
Abstract
We report a male patient with developmental delay carrying an interstitial 4p16.3 deletion of 287 kb, disclosed by oligo array-CGH and inherited from his father with a similar but milder phenotype. This deletion is distal to the Wolf-Hirschhorn syndrome critical regions, but includes the FGFRL1 gene proposed to be a plausible candidate for part of the craniofacial characteristics of Wolf-Hirschhorn syndrome patients. However, the proband lacks the typical facial appearance of the syndrome, but exhibits overgrowth, dysfunction of temporomandibular articulation and a bicuspid aortic valve. Given the pattern of expression of the fibroblast growth factor receptor-like 1 and its involvement in bone and cartilage formation as well as in heart valve morphogenesis, we discuss the impact of its haploinsufficiency in the phenotype.
Similar content being viewed by others


Background
Wolf-Hirschhorn syndrome (WHS; OMIM#194190) is a contiguous gene syndrome caused by deletion of the short arm of chromosome 4. There is a considerable variation in the phenotypic spectrum and a correlation between the size of the deletion and severity of the phenotype has been recognized [1]. Two Wolf-Hirschhorn critical regions, WHCR1 and WHCR2, have been identified within 4p16.3 [2],[3]. Beyond this region, the loss of additional critical genes appears to be responsible for variable present features, the identification of patients with distal 4p deletions could help to identify for pathogenic genes that may be involved in components of the WHS phenotype.
Here we report a patient with a mild phenotype carrying an interstitial 287 kb deletion at 4p16.3, distal to the already described WHS critical regions, and encompassing FGFRL1 gene proposed to be involved in the craniofacial characteristics. We discuss the involvement of this gene in the features displayed by the patient and compare with other patients reported with overlapping imbalances.
Case presentation
Clinical report
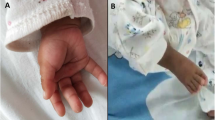
The 11-years-old male proband is the first child of unrelated parents. He was born at 38 weeks of gestation after uneventful pregnancy. His weight was 3900 g (90th percentile), length of 51 cm (50th percentile), and occipitofrontal circumference (OFC) of 36 cm (75th percentile). Apgar scores were 9 and 10 at 1 and 5 minutes, respectively. He started to talk and walk independently at 2 years of age. It was noted to have difficulties in language skills, so he has been having language therapy since the age of 3. At 6 years he started to have pedo-psychiatric support. At the age of 11 years he was referred to genetics evaluation after diagnosis of bicuspid aortic valve in routine echocardiography. On physical examination, the patient showed overgrowth with a weight of 51,5 kg (95th percentile), height of 168 cm (>95th percentile), and OFC of 53,5 cm (50th-75th percentiles). Craniofacial findings included an asymmetric long face, with retrognathia, short philtrum (Figure 1a), high palate with misaligned teeth, dysfunction of temporomandibular articulation, long prominent nose and prominent ears (Figure 1b). The hands have long fingers (Figure 1c), the feet display bilateral sandal gap of the first toes with large halluces (Figure 1d). Because of the developmental delay and overgrowth, homocysteine levels were investigated but showed a normal result. Electroencephalogram was normal, but MRI of the brain revealed a slight diminish of the frontal lobe mainly on the right side. Although in orthopedic examination he didn’t reveal any remarkable alterations, he has a postural kyphosis with sloping shoulders (Figure 1e) and asymmetric position of scapulas. He has a mild global cognitive impairment, but a more pronounced dysfunction in expressive language and articulation of speech.

Phenotypic characteristics of the patient at 14 years of age. (a) Asymmetric long face, with retrognathia and short philtrum, (b) long and prominent nose, large or prominent ears, (c) hands with long fingers, (d) bilateral sandal gap of the first toes with large halluces, (e) postural kyphosis with sloping shoulders.
His father has similar but milder phenotypic features and had strong learning difficulties at school, also with limitations in language articulation. He exhibits a tall stature, the facial appearance displays a slight asymmetry with a long prominent nose, short philtrum and retrognathia. Neither the patient nor his father have had seizures.
In the father’s family there is a history of remarkable delay in his sister, brothers and nephews with learning difficulties. Unfortunately, it was not possible to evaluate any other of the affected family members.
The proband had already normal investigation results of conventional cytogenetics analysis, fragile-X syndrome testing and subtelomeric screening.
Methods
High-resolution comparative genomic hybridization (array-CGH) analysis was performed using Agilent SurePrint G3 Human Genome microarray 180 K (Agilent Technologies, Santa Clara, CA), an oligonucleotide microarray containing approximately 180,000 sixty-mer probes with a 17 kb average probe spacing, as described before [4]. In order to confirm the array-CGH result, MLPA (MRC Holland, Amsterdam, Netherlands) using the set of probes P373, was performed according to the manufacturers’ instructions. The study of the parents was done with the same MLPA probemix.
Results
The oligo array-CGH showed an interstitial deletion at 4p16.3 spanning about 287 kb of genomic DNA from position 927,780 bp (clone A_16_P16565674) to 1,214,915 bp (clone A_16_P00907084) flanked by probe A_16_P00906727 (920,051 bp) and probe A_18_P23419088 (1,228,462 bp) according to UCSC Genome Browser (hg19; GRChBuild 37.1, February 2009) (Figure 2). The deleted segment contains 11 RefSeq genes, 7 genes are described in the OMIM database: DGKQ (#601207) a diacylglycerol kinase, SLC26A1 (# 610130) a sulfate anion transporter 1, IDUA (# 252800) a alpha-L-iduronidase (associated to a lysosomal storage disease by homozygous or compound heterozygous mutation), FGFRL1 (#605830) the fibroblast growth factor receptor-like 1, RNF212 (#612041) a ring finger protein that plays a role in meiotic recombination, SPON2 (#605918) the spondin 2, CTBP1 (#602618) the C-terminal-binding protein 1.

Genomic region of 4p16.3 involved in the deletions of patients with distal imbalances to WHS critical regions (adapted from UCSC genome browser, GRCh37/hg19), displaying the RefSeq genes. A comparison of the extension of the deletions with previously reported patients is also shown.
MLPA confirmed the array result with two probes for genes SPON2 and FGFRL1. Parental testing was performed with MLPA, showing the father to be carrier of the same imbalance.
Discussion
We report a patient with an interstitial deletion on 4p16.3, inherited from the father, this deletion is distal to de WHS critical regions. However, this imbalance encompasses the fibroblast growth factor receptor-like 1 (FGFRL1) gene proposed to be an important candidate contributing to the craniofacial phenotype [5],[6]. The patient described lacks the typical facial features of the WHS, only exhibiting prominent nose and short philtrum. This gene has also been associated with intrauterine growth retardation and postnatal short stature in WHS patients [7], opposite to the finding in our patient that displays overgrowth since the neonatal period. Delayed in speech is a frequent problem in patients with 4p16.3 deletions [8], even among patients with non overlapping imbalances. Probably more than one gene contributes to the language impairment, that in the proband and his father could be associated with the dysfunction of the temporomandibular articulation.
FGFRL1 inhibits cell proliferation, but promotes cell differentiation and induces cell adhesion (involved in cell-cell fusion), it is primarily expressed in cartilaginous tissues and in bone primordia (like the maxillae, the mandibles, the ribs and the nose), the muscles of the tongue and the diaphragm express FGRFL1 at a relatively high level, but the heart, aorta, skeletal muscle, brain, lung, liver, kidney and gut it is expressed at a moderate level [9]. Taking into account its function and pattern of expression, the haploinsufficiency of FGFRL1 could be associated with the craniofacial phenotype of proband including the dysfunction of temporomandibular articulation and the overgrowth could be a consequence of the partial lack of the negative effect of FGFRL1 in cell proliferation particularly in bone tissues. However, in the research work of Catela et al.[6] with an Fgfrl1 −/− null mice, an animal model to dissect the aetiology of WHS, as far as we know overgrowth has never been detected, only short stature is described in mutants that develop to term, but most homozygous embryos suffer prenatal death. Since the heterozygous mutant Fgfrl1 +/− did not display a discernible phenotype [6], these contradictory results are difficult to interpret. Probably in humans the haploinsufficiency of other neighbouring genes plays a critical role in the physiological impact of FGFRL1 heterozygous deletion. The presence of cardiac defects is observed in some patients with WHS [7], but a patient described by Okamoto et al.[10], with a small deletion encompassing WHSC1 but not including the FGFRL1, didn’t exhibit cardiac malformation. On the other hand, in the report of South et al. [11] the echocardiogram of both patients didn’t reveal any alteration although the deletions involved include the FGFRL1 gene. In the patient described the presence of a bicuspid aortic valve could be a coincidental finding or maybe suggests the contribution of FGFRL1 in heart valvulogenesis. This observation is supported by the experimental results with the Fgfrl1 −/− null mice, suggesting a role for Fgfrl1 in valvulogenesis and ventricular septation [6].
In the work reported by Hammond et al.[12] a correlation between the genes involved in different deletions within 4p16.3 and the craniofacial phenotype is done. One of the patients was reported by Faravelli et al.[13], mother of the propositus, she carries a terminal deletion distal to both critical regions of WHS, but including FGFRL1, her facial features included hypertelorism, high nasal bridge, down-slanting of palpebral fissures and large protruding eyes, but our patient with a smaller deletion also harbouring FGFRL1 doesn’t display neither of these characteristics. South et al. [11] described two patients with 4p terminal deletions distal to WHS critical regions but including FGFRL1 and a portion of LETM1 in one of them (Figure 2), they didn’t exhibit the typical facial features of the syndrome, but have prominent forehead, mild telecanthus and normal philtral length, in both it was observed growth delay and mild developmental delay. On the other hand the patient reported by Engbers et al. [5] with a 1.6 Mb deletion have typical facial signs of the WHS, this deletion includes FGFRL1 but also other proximal genes that could be involved in the displayed phenotype (Figure 2). Our report supports the hypothesis that multiple genes are involved in the orchestration, not only of the facial phenotypic features but in the other malformations and cognitive skills observed in WHS patients in a contiguous manner. As it was confirmed in the study of Maas et al.[7], of a cohort of patients with different size deletions and genotype-phenotype correlation, several clinical features of WHS have considerable variable expressivity or penetrance, so it is difficult to pinpoint the genes involved in the more rare aspects of the WHS phenotype. The identification of distal 4p deletions covering about 0.4-0.5 Mb in individuals without a pathogenic phenotype suggests that monosomy of this region is likely benign [14],[15]. But deletions extending to the 1.4 Mb region appear to be pathogenic (Figure 2), although with a mild phenotype [7],[11],[13] and no typical facial appearance of WHS. Deletions extending to 1.6 Mb can however display some overlapping features to the phenotype of WHS [5], suggesting the presence of other genes contributing to the displayed features.
Conclusions
The present report could help to unravel the phenotypic manifestations underlying the haploinsufficiency of the FGFRL1 gene. Its involvement in the bone primordia formation and in heart morphogenesis has been described even in animal models [6]. However, the observation of a tall stature in the proband and his father is a specific finding of this report. A better characterization of patients with atypical and smaller interstitial deletions could contribute to the knowledge of the biological function of neighbouring genes out of the core critical regions of the WHS.
Consent
Written informed consent was obtained for publication of this paper and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, Neri G: On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Sem Med Genet 2008, 148C: 257–269. 10.1002/ajmg.c.30190
Rauch A, Schellmoser S, Kraus C, Dörr HG, Trautmann U, Altherr MR, Pfeiffer RA, Reis A: First known microdeletion within the Wolf-Hirschhorn syndrome critical region refines genotype-phenotype correlation. Am J Med Genet 2001, 99: 338–342. 10.1002/ajmg.1203
Zollino M, Lecce R, Fischetto R, Murdolo M, Faravelli F, Selicorni A, Buttè C, Memo L, Capovilla G, Neri G: Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am J Hum Genet 2003, 72: 590–597. 10.1086/367925
Matoso E, Melo JB, Ferreira SI, Jardim A, Castelo TM, Weise A, Carreira IM: Insertional translocation leading to a 4q13 duplication including the EPHA5 gene in two siblings with attention-deficit hyperactivity disorder. Am J Med Genet Part A 2013, 161A: 1923–1928. 10.1002/ajmg.a.36032
Engbers H, van der Smagt JJ, van 't Slot R, Vermeesch JR, Hochstenbach R, Poot M: Wolf-Hirschhorn syndrome facial dysmorphic features in a patient with a terminal 4p16.3 deletion telomeric to the WHSCR and WHSCR 2 regions. Eur J Hum Genet 2009, 17: 129–132. 10.1038/ejhg.2008.168
Catela C, Bilbao-Cortes D, Slonimsky E, Kratsios P, Rosenthal N, Te Welscher P: Multiple congenital malformations of Wolf-Hirschhorn syndrome are recapitulated in Fgfrl1 null mice. Dis Model Mech 2009, 2: 283–294. 10.1242/dmm.002287
Maas NM, Van Buggenhout G, Hannes F, Thienpont B, Sanlaville D, Kok K, Midro A, Andrieux J, Anderlid BM, Schoumans J, Hordijk R, Devriendt K, Fryns JP, Vermeesch JR: Genotype-phenotype correlation in 21 patients with Wolf-Hirschhorn syndrome using high resolution array comparative genome hybridisation (CGH). J Med Genet 2008, 45: 71–80. 10.1136/jmg.2007.052910
Andersen EF, Carey JC, Earl DL, Corzo D, Suttie M, Hammond P, South ST: Deletions involving genes WHSC1 and LETM1 may be necessary, but are not sufficient to cause Wolf-Hirschhorn syndrome. Eur J Hum Genet 2014, 22: 464–470. 10.1038/ejhg.2013.192
Trueb B, Taeschler S: Expression of FGFRL1, a novel fibroblast growth factor receptor, during embryonic development. Int J Mol Med 2006, 17: 617–620.
Okamoto N, Ohmachi K, Shimada S, Shimojima K, Yamamoto T: 109 kb deletion of chromosome 4p16.3 in a patient with mild phenotype of Wolf-Hirschhorn syndrome. Am J Med Genet A 2013, 161A: 1465–1469. 10.1002/ajmg.a.35910
South ST, Bleyl SB, Carey JC: Two unique patients with novel microdeletions in 4p16.3 that exclude the WHS critical regions: Implications for critical region designation. Am J Med Genet A 2007, 143A: 2137–2142. 10.1002/ajmg.a.31900
Hammond P, Hannes F, Suttie M, Devriendt K, Vermeesch JR, Faravelli F, Forzano F, Parekh S, Williams S, McMullan D, South ST, Carey JC, Quarrell O: Fine-grained facial phenotype-genotype analysis in Wolf-Hirschhorn syndrome. Eur J Hum Genet 2012, 20: 33–40. 10.1038/ejhg.2011.135
Faravelli F, Murdolo M, Marangi G, Bricarelli FD, Di Rocco M, Zollino M: Mother to son amplification of a small subtelomeric deletion: a new mechanism of familial recurrence in microdeletion syndromes. Am J Med Genet A 2007, 143A: 1169–1173. 10.1002/ajmg.a.31723
Van Buggenhout G, Melotte C, Dutta B, Froyen G, Van Hummelen P, Marynen P, Matthijs G, de Ravel T, Devriendt K, Fryns JP, Vermeesch JR: Mild Wolf–Hirschhorn syndrome: microarray CGH analysis of atypical 4p16.3 deletions enables refinement of the genotype–phenotype map. J Med Genet 2004, 41: 691–698. 10.1136/jmg.2003.016865
Ravnan JB, Tepperberg JH, Papenhausen P, Lamb AN, Hedrick J, Eash D, Ledbetter DH, Martin CL: Subtelomere FISH analysis of 11 688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet 2006, 43: 478–489. 10.1136/jmg.2005.036350
Acknowledgments
The authors would like to gratefully acknowledge the patient and his parents for their collaboration.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EM drafted the manuscript and was involved in the interpretation of array-CGH results; FB provided the clinical data, the biological samples and revised the manuscript critically; JF and LMP carried out the microarray and molecular genetic studies; AM performed the cytogenetic studies; JBM was involved in array-CGH analysis and revised the manuscript critically; IMC coordinated the study, made substantial contribution to conception design and interpretation of data and revised the manuscript critically. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Matoso, E., Ramos, F., Ferrão, J. et al. Interstitial 287 kb deletion of 4p16.3 including FGFRL1 gene associated with language impairment and overgrowth. Mol Cytogenet 7, 87 (2014). https://doi.org/10.1186/s13039-014-0087-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-014-0087-2