of LD borreliae was 43.8% (1164/2656), and the percentages of ticks positive for LD borreliae by district were 59.5% in Genhe, 45.0% in Yakeshi, 31.3% in Arong Banner, and 55.8% in Arxan (Table 1). Co-infection with RF and LB borreliae was observed in 46 ticks (1.7%) in Hulun Buir.
Identification of B. miyamotoi in ticks
To identify the RF borrelia in ticks from Hulun Buir, we performed sequence analysis followed by glpQ qPCR of RF-borrelia-positive samples (134 samples). Of these 134 tick samples, we successfully sequenced partial glpQ from 105, and these sequences were 100% identical to each another and to that of the B. miyamotoi strain FR64b (accession number: CP004217) (Fig. 2). Nucleotide sequences of the representative B. miyamotoi isolate from Hulun Buir were deposited in the DDBJ/GenBank DNA database with accession numbers LC570864–LC570882. In 29 of the tick specimens, weak or no amplification of glpQ was seen. The discrepancy between qPCR and glpQ-PCR remains unclear; however, it may be that the sensitivity of the 16S rDNA qPCR reaction was higher than that for conventional PCR targeting the glpQ gene.

Phylogenetic analysis of RF borreliae based on glpQ. The tree was constructed based on glpQ sequences by the maximum likelihood method based on the Kimura two-parameter model with 1000-bootstrap resampling. The bar indicates the percentage of sequence divergence. Borrelia miyamotoi found in this study is indicated by black arrows. The origin of isolation (country and source) for each B. miyamotoi is listed after each lineage. The number in parentheses indicates the GenBank accession number. *Uncultured Borrelia miyamotoi. **Uncultured B. miyamotoi from Ixodes persulcatus in northeastern Inner Mongolia, China. Accession numbers of B. miyamotoi “Ticks” were from LC557142 to LC557151
Genetic characterization of B. miyamotoi DNA from cultured isolates and ticks using glpQ genes
We successfully cultured one B. miyamotoi isolate from an I. persulcatus tick using BSK-M medium. This isolate was used in the initial qPCR confirmation of the pathogen and for analyzing the glpQ sequences. Based on the amplified region of the glpQ gene, the Hetao-1 isolated in this study (accession number: LC557152) clustered together with Siberian B. miyamotoi strains isolated in Japan and Russia (Fig. 2).
MLSA by draft genome sequence
A draft genomic sequence of the B. miyamotoi isolated from ticks sampled in Inner Mongolia was obtained to characterize the Hetao-1 strain. The chromosome of the strain was estimated to be approximately 906.1 kbp in length, with GC content of 28.9%. The chromosome sequence showed 46 single-nucleotide polymorphisms (SNPs) without Ins/Del compared with the B. miyamotoi strain Izh-4 (Accession number: CP024390) [36]. Using the genome assembly data, MLSA was carried out using eight genes (clpA, clpX, nifS, pepX, pyrG, recG, rplB, and uvrA) isolated from the draft genome sequence. Analysis of the eight concatenated housekeeping gene sequences (4776 nucleotides) identified the Chinese Hetao-1 isolate from I. persulcatus as ST633 and as identical to the B. miyamotoi Japanese isolate HT31 (Japan) and M12C4 (Mongolia) (Fig. 3).

MLSA of B. miyamotoi Hetao-1 and other relapsing fever borreliae. A phylogenetic inference of the concatenated housekeeping gene sequences of the representative relapsing fever borreliae is shown. The arrow indicates the B. miyamotoi strain Hetao-1 isolated in this study. Consensus sequences for the eight housekeeping genes were isolated from the draft genome sequence of B. miyamotoi strains Hetao-1, trimmed to lengths and concatenated in the order: clpA, clpX, nifS, pepX, pyrG, recG, rplB, and uvrA according to the Borrelia PubMLST database. For phylogenetic reconstruction, the maximum likelihood model based on the Kimura two-parameter model with MEGA 6.0 was used with 1000 bootstrap replicates. Borrelia turcica IST7 was used as outgroup. The ST number designated in each strain indicates the “sequence type” registered in the Borrelia PubMLST database
DNA accession numbers
Accession numbers obtained in this study are as follow: B. miyamotoi strain Hetao-1 CTP synthase gene (pyrG), DNA helicase gene (recG), ATP-dependent endopeptidase clp ATP-binding subunit gene (clpX), aspartyl aminopeptidase gene (pepX), excinuclease ABC subunit A gene (uvrA), cysteine desulfurase gene (nifS), ATP-dependent Clp protease subunit A gene (clpA), LSU ribosomal protein L2P gene (rplB), 16SrRNA, and flagellin gene (flaB) are LC557142, LC557143, LC557144, LC557145, LC557146, LC557147, LC557148, LC557149, LC557150, and LC557151, respectively.
Discussion
Borrelia miyamotoi is a newly described emerging pathogen. Before this study, no isolates of this spirochete from China had been cultured, and there was little information on B. miyamotoi infections in humans and ticks [12, 37, 38]. In this study, we detected B. miyamotoi in I. persulcatus ticks and successfully isolated a B. miyamotoi strain from I. persulcatus collected in Greater Khingan, Inner Mongolia, China. Our findings demonstrate that B. miyamotoi infection of I. persulcatus is widespread across the regions examined. Similar to Russia and Japan, I. persulcatus ticks in Greater Khingan, China, are abundant mainly in forested regions. To date, there have been no reports on the prevalence of B. miyamotoi infections in humans or ticks in Greater Khingan; however, human tick bites and tick-borne LD or tick-borne encephalitis (TBE) are known to occur frequently in this region. We conducted large-scale tick surveillance for B. miyamotoi in Greater Khingan, as human cases of B. miyamotoi infection were confirmed in Jilin and Heilongjiang, China, in 2018, and B. miyamotoi has previously been found in I. persulcatus ticks [12]. From our research, the prevalence of B. miyamotoi among I. persulcatus was shown to be approximately 5%, which is similar to the prevalence shown by the most recent study in China (approximately 3% in I. persulcatus) [12, 38]. Most previous reports suggest that Ixodes ticks are transmission vectors of B. miyamotoi in North America, Europe, and in other Asian countries. Our results support the hypothesis that I. persulcatus is an important vector of B. miyamotoi in Inner Mongolia, China, as well.
While other tick species (Haemaphysalis and Dermacentor ticks) have been found to carry B. miyamotoi in China [12, 37, 38], the potential for Haemaphysalis and Dermacentor ticks to act as vectors of B. miyamotoi remains unclear. Haemaphysalis, however, is suggested to be a vector of Borrelia species related to B. theileri in Japan [39, 40]. The Borrelia species (i.e., Borrelia sp. HL) was classified as a hard-tick-borne RF borrelia, but it is clearly distinguishable from B. miyamotoi by sequencing of several housekeeping genes [39]. Thus, further study may be required on the competency of these tick species as vectors of B. miyamotoi to assess the risk of BMD in China.
The data collated in this study provide information on the risk of B. miyamotoi human infection (Table 1). Additionally, we detected LD borreliae from 43.8% of I. persulcatus ticks, which are thought to be the vectors of B. miyamotoi and LD borreliae in Inner Mongolia. Although the prevalence of B. miyamotoi is lower than that of LD borreliae in Greater Khingan, B. miyamotoi, as a cause of fever and various other symptoms, is also a risk to public health.
It is known that B. miyamotoi found in Russia and other Asian countries is widely distributed in habitat areas of the Ixodes persulcatus tick. Using MLSA, we revealed that B. miyamotoi ST633, which has previously been found in Mongolia and Japan [18], is distributed in several regions of Inner Mongolia. Furthermore, the draft genome sequence revealed that the Inner Mongolia isolate has only 46 chromosomal SNPs compared with the B. miyamotoi strain Izh-4, although no geographical relationship was observed between these strains. This suggests that clonal expansion of B. miyamotoi may have occurred with the migration of vectors/reservoirs throughout Asian countries, including Russia. To resolve this question, further epidemiological studies of B. miyamotoi infection are required.
Conclusion
In this study, we detected B. miyamotoi in I. persulcatus ticks from Inner Mongolia, China, and successfully isolated a strain of B. miyamotoi. To our knowledge, this is the first report of isolation of B. miyamotoi from China. Further epidemiological studies investigating the prevalence of B. miyamotoi and other borreliae in I. persulcatus ticks will provide new insights into the epidemiological aspects of B. miyamotoi infection in Inner Mongolia, China.
Availability of data and materials
The datasets used and/or analyzed in the current study are available from Gaowa on reasonable request.
References
Mather TN, Mather ME. Intrinsic competence of three ixodid ticks (Acari) as vectors of the lyme disease spirochete. J Med Entomol. 1990;4:646–50.
Niu Q, Guan G, Yang J, Fu Y. Detection and differentiation of Borrelia burgdorferi sensu lato in ticks collected from sheep and cattle in China. BMC Vet Res. 2011;7:56.
Fukunaga M, Takahashi Y, Tsuruta Y, Matsushita O, Ralph D, McClelland M, et al. Genetic and phenotypic analysis of Borrelia miyamotoi sp. nov., isolated from the ixodid tick Ixodes persulcatus, the vector for Lyme disease in Japan. Int J Syst Bacteriol. 1995;45(4):804–10.
Platonov AE, Karan LS, Kolyasnikova NM, Makhneva NA, Toporkova MG, Maleev VV, et al. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg Infect Dis. 2011;17:1816–23.
Krause PJ, Fish D, Narasimhan S, Barbour GA. Borrelia miyamotoi infection in nature and in humans. Clin Microbiol Infect. 2015;21:631–9.
Krause PJ, Narasimhan S, Wormser GP, Rollend L, Fikrig E, Lepore T, et al. Human Borrelia miyamotoi infection in the United States. N Engl J Med. 2013;368(3):291–3.
Hovius JW, de Wever B, Sohne M, Brouwer MC, Coumou J, Wagemakers A, et al. A case of meningoencephalitis by the relapsing fever spirochaete Borrelia miyamotoi in Europe. Lancet. 2013;382(9892):658.
Boden K, Lobenstein S, Hermann B, Margos G, Fingerle V. Borrelia miyamotoi-associated neuroborreliosis in immunocompromised person. Emerg Infect Dis. 2016;22(9):1617–20.
Henningsson AJ, Asgeirsson H, Hammas B, Karlsson E, Parke Å, Hoornstra D, et al. Two cases of Borrelia miyamotoi Meningitis, Sweden, 2018. Emerg Infect Dis. 2019;25(10):1965–8.
Tobudic S, Burgmann H, Stanek G, Winkler S, Schötta AM, Obermüller M, et al. Human Borrelia miyamotoi infection, Austria. Emerg Infect Dis. 2020;26(9):2201–4.
Sato K, Takano A, Konnai S, Nakao M, Ito T, Koyama K, et al. Human infections with Borrelia miyamotoi, Japan. Emerg Infect Dis. 2014;20(8):1391–3.
Jiang BG, Jia N, Jiang JF, Zheng YC, Chu YL, Jiang RR, et al. Borrelia miyamotoi infections in humans and ticks, Northeastern China. Emerg Infect Dis. 2018;24(2):236–41.
Gugliotta JL, Goethert HK, Berardi VP, Telford SR 3rd. Meningoencephalitis from Borrelia miyamotoi in an immunocompromised patient. N Engl J Med. 2013;368(3):240–5.
Mun J, Eisen RJ, Eisen L, Lane RS. Detection of a Borrelia miyamotoi sensu lato relapsing-fever group spirochete from Ixodes pacificus in California. J Med Entomol. 2006;43(1):120–3.
Graham CB, Pilgard MA, Maes SE, Hojgaard A, Eisen RJ. Paired real-time PCR assays for detection of Borrelia miyamotoi in North American Ixodes scapularis and Ixodes pacificus (Acari: Ixodidae). Ticks Tick-borne Dis. 2016;7:1230–5.
Geller J, Nazarova L, Katargina O, Järvekülg L, Fomenko N, Golovljova I. Detection and genetic characterization of relapsing fever spirochete Borrelia miyamotoi in Estonian ticks. PLoS ONE. 2012;7(12):e51914.
Takano A, Toyomane K, Konnai S, Ohashi K, Nakao M, Ito T, et al. Tick surveillance for relapsing fever spirochete Borrelia miyamotoi in Hokkaido, Japan. PLoS ONE. 2014;9(8):e104532.
Iwabu-Itoh Y, Bazartseren B, Naranbaatar O, Yondonjamts E, Furuno K, Lee K, et al. Tick surveillance for Borrelia miyamotoi and phylogenetic analysis of isolates in Mongolia and Japan. Ticks Tick Borne Dis. 2017;8:850–7.
Li ZY, Liu HH, Liu Q, Ma HY, Wei F. Molecular detection of Hepatozoon sp in ticks from northeastern China. Chin J Vet Sci. 2018;38:1720–4.
Chu CY, Jiang BG, He J, Gao Y, Zhang PH, Wu XM, et al. Genetic diversity of Borrelia burgdorferi sensu lato isolates from Northeastern China. Vector Borne Zoonotic Dis. 2011;11(7):877–82.
Iwao K, Nishida K, Kinoshita T, Yamagata Y. Validating land cover maps with Degree Confluence Project information. Geophys Res Lett. 2006;33:L23404. https://warp.da.ndl.go.jp/info:ndljp/pid/11556569/db.cger.nies.go.jp/dataset/landuse/ja/.
Filippova NA. Ixodid ticks of the subfamily Ixodinae. Fauna of the USSR. Leningrad: Publishing House Nauka; 1977. (In Russian).
Yamazaki-Matsune W, Taguchi M, Seto K, Kawahara R, Kawatsu K, Kumeda Y, et al. Development of a multiplex PCR assay for identification of Campylobacter coli, Campylobacter fetus, Campylobacter hyointestinalis subsp. hyointestinalis, Campylobacter jejuni, Campylobacter lari and Campylobacter upsaliensis. J Med Microbiol. 2007;56:1467–73.
Barbour AG. Isolation and cultivation of Lyme disease spirochetes. Yale J Biol Med. 1984;57:521–5.
Barbour AG, Bunikis J, Travinsky B, Hoen AG, Diuk-Wasser MA, Fish D, et al. Niche partitioning of Borrelia burgdorferi and Borrelia miyamotoi in the same tick vector and mammalian reservoir species. Am J Trop Med Hyg. 2009;81:1120–31.
Espy MJ, Uhl JR, Sloan LM, Buckwalter SP, Jones MF, Vetter EA, Yao JD, Wengenack NL, Rosenblatt JE, Cockerill FR 3rd, Smith TF. Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin Microbiol Rev. 2006;19:165–256.
Takano A, Fujita H, Kadosaka T, Konnai S, Tajima T, Watanabe H, et al. Characterization of reptile-associated Borrelia sp. in the vector tick, Amblyomma geoemydae, and its association with Lyme disease and relapsing fever Borrelia spp. Environ Microbiol Rep. 2011;3:632–7.
Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Lim HJ, Lee EH, Yoon Y, Cha B, Son A. Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J Appl Microbiol. 2016;120:379–87.
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–72.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117–23.
Lin SH, Liao YC. CISA: Contig integrator for sequence assembly of bacterial genomes. PLoS ONE. 2013;8(3):e60843.
Margos G, Gatewood AG, Aanensen DM, Hanincová K, Terekhova D, Vollmer SA, et al. MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. Proc Natl AcadSci USA. 2008;105:8730–5.
Kuleshov KV, Margos G, Fingerle V, Koetsveld J, Goptar IA, Markelov ML, et al. Whole genome sequencing of Borrelia miyamotoi isolate Izh-4: reference for a complex bacterial genome. BMC Genomics. 2020;21:16.
Yang Y, Yang Z, Kelly P, Li J, Ren Y, Wang C. Borrelia miyamotoi sensu lato in Pere David Deer and Haemaphysalis longicornis ticks. Emerg Infect Dis. 2018;24:928–31.
Gao Y, Lv XL, Han SZ, Wang W, Liu Q, Song M. First detection of Borrelia miyamotoi infections in ticks and humans from the northeast of Inner Mongolia, China. Acta Trop. 2021;217:105857.
Lee K, Takano A, Taylor K, Sashika M, Shimozuru M, Konnai S, et al. A relapsing fever group Borrelia sp. similar to Borrelia lonestari found among wild sika deer (Cervus nippon yesoensis) and Haemaphysalis spp. ticks in Hokkaido, Japan. Ticks Tick Borne Dis. 2014;5:841–7.
Kumagai Y, Sato K, Taylor KR, Zamoto-Niikura A, Imaoka K, Morikawa S, et al. A relapsing fever group Borrelia sp. is widely distributed among wild deer in Japan. Ticks Tick Borne Dis. 2018;9:465–70.
Acknowledgements
We thank Suzanne Leech, Ph.D., from Liwen Bianji, Edanz Editing China (http://www.liwenbianji.cn/ac), and Kyle R. Taylor, Ph.D. (Washington State University), for editing the English text of a draft of this manuscript.
Funding
The research was supported by the following grants: National Natural Science Foundation of China (nos. 31660032 and 31660044), Inner Mongolia Science and Technology Talent Project for Youth (NJYT-18-A19), Science and Technology Program of Inner Mongolia; Bayan Nur Doctoral Scientific Research Station (no. BKZ2016), The Grassland Elite Program of Inner Mongolia, The Hetao Talent Program of Bayan Nur (to Gaowa), AMED under grant numbers JP20wm0225016, JP20fk0108068, and 21fk0108614.
Author information
Authors and Affiliations
Contributions
G designed the study and wrote the manuscript. All authors contributed to tick collection. All PCR analyses were performed by G. KS and HK supported borrelial culture in Inner Mongolia. HK provided helpful advice and discussion. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gaowa, Wulantuya, Sato, K. et al. Surveillance of Borrelia miyamotoi-carrying ticks and genomic analysis of isolates in Inner Mongolia, China. Parasites Vectors 14, 368 (2021). https://doi.org/10.1186/s13071-021-04809-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-04809-z