
Abstract
Many human cancers exhibit elevated prostaglandin (PG) levels due to upregulation of cyclooxygenase-2 (COX-2), a key enzyme in eicosanoid biosynthesis. COX-2 over-expression has been observed in about 40% of cases of invasive breast carcinoma and at a higher frequency in preinvasive ductal carcinoma in situ tumors, Extensive pharmacologic and genetic evidence implicates COX enzymes in neoplasia. Epidemiologic analyses demonstrate a protective effect of COX-inhibiting nonsteroidal anti-inflammatory drugs with respect to human cancer. Complementary experimental studies have established that both conventional nonsteroidal anti-inflammatory drugs and selective COX-2 inhibitors suppress mammary tumor formation in rodent breast cancer models. Furthermore, knocking out Cox-2 reduces mammary tumorigenesis and angiogenesis, and, conversely, transgenic COX-2 over-expression induces tumor formation. The utility of COX/PG signaling as a target for chemoprevention has been established by randomized controlled clinical trials. However, these studies also identified increased cardiovascular risk associated with use of selective COX-2 inhibitors. Thus, current efforts are directed toward identifying safer approaches to antagonizing COX/PG signaling for cancer prevention and treatment, with a particular focus on PGE2 regulation and signaling, because PGE2 is a key protumorigenic prostanoid.
Similar content being viewed by others


Introduction
The past few years have witnessed intense interest in the role played by the cyclooxygenase (COX) family of prostaglandin (PG) synthases in cancer. Upregulation of the inducible isoform COX-2 has been identified in many human cancers and precancerous lesions. Initially recognized in the context of colorectal cancer, COX-2 over-expression has also been detected in approximately 40% of cases of human breast carcinoma as well as in preinvasive ductal carcinoma in situ (DCIS) lesions. Furthermore, epidemiologic analyses suggest a protective effect of COX inhibitory drugs with respect to both colon and breast cancer. Together, these observations have stimulated widespread enthusiasm for COX-2 as a molecular target for cancer prevention.
Substantial data support the validity of COX-2 as an anti-cancer target. Transgenic COX-2 over-expression drives mammary tumor formation, and, conversely, knocking out Cox-2 reduces tumor formation in rodent models of intestinal, breast, and skin cancer. Consistent with these genetic studies, selective COX-2 inhibitors (COXibs) have proven to be effective in suppressing experimental tumorigenesis. Furthermore, several recently reported randomized clinical trials have shown that COXibs significantly reduce the incidence of colorectal adenomas in humans. Dismayingly, these trials also identified an increased risk for cardiovascular events associated with COXib use, suggesting that COXibs may not be sufficiently safe for general use as cancer chemopreventive agents. Nevertheless, the demonstrated role of COX/PG signaling in neoplasia identifies this pathway as an important anticancer target. Therefore, it behooves us to identify alternative components of the COX/PG signaling pathway, antagonism of which will achieve protection comparable to that afforded by COXibs but with minimal collateral toxicity.
Here, I review the data implicating COX/PG signaling in breast cancer, and consider alternative approaches to suppressing this pathway that may have clinical utility.
Cyclooxygenases, prostaglandins, and cancer
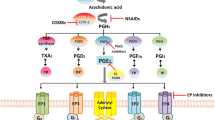
The COX family of enzymes comprises two members. COX-1 (more properly PTGS1 [PG-endoperoxide synthase [1]]) is generally considered to be ubiquitously expressed, whereas COX-2 (or PTGS2 [PG-endoperoxide synthase [2]]) is constitutively expressed in only a limited range of tissues, including placenta, brain, and kidney [1, 2]. However, COX-2 upregulation is elicited by numerous stimuli, including cytokines, growth factors and oncogenes, and is both an important component of the inflammatory response as well as an early response gene. Both COX enzymes catalyze the conversion of arachidonic acid to PGG2 and subsequently to PGH2, which acts as a substrate for multiple isomerases that are individually responsible for the generation of eicosanoid products, including PGE2, prostacyclin (PGI2), and thromboxane A2 (Figure 1). COX-derived prostanoids contribute to many bodily functions, including hemostasis, platelet aggregation, kidney and gastric function, and several female reproductive processes [1, 2]. Eicosanoids are also key mediators of pain, fever, and inflammation. Hence, COX enzymes are the targets for common painkillers of the non-steroidal anti-inflammatory drug (NSAID) class. Inhibitors that are selective for COX-2 have also been developed as anti-inflammatory agents, with the goal of minimizing the gastrointestinal complications associated with traditional NSAIDs.

Eicosanoid metabolism and signaling. Cyclooxygenase (COX) enzymes convert arachidonic acid to the intermediate prostaglandin PGG2, and thence to PGH2. Subsequent enzymatic steps, catalyzed by specific isomerases, generate a variety of eicosanoid products. Thromboxane (TX) A2 and prostacyclin (PGI2), products of platelet COX-1 and endothelial COX-2, respectively, are thought to play opposing roles in cardiovascular biology. Most important in the context of epithelial tumorigenesis, PGE2 is generated from PGH2 through the action of PGE synthases. Signaling downstream of PGE2 is initiated via interaction of PGE2 with cognate PGE2 receptors EP1 to EP4. PGE2 signaling can be terminated via catabolism mediated by 15-hydroxyprostaglandin dehydrogenase (15-PGDH). Elevated PGE2 levels in neoplastic tissues can thus be a consequence of COX-2 over-expression, PGE synthase modulation, and/or loss of 15-PGDH expression.
Aberrant activation of COX/PG signaling is widespread in human neoplasia [1, 3]. The first indication of this was provided by the detection of elevated PG levels in cancer samples. Subsequently, it became apparent that cancer-associated increases in PG synthesis correlated with COX-2 over-expression. COX-2 upregulation is particularly striking in colon cancer. Thus, COX-2 protein is virtually undetectable in normal colonic mucosa, but more than 85% of colorectal adenocarcinomas have elevated COX-2 levels [1, 4]. Similarly, COX-2 is undetectable in normal breast tissue by immuno-histochemistry, but it is over-expressed in about 40% of human breast carcinomas (discussed below). These and similar data from cancers of other organ sites have identified COX-2 upregulation as a common event in human cancer, and thus they suggest that COX-2 may play a role in tumorigenesis.
A protumorigenic role for COX enzymes and COX-derived PGs has also been inferred from epidemiologic analyses. Several studies have identified an inverse relationship between colon cancer incidence and use of NSAIDs, which inhibit COX activity [4]. Similar data are also available for breast cancer [5–12]. Discrepant findings in some reports may reflect the fact that human breast cancers do not uniformly over-express COX-2. Nevertheless, epidemiologic data broadly support a protumorigenic role for COX enzymes in breast cancer.
Cyclooxygenase-2 expression in breast cancer
Elevated COX-2 protein levels have been detected immunohistochemically in approximately 40% of invasive breast carcinomas, with individual studies reporting frequencies ranging from 17% to 84% [13–26]. COX-2 protein is predominantly confined to the tumor epithelium, with negligible expression in normal epithelium (Figure 2). In contrast, COX-1 appears to be ubiquitously expressed in mammary tissues [21, 25]. Cox-2 upregulation has also been detected in rodent mammary tumors, including those from both transgenic mouse and carcinogen-dependent breast cancer models [27–31]. Consistent with human observations, Cox-2 protein is present in epithelial cells in rodent tumors [30, 31]. These localization data provide a marked contrast to those from colorectal cancer studies, which have identified substantial COX-2 expression in the stromal component of intestinal adenomas [32]. It remains to be determined whether stromal COX-2 expression plays a significant role in breast neoplasia.

COX-2 expression in human breast tumors. Cyclooxygenase (COX)-2 protein has been detected in human breast biopsies in both (a) ductal carcinoma in situ and (b) infiltrating mammary carcinoma using immunohistochemistry on formalin-fixed tissue sections. Representative data are reproduced from [21] by permission of Wiley-Liss Inc., a subsidiary of John Wiley and Sons Inc. (Copyright (2000) American Cancer Society.)
COX-2 over-expression in human breast cancers correlates with several parameters that are characteristic of aggressive breast disease, including large tumor size, high grade, high proliferation, hormone receptor negative status, and over-expression of HER2 (human epidermal growth factor receptor 2; also called neu and c-ERBB2) [13, 24, 26, 33]. Consistent with these findings, Ristimaki and colleagues [26] have identified an inverse relationship between COX-2 protein levels and disease-free survival (Figure 3). Because HER2/neu can induce COX-2 transcription in vitro, the correlation between HER2/neu and COX-2 expression in breast carcinomas probably reflects a causal relationship [13, 24, 26, 33, 34]. Interestingly, both HER2/neu and COX-2 are expressed at higher frequencies in DCIS (50% to 60% and 63% to 85%, respectively), which again is suggestive of a potential interrelationship. The high frequency of COX-2 over-expression in DCIS, a common precursor to invasive breast cancer, identifies COX/PG signaling as a potentially useful target for preventing progression of DCIS to invasive disease [13, 17, 20, 21, 23, 35, 36]. Intriguingly, COX-2 expression has also been detected in focal regions of normal breast in association with silencing of CDKN2A (p16INK4a), suggesting that COX-2 upregulation may be a very early event in breast neoplasia [37].

COX-2 expression in human breast cancer correlates with decreased disease-free survival. Distant disease-free survival of breast cancer patients was plotted as a function of cyclooxygenase (COX)-2 expression: score 0 = no COX-2 expression (n = 133); score 1 = weak COX-2 expression (n = 854); score 2 = moderate COX-2 expression (n = 511); and score 3 = strong COX-2 expression (n = 78). Elevated expression of COX-2 protein correlated with reduced survival (P < 0.0001; log rank test). Reproduced from [26] with permission from the American Association of Cancer Research.
Cyclooxygenase-2 contributes to breast cancer: experimental evidence
Since Cox-2 is over-expressed in mammary tumors from rodent breast cancer models, these animals provide useful experimental systems in which to evaluate the role of COX enzymes. Numerous studies have shown that experimental breast cancer can be suppressed by inhibiting Cox activity with either conventional NSAIDs or COXibs [38, 39]. Furthermore, genetic ablation of Cox-2 decreases mammary tumor formation [40]. Strikingly, transgenic over-expression of COX-2 is sufficient to induce mammary neoplasia in multiparous animals, providing direct evidence of the in vivo oncogenicity of COX-2 [41]. Thus, animal-based approaches have played a pivotal role in definitively establishing that COX-2 contributes to breast cancer.
Cyclooxygenase inhibitors suppress experimental breast cancer
The efficacy of COX inhibitors as anticancer agents has been tested in a variety of animal models (for detailed reviews, see Howe [38], Howe and coauthors [39], Reddy [42], and Corpet and Pierre [43]). The ability of conventional NSAIDs such as indomethacin and flurbiprofen to suppress carcinogen-induced mammary tumor formation was first demonstrated more than 20 years ago. More recently, following the development of COXibs, these agents have also been tested in animal breast cancer models. Several COXibs, including celecoxib, nimesulide and rofecoxib, have demonstrated chemopreventive efficacy in chemical carcinogenesis models [30, 38, 39, 44, 45]. COX inhibitors also reduce the growth rate of implanted tumors, suggesting potential therapeutic utility. Chemically-induced tumors tend to be hormone-dependent, providing a valuable model for human breast cancers, of which approximately two-thirds are estrogen dependent. Additionally, we considered it relevant to evaluate COXib efficacy in an estrogen receptor (ER)-negative model, because several groups had reported a correlation between COX-2 over-expression and ER-negative status [13, 16, 24, 26]. HER2/neu transgenic mice offer a compelling test system, because the tumors not only lack ER but also express both HER2/neu and Cox-2, thus recapitulating the relationship between HER2/neu and COX-2 in human breast cancer. Using HER2/neu transgenic mice, both we and others have shown that ER-negative tumor formation is significantly delayed by celecoxib administration [29, 46]. These data suggest that antagonism of COX/PG signaling could be useful with respect to both ER-negative and HER2/neu-over-expressing breast cancers. The demonstrated chemopreventive efficacy of NSAIDs and COXibs in animal models is consistent with epidemiologic studies that show reduced breast cancer incidence in association with NSAID use [5–12].
Mammary tumorigenesis is reduced in cyclooxygenase-2-null mice
While the anticancer effects of conventional NSAIDs and COXibs strongly implicate COX enzymes in breast cancer, numerous COX-independent effects have been ascribed to NSAIDs [47, 48]. Therefore, we also used a complementary genetic approach to definitively address the involvement of COX-2 in mammary tumorigenesis. Mice with targeted disruption of the Cox-2 gene were first utilized to establish the contribution of COX-2 to tumorigenesis by Taketo and colleagues, using an intestinal cancer model [32]. We have adopted a parallel approach, crossing Cox-2 knockout mice with the HER2/neu transgenic mouse mammary tumor virus (MMTV)/neu deletion mutant (NDL) strain, to test the role of COX-2 in breast cancer [40].
MMTV/NDL mice express a mutationally activated HER2/neu transgene that drives formation of multiple DCIS-like tumors in each mammary gland. These tumors subsequently progress to invasive carcinomas and ultimately metastasize to the lung, thus recapitulating the human disease [49]. Hence, we employed the MMTV/NDL strain as a breast cancer model system in which to examine the consequences of knocking out Cox-2. MMTV/NDL mice were crossed with Cox-2-deficient mice, and tumor multiplicity was compared in HER2/neu transgenic mice that were Cox-2 wild type, heterozygous, and null. We found that tumor multiplicity was significantly reduced in both Cox-2 heterozygous and null animals relative to Cox-2 wild-type control animals (P < 0.001; Figure 4a). Complete ablation of Cox-2 reduced the mean tumor multiplicity by approximately 50%. Additionally, an overall shift toward a higher proportion of smaller tumors in Cox-2 null animals relative to Cox-2 wild-type animals was observed (P = 0.02; Figure 4b), suggesting that Cox-2 contributes not only to mammary tumor formation but also to tumor growth. PGE2 levels in MMTV/NDL mammary glands correlated with Cox-2 gene dosage. Thus, PGE2 levels (ng/mg protein) in Cox-2 wild-type, heterozygous, and null mammary tissues were 0.69 ± 0.11 (n = 7), 0.53 ± 0.15 (n = 5; P = 0.043), and 0.35 ± 0.07 (n = 5; P = 0.0001), respectively. These data provide the first genetic evidence that Cox-2 contributes to HER2/neu-induced mammary tumorigenesis [40].

Knocking out Cox-2 reduces mammary tumorigenesis. Mouse mammary tumor virus (MMTV)/neu deletion mutant (NDL) mice, which express a mammary-targeted HER2/neu transgene, were crossed with Cox-2-deficient mice, and mammary tumor formation was evaluated in age-matched virgin MMTV/NDL females that were Cox-2 wild type (WT; n = 72), heterozygous (HET; n = 42), and null (NULL; n = 18). (a) Tumor multiplicity was significantly reduced in Cox-2 deficient mice (data shown are mean ± SEM. *P < 0.001, by likelihood ratio test. (b) The percentage of tumors in each of the indicated size categories was calculated for each genotype. The proportion of large tumors was significantly reduced in Cox-2 deficient MMTV/NDL animals relative to Cox-2 wild-type controls (P = 0.02). Reproduced with permission from [40].
Intriguingly, our Cox-2 knockout experiment also suggested a novel role for Cox-2 in mammary gland vascularization. Specifically, we observed a striking reduction in mammary vasculature in Cox-2 null animals relative to wild-type controls. Blood vessels were virtually absent from both dysplastic regions and areas of normal looking epithelium (Figure 5a). Consistent with the marked reduction in mammary vasculature in Cox-2 null mammary tissues, the expression of several angiogenesis-associated genes was decreased (Figure 5b), including VEGF (which encodes vascular endothelial growth factor), Ang1 and Ang2 (which encode Tie-2 ligands), and Flk-1 and Flt-1 (which encode vascular endothelial growth factor receptors). Our data contrast with those obtained in studies using mouse colorectal cancer models, which suggest that Cox-2 contributes primarily to the growth and vascularization of intestinal tumors beyond 1 mm in diameter [50, 51]. Thus, in addition to its previously described role in tumor angiogenesis, Cox-2 may also contribute to blood vessel formation in nontumorous mammary tissues.

Mammary gland vascularization is reduced in Cox-2 knockout mice. (a) Mammary gland tissue sections from age-matched virgin mouse mammary tumor virus (MMTV)/neu deletion mutant (NDL) females that were Cox-2 wild type (subpanels a to f) and Cox-2 null (subpanels g to l) were subjected to anti-CD31 immunohistochemistry, and counterstained with methyl green. Both the number and size of blood vessels were strikingly reduced in Cox-2 null samples. (b) Expression levels of angiogenesis-related genes were compared by quantitative reverse transcription polymerase chain reaction in MMTV/NDL mammary glands from Cox-2 wild-type (blue columns) and Cox-2 null females (yellow columns). The height of the columns indicate means normalized to the mean expression level of that gene in MMTV/NDL, Cox-2 wild-type samples; the bars indicate the standard error. Expression of VEGF, Ang1, and Flt1 was significantly reduced (P = 0.016, 0.049 and 0.010, respectively). The average of log values across all six genes for each mouse, representing a global effect, was significantly higher in wild-type tissues than in null tissues at P = 0.025. Reproduced with permission from [40].
Cyclooxygenase-2 acts as an oncogene in vivo
As described above, mammary tumorigenesis can be suppressed by both genetic and pharmacologic ablation of Cox-2, thus clearly identifying a role for COX-2 in breast neoplasia. Furthermore, Hla and colleagues have provided definitive evidence for in vivo oncogenic action of COX-2 through generation of an MMTV/COX-2 transgenic mouse strain [41]. COX-2 over-expression in mouse mammary gland induced tumor formation in more than 85% of multiparous mice [41]. Prior to visible tumor formation, COX-2 induced angiogenesis, as evidenced by increased microvessel density and expression of proangiogenic genes [52]. Furthermore, mammary gland involution after weaning was delayed in transgenic animals relative to wild-type littermates, with an accompanying decrease in apoptosis [41]. Therefore, these data suggest that COX-2 may drive tumor formation both by increasing angiogenesis and by suppressing apoptotic cell death.
Together, these genetic and pharmacologic approaches provide irrefutable evidence that COX-2 contributes to breast cancer. Furthermore, these studies offer mechanistic insights into the role of COX-2 in mammary neoplasia, indicating that COX-2 is important for angiogenesis and may also play a critical role in suppressing apoptosis. These observations are consistent with earlier studies in cell culture and in intestinal tumor models. Multiple additional protumorigenic roles have been described for COX-derived prostanoids, including as proproliferative stimuli, immune system depressants, and promoters of cell invasiveness. COX-2 is also believed to contribute to the establishment of bone metastases. Of particular relevance to breast cancer, PGs can increase estrogen biosynthesis via upregulation of aromatase transcription (discussed below). Thus, numerous mechanisms are likely to contribute to the protumorigenic and metastasis-promoting effects of eicosanoids.
The cyclooxygenase-aromatase connection
The relationship between COX and aromatase enzymes is currently attracting considerable interest. The aromatase cytochrome P450, encoded by the CYP19 gene, is responsible for estrogen biosynthesis and thus is extremely relevant to breast carcinogenesis, since 60% to 70% of breast cancers are hormone-dependent. Interestingly, correlations between COX and aromatase expression have been observed in human breast carcinomas [53, 54]. These correlations are thought to reflect a causal link, because PG signaling can stimulate transcription of the CYP19 gene [55–60]. PG-dependent CYP19 induction is achieved through cAMP accumulation. At least two PGE2 receptor (EP) isoforms signal by increasing adenylate cyclase activity [61], and CYP19 is transcribed from cAMP-responsive promoters in breast tumor-proximal stromal tissue. Thus, both paracrine (tumor cell derived) and autocrine (stromally produced) PGE2 may contribute to aromatase upregulation in breast cancers. Recent animal studies have demonstrated that mammary aromatase activity is significantly reduced in Cox-2 knockout mice, and, conversely, that aromatase expression and activity are increased by transgenic COX-2 over-expression. Together these datasets definitively establish that Cox-2 can regulate aromatase in vivo in mammary tissues [56, 62].
Peripheral aromatization is largely responsible for estrogen production in postmenopausal women, and mammary adipose tissue is a particularly important local estrogen source. Therefore, regulation of mammary aromatase synthesis by COX/PG signaling is most likely to be important in the context of postmenopausal breast cancer. The ability of COX-derived PGs to increase aromatase expression and hence local estrogen levels may provide a partial explanation for reports of reduced incidence of breast cancer associated with NSAID use [5–12], because COX inhibition is predicted to decrease mammary estrogen levels and hence restrict the growth of estrogen-dependent tumors. This mechanism is expected to be operative irrespective of COX-2 expression status because COX-1 is constitutively expressed in human breast tissues [21, 25]. Importantly, the idea that both COX isoforms can impact on tumorigenesis is supported by genetic evidence obtained using Cox-1 and Cox-2 knockout mouse strains [32, 40, 63, 64]. Intriguingly, Neugut and colleagues [65] have identified differential sensitivity of breast cancers to NSAID-mediated protection according to hormone receptor status. Specifically, they found that aspirin use was associated with a decreased risk of hormone receptor positive breast cancer, but did not affect the incidence of hormone receptor-negative disease, in a population-based case-control study. These data support the concept that COX inhibition reduces mammary neoplasia at least in part through suppression of estrogen biosynthesis.
Cyclooxygenase/prostaglandin signaling as an anticancer target
In combination, the data from COX-2 expression analyses, NSAID-related epidemiology, animal studies, and in vitro experiments strongly support a protumorigenic role for COX-2 with respect to breast and other cancers. The considerable weight of evidence linking COX/PG signaling with colorectal neoplasia has stimulated evaluation of NSAIDs as preventive agents in individuals at risk for colorectal cancer. Positive results with conventional NSAIDs led to the development of similar trials to test the efficacy of COXibs, based on the expectation that these agents would have reduced gastrointestinal complications relative to conventional NSAIDs. Importantly, COXibs were found to reduce the incidence of both familial and sporadic disease, providing important proof-of-principle for targeting COX/PG signaling [66–69]. Less propitiously, some trials also identified increased cardiovascular risk associated with COXib use [70, 71].
Cardiovascular toxicity of selective cyclooxygenase-2 inhibitors
The cardiovascular toxicity of COXibs has been ascribed to their selective depression of prostacyclin levels [72]. Prostacyclin (PGI2) derived from endothelial COX-2 confers constraints on thrombogenesis, hypertension, and atherogenesis. COX-2 inhibition decreases the cardioprotection afforded by PGI2, but, in contrast, the prothrombotic effects of thromboxane A2, emanating from COX-1 activity in platelets, are unaffected. The cardiovascular toxicity of COXibs decreases the desirability of using this class of drugs in cancer prevention. Nevertheless, the demonstrated efficacy of COXibs as anticancer agents identifies the COX/PG signaling axis as an important target. Thus, it is incumbent on us to identify alternative components of this signaling pathway that offer safer targets for preventing cancer.
Alternative targets on the cyclooxygenase/prostaglandin signaling axis
Several potential targets have been identified through examination of eicosanoid metabolic pathways (Figure 1). Importantly in this respect, substantial data support PGE2 as being the predominant protumorigenic prostanoid. Thus, it seems reasonable to hypothesize that selective targeting of PGE2 synthases or receptors might be useful with respect to neoplasia, and more broadly for analgesia and anti-inflammatory applications. Substantial interest is focused on microsomal prostaglandin E synthase (mPGES)-1, which is up-regulated in numerous human cancers, including breast carcinomas [73, 74]. mPGES-1 ablation does not increase thrombogenesis or blood pressure [75], consistent with the hypothesis that prostacyclin suppression is a key component of COXib-induced cardiotoxicity. Thus, mPGES-1 may offer a useful alternative target to COX-2 for combating inflammation and cancer.
The role of individual PGE2 receptors (prostaglandin E receptors [PTGERs] 1 to 4; more usually called EP1 to EP4) in cancer is also under investigation. Expression of all four EPs has been identified in mouse mammary tumors [29, 52]. Genetic and pharmacologic ablation approaches have been used to analyze the contributions of individual EPs to tumorigenesis in multiple animal models. No single EP has emerged as a clear favorite, with different receptors being implicated depending on the experimental system [76]. EP1, EP2, and EP4 all appear to have protumorigenic activity in at least one breast cancer model. Thus, it remains to be established which PGE2 receptor(s) is the optimal candidate for anticancer applications.
Enhancing PGE2 inactivation may provide an alternative mechanism for ameliorating COX-associated neoplasia. PGE2 is metabolized to relatively inactive 15-keto-PGs and 15-keto-lipoxins by the enzyme hydroxyprostaglandin dehydrogenase 15-NAD, more commonly called 15-hydroxyprostaglandin dehydrogenase (15-PGDH; Figure 1). Strikingly, reduced levels of 15-PGDH have been observed in multiple tumors, including non-small-cell lung cancers, colorectal carcinomas, and breast cancers, and substantial evidence suggests that 15-PGDH acts as a tumor suppressor [77–82]. These observations suggest the intriguing possibility that PGE2 signaling could be terminated by reversing epigenetic inactivation of the 15-PGDH locus, and that this could offer a novel approach to targeting PGE2-driven neoplasia.
Conclusion
The COX/PG signaling pathway offers a useful target for anti-breast cancer strategies. COX-2 is over-expressed in a significant proportion of invasive breast carcinomas, and at a higher frequency in breast pre-cancers. Both pharmacologic and genetic ablation of Cox-2 suppress experimental breast cancer, and transgenic COX-2 over-expression drives tumor formation. Together these data strongly support the validity of COX/PG signaling as an anticancer target. The recently appreciated cardiovascular toxicity of COXibs diminishes the likely utility of this class of drugs for preventing cancer. However, analysis of the pathways by which COX-derived PGE2 drives tumorigenesis is expected to lead to identification of novel drug targets for cancer treatment and prevention.
Note
This article is part of a review series on Inflammation and breast cancer, edited by Mina J Bissell and Jeffrey W Pollard.
Other articles in the series can be found online at http://breast-cancer-research.com/articles/review-series.asp?series=bcr_Inflammation
Abbreviations
- COX:
-
cyclooxygenase
- COXib:
-
selective cyclooxygenase-2 inhibitor
- DCIS:
-
ductal carcinoma in situ
- EP:
-
prostaglandin E2 receptor
- ER:
-
estrogen receptor
- HER:
-
human epidermal growth factor receptor
- MMTV:
-
mouse mammary tumor virus
- mPGES:
-
microsomal prostaglandin E synthase
- NDL:
-
neu deletion mutant
- NSAID:
-
nonsteroidal anti-inflammatory drug
- PG:
-
prostaglandin
- 15-PGDH:
-
15-hydroxyprostaglandin dehydrogenase.
References
Williams CS, Mann M, DuBois RN: The role of cyclooxygenases in inflammation, cancer, and development. Oncogene. 1999, 18: 7908-7916. 10.1038/sj.onc.1203286.
Herschman HR: Prostaglandin synthase 2. Biochim Biophys Acta. 1996, 1299: 125-140.
Dannenberg AJ, Altorki NK, Boyle JO, Dang C, Howe LR, Weksler BB, Subbaramaiah K: Cyclo-oxygenase 2: a pharmacological target for the prevention of cancer. Lancet Oncol. 2001, 2: 544-551. 10.1016/S1470-2045(01)00488-0.
Brown JR, DuBois RN: COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol. 2005, 23: 2840-2855. 10.1200/JCO.2005.09.051.
Friedman GD, Ury HK: Initial screening for carcinogenicity of commonly used drugs. J Natl Cancer Inst. 1980, 65: 723-733.
Harris RE, Chlebowski RT, Jackson RD, Frid DJ, Ascenseo JL, Anderson G, Loar A, Rodabough RJ, White E, McTiernan A: Breast cancer and nonsteroidal anti-inflammatory drugs: prospective results from the Women's Health Initiative. Cancer Res. 2003, 63: 6096-6101.
Harris RE, Namboodiri KK, Farrar WB: Nonsteroidal antiinflammatory drugs and breast cancer. Epidemiology. 1996, 7: 203-205. 10.1097/00001648-199603000-00017.
Khuder SA, Mutgi AB: Breast cancer and NSAID use: a meta-analysis. Br J Cancer. 2001, 84: 1188-1192. 10.1054/bjoc.2000.1709.
Schreinemachers DM, Everson RB: Aspirin use and lung, colon, and breast cancer incidence in a prospective study. Epidemiology. 1994, 5: 138-146. 10.1097/00001648-199403000-00003.
Sharpe CR, Collet JP, McNutt M, Belzile E, Boivin JF, Hanley JA: Nested case-control study of the effects of non-steroidal anti-inflammatory drugs on breast cancer risk and stage. Br J Cancer. 2000, 83: 112-120. 10.1054/bjoc.2000.1119.
Harris RE, Beebe-Donk J, Alshafie GA: Reduction in the risk of human breast cancer by selective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer. 2006, 6: 27-10.1186/1471-2407-6-27.
Rahme E, Ghosn J, Dasgupta K, Rajan R, Hudson M: Association between frequent use of nonsteroidal anti-inflammatory drugs and breast cancer. BMC Cancer. 2005, 5: 159-10.1186/1471-2407-5-159.
Boland GP, Butt IS, Prasad R, Knox WF, Bundred NJ: COX-2 expression is associated with an aggressive phenotype in ductal carcinoma in situ. Br J Cancer. 2004, 90: 423-429. 10.1038/sj.bjc.6601534.
Costa C, Soares R, Reis-Filho JS, Leitao D, Amendoeira I, Schmitt FC: Cyclo-oxygenase 2 expression is associated with angiogenesis and lymph node metastasis in human breast cancer. J Clin Pathol. 2002, 55: 429-434.
Davies G, Salter J, Hills M, Martin LA, Sacks N, Dowsett M: Correlation between cyclooxygenase-2 expression and angiogenesis in human breast cancer. Clin Cancer Res. 2003, 9: 2651-2656.
Denkert C, Winzer KJ, Muller BM, Weichert W, Pest S, Kobel M, Kristiansen G, Reles A, Siegert A, Guski H, et al: Elevated expression of cyclooxygenase-2 is a negative prognostic factor for disease free survival and overall survival in patients with breast carcinoma. Cancer. 2003, 97: 2978-2987. 10.1002/cncr.11437.
Half E, Tang XM, Gwyn K, Sahin A, Wathen K, Sinicrope FA: Cyclooxygenase-2 expression in human breast cancers and adjacent ductal carcinoma in situ. Cancer Res. 2002, 62: 1676-1681.
Kelly LM, Hill AD, Kennedy S, Connolly EM, Ramanath R, Teh S, Dijkstra B, Purcell R, McDermott EW, O'Higgins N: Lack of prognostic effect of Cox-2 expression in primary breast cancer on short-term follow-up. Eur J Surg Oncol. 2003, 29: 707-710. 10.1016/j.ejso.2003.07.001.
Lim SC: Role of COX-2, VEGF and cyclin D1 in mammary infiltrating duct carcinoma. Oncol Rep. 2003, 10: 1241-1249.
Shim JY, An HJ, Lee YH, Kim SK, Lee KP, Lee KS: Overexpression of cyclooxygenase-2 is associated with breast carcinoma and its poor prognostic factors. Mod Pathol. 2003, 16: 1199-1204. 10.1097/01.MP.0000097372.73582.CB.
Soslow RA, Dannenberg AJ, Rush D, Woerner BM, Khan KN, Masferrer J, Koki AT: COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer. 2000, 89: 2637-2645. 10.1002/1097-0142(20001215)89:12<2637::AID-CNCR17>3.0.CO;2-B.
Spizzo G, Gastl G, Wolf D, Gunsilius E, Steurer M, Fong D, Amberger A, Margreiter R, Obrist P: Correlation of COX-2 and Ep-CAM overexpression in human invasive breast cancer and its impact on survival. Br J Cancer. 2003, 88: 574-578. 10.1038/sj.bjc.6600741.
Watanabe O, Shimizu T, Imamura H, Kinoshita J, Utada Y, Okabe T, Kimura K, Hirano A, Yoshimatsu K, Aiba M, et al: Expression of cyclooxygenase-2 in malignant and benign breast tumors. Anticancer Res. 2003, 23: 3215-3221.
Wulfing P, Diallo R, Muller C, Wulfing C, Poremba C, Heinecke A, Rody A, Greb RR, Bocker W, Kiesel L: Analysis of cyclooxygenase-2 expression in human breast cancer: high throughput tissue microarray analysis. J Cancer Res Clin Oncol. 2003, 129: 375-382. 10.1007/s00432-003-0459-1.
Yoshimura N, Sano H, Okamoto M, Akioka K, Ushigome H, Kadotani Y, Yoshimura R, Nobori S, Higuchi A, Ohmori Y, et al: Expression of cyclooxygenase-1 and -2 in human breast cancer. Surg Today. 2003, 33: 805-811. 10.1007/s00595-003-2606-3.
Ristimaki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, Joensuu H, Isola J: Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002, 62: 632-635.
Hamid R, Singh J, Reddy BS, Cohen LA: Inhibition by dietary menhaden oil of cyclooxygenase-1 and -2 in N-nitrosomethylurea-induced rat mammary tumors. Int J Oncol. 1999, 14: 523-528.
Howe LR, Crawford HC, Subbaramaiah K, Hassell JA, Dannenberg AJ, Brown AMC: PEA3 is upregulated in response to Wnt1 and activates the expression of cyclooxygenase-2. J Biol Chem. 2001, 276: 20108-20115. 10.1074/jbc.M010692200.
Howe LR, Subbaramaiah K, Patel J, Masferrer JL, Deora A, Hudis C, Thaler HT, Muller WJ, Du B, Brown AMC, et al: Celecoxib, a selective cyclooxygenase-2 inhibitor, protects against human epidermal growth factor receptor 2 (HER-2)/neu-induced breast cancer. Cancer Res. 2002, 62: 5405-5407.
Nakatsugi S, Ohta T, Kawamori T, Mutoh M, Tanigawa T, Watanabe K, Sugie S, Sugimura T, Wakabayashi K: Chemoprevention by nimesulide, a selective cyclooxygenase-2 inhibitor, of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced mammary gland carcinogenesis in rats. Jpn J Cancer Res. 2000, 91: 886-892.
Robertson FM, Parrett ML, Joarder FS, Ross M, Abou-Issa HM, Alshafie G, Harris RE: Ibuprofen-induced inhibition of cyclooxygenase isoform gene expression and regression of rat mammary carcinomas. Cancer Lett. 1998, 122: 165-175. 10.1016/S0304-3835(97)00387-X.
Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM: Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell. 1996, 87: 803-809. 10.1016/S0092-8674(00)81988-1.
Subbaramaiah K, Norton L, Gerald W, Dannenberg AJ: Cyclooxygenase-2 is Overexpressed in HER-2/neu-positive breast cancer. Evidence for involvement of AP-1 and PEA3. J Biol Chem. 2002, 277: 18649-18657. 10.1074/jbc.M111415200.
Vadlamudi R, Mandal M, Adam L, Steinbach G, Mendelsohn J, Kumar R: Regulation of cyclooxygenase-2 pathway by HER2 receptor. Oncogene. 1999, 18: 305-314. 10.1038/sj.onc.1202307.
Shim V, Gauthier ML, Sudilovsky D, Mantei K, Chew KL, Moore DH, Cha I, Tlsty TD, Esserman LJ: Cyclooxygenase-2 expression is related to nuclear grade in ductal carcinoma in situ and is increased in its normal adjacent epithelium. Cancer Res. 2003, 63: 2347-2350.
Tan KB, Yong WP, Putti TC: Cyclooxygenase-2 expression: a potential prognostic and predictive marker for high-grade ductal carcinoma in situ of the breast. Histopathology. 2004, 44: 24-28. 10.1111/j.1365-2559.2004.01774.x.
Crawford YG, Gauthier ML, Joubel A, Mantei K, Kozakiewicz K, Afshari CA, Tlsty TD: Histologically normal human mammary epithelia with silenced p16(INK4a) overexpress COX-2, promoting a premalignant program. Cancer Cell. 2004, 5: 263-273. 10.1016/S1535-6108(04)00023-6.
Howe LR: Cyclooxygenase-2 and breast cancer. Trends in Breast Cancer Research. Edited by: Yao AP. 2005, New York: Nova, 9: 1-38.
Howe LR, Subbaramaiah K, Brown AMC, Dannenberg AJ: Cyclooxygenase-2: a target for the prevention and treatment of breast cancer. Endocr Relat Cancer. 2001, 8: 97-114. 10.1677/erc.0.0080097.
Howe LR, Chang SH, Tolle KC, Dillon R, Young LJ, Cardiff RD, Newman RA, Yang P, Thaler HT, Muller WJ, et al: HER2/neu-induced mammary tumorigenesis and angiogenesis are reduced in cyclooxygenase-2 knockout mice. Cancer Res. 2005, 65: 10113-10119. 10.1158/0008-5472.CAN-05-1524.
Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, Haudenschild C, Lane TF, Hla T: Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem. 2001, 276: 18563-18569. 10.1074/jbc.M010787200.
Reddy BS: Studies with the azoxymethanerat preclinical model for assessing colon tumor development and chemoprevention. Environ Mol Mutagen. 2004, 44: 26-35. 10.1002/em.20026.
Corpet DE, Pierre F: Point: from animal models to prevention of colon cancer. Systematic review of chemoprevention in min mice and choice of the model system. Cancer Epidemiol Biomarkers Prev. 2003, 12: 391-400.
Harris RE, Alshafie GA, Abou-Issa H, Seibert K: Chemoprevention of breast cancer in rats by celecoxib, a cyclooxygenase 2 inhibitor. Cancer Res. 2000, 60: 2101-2103.
Kubatka P, Ahlers I, Ahlersova E, Adamekova E, Luk P, Bojkova B, Markova M: Chemoprevention of mammary carcinogenesis in female rats by rofecoxib. Cancer Lett. 2003, 202: 131-136. 10.1016/j.canlet.2003.08.006.
Lanza-Jacoby S, Miller S, Flynn J, Gallatig K, Daskalakis C, Masferrer JL, Zweifel BS, Sembhi H, Russo IH: The cyclooxygenase-2 inhibitor, celecoxib, prevents the development of mammary tumors in Her-2/neu mice. Cancer Epidemiol Biomarkers Prev. 2003, 12: 1486-1491.
Grosch S, Maier TJ, Schiffmann S, Geisslinger G: Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J Natl Cancer Inst. 2006, 98: 736-747.
Soh JW, Weinstein IB: Role of COX-independent targets of NSAIDs and related compounds in cancer prevention and treatment. Prog Exp Tumor Res. 2003, 37: 261-285.
Siegel PM, Ryan ED, Cardiff RD, Muller WJ: Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. Embo J. 1999, 18: 2149-2164. 10.1093/emboj/18.8.2149.
Seno H, Oshima M, Ishikawa TO, Oshima H, Takaku K, Chiba T, Narumiya S, Taketo MM: Cyclooxygenase 2- and prostaglandin E2 receptor EP2-dependent angiogenesis in ApcΔ716 mouse intestinal polyps. Cancer Res. 2002, 62: 506-511.
Takeda H, Sonoshita M, Oshima H, Sugihara K, Chulada PC, Langenbach R, Oshima M, Taketo MM: Cooperation of cyclooxygenase 1 and cyclooxygenase 2 in intestinal polyposis. Cancer Res. 2003, 63: 4872-4877.
Chang SH, Liu CH, Conway R, Han DK, Nithipatikom K, Trifan OC, Lane TF, Hla T: Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci USA. 2004, 101: 591-596. 10.1073/pnas.2535911100.
Brodie AM, Lu Q, Long BJ, Fulton A, Chen T, Macpherson N, DeJong PC, Blankenstein MA, Nortier JW, Slee PH, et al: Aromatase and COX-2 expression in human breast cancers. J Steroid Biochem Mol Biol. 2001, 79: 41-47. 10.1016/S0960-0760(01)00131-5.
Brueggemeier RW, Quinn AL, Parrett ML, Joarder FS, Harris RE, Robertson FM: Correlation of aromatase and cyclooxygenase gene expression in human breast cancer specimens. Cancer Lett. 1999, 140: 27-35. 10.1016/S0304-3835(99)00050-6.
Diaz-Cruz ES, Shapiro CL, Brueggemeier RW: Cyclooxygenase inhibitors suppress aromatase expression and activity in breast cancer cells. J Clin Endocrinol Metab. 2005, 90: 2563-2570. 10.1210/jc.2004-2029.
Subbaramaiah K, Howe LR, Port ER, Brogi E, Fishman J, Liu CH, Hla T, Hudis C, Dannenberg AJ: HER-2/neu status is a determinant of mammary aromatase activity in vivo: evidence for a cyclooxygenase-2-dependent mechanism. Cancer Res. 2006, 66: 5504-5511. 10.1158/0008-5472.CAN-05-4076.
Prosperi JR, Robertson FM: Cyclooxygenase-2 directly regulates gene expression of P450 Cyp19 aromatase promoter regions pII, pI.3 and pI.7 and estradiol production in human breast tumor cells. Prostaglandins Other Lipid Mediat. 2006, 81: 55-70. 10.1016/j.prostaglandins.2006.07.003.
Agarwal VR, Bulun SE, Leitch M, Rohrich R, Simpson ER: Use of alternative promoters to express the aromatase cytochrome P450 (CYP19) gene in breast adipose tissues of cancer-free and breast cancer patients. J Clin Endocrinol Metab. 1996, 81: 3843-3849. 10.1210/jc.81.11.3843.
Chen S, Zhou D, Okubo T, Kao YC, Yang C: Breast tumor aromatase: functional role and transcriptional regulation. Endocr Relat Cancer. 1999, 6: 149-156. 10.1677/erc.0.0060149.
Zhao Y, Agarwal VR, Mendelson CR, Simpson ER: Estrogen biosynthesis proximal to a breast tumor is stimulated by PGE2 via cyclic AMP, leading to activation of promoter II of the CYP19 (aromatase) gene. Endocrinology. 1996, 137: 5739-5742. 10.1210/en.137.12.5739.
Narumiya S, Sugimoto Y, Ushikubi F: Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999, 79: 1193-1226.
Muller-Decker K, Berger I, Ackermann K, Ehemann V, Zoubova S, Aulmann S, Pyerin W, Furstenberger G: Cystic duct dilatations and proliferative epithelial lesions in mouse mammary glands upon keratin 5 promoter-driven overexpression of cyclooxygenase-2. Am J Pathol. 2005, 166: 575-584.
Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O, Langenbach R: Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000, 60: 4705-4708.
Tiano HF, Loftin CD, Akunda J, Lee CA, Spalding J, Sessoms A, Dunson DB, Rogan EG, Morham SG, Smart RC, et al: Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 2002, 62: 3395-3401.
Terry MB, Gammon MD, Zhang FF, Tawfik H, Teitelbaum SL, Britton JA, Subbaramaiah K, Dannenberg AJ, Neugut AI: Association of frequency and duration of aspirin use and hormone receptor status with breast cancer risk. JAMA. 2004, 291: 2433-2440. 10.1001/jama.291.20.2433.
Arber N, Eagle CJ, Spicak J, Racz I, Dite P, Hajer J, Zavoral M, Lechuga MJ, Gerletti P, Tang J, et al: Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006, 355: 885-895. 10.1056/NEJMoa061652.
Baron JA, Sandler RS, Bresalier RS, Quan H, Riddell R, Lanas A, Bolognese JA, Oxenius B, Horgan K, Loftus S, et al: A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006, 131: 1674-1682. 10.1053/j.gastro.2006.08.079.
Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, et al: Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006, 355: 873-884. 10.1056/NEJMoa061355.
Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y, Fujimura T, et al: The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000, 342: 1946-1952. 10.1056/NEJM200006293422603.
Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, et al: Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005, 352: 1092-1102. 10.1056/NEJMoa050493.
Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, Anderson WF, Zauber A, Hawk E, Bertagnolli M: Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005, 352: 1071-1080. 10.1056/NEJMoa050405.
Grosser T, Fries S, FitzGerald GA: Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006, 116: 4-15. 10.1172/JCI27291.
Yoshimatsu K, Altorki NK, Golijanin D, Zhang F, Jakobsson PJ, Dannenberg AJ, Subbaramaiah K: Inducible prostaglandin E synthase is overexpressed in non-small cell lung cancer. Clin Cancer Res. 2001, 7: 2669-2674.
Mehrotra S, Morimiya A, Agarwal B, Konger R, Badve S: Microsomal prostaglandin E2 synthase-1 in breast cancer: a potential target for therapy. J Pathol. 2006, 208: 356-363. 10.1002/path.1907.
Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA: Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006, 116: 1391-1399. 10.1172/JCI27540.
Fulton AM, Ma X, Kundu N: Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. 2006, 66: 9794-9797. 10.1158/0008-5472.CAN-06-2067.
Backlund MG, Mann JR, Holla VR, Buchanan FG, Tai HH, Musiek ES, Milne GL, Katkuri S, DuBois RN: 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem. 2005, 280: 3217-3223. 10.1074/jbc.M411221200.
Ding Y, Tong M, Liu S, Moscow JA, Tai HH: NAD+-linked 15-hydroxyprostaglandin dehydrogenase (15-PGDH) behaves as a tumor suppressor in lung cancer. Carcinogenesis. 2005, 26: 65-72. 10.1093/carcin/bgh277.
Myung SJ, Rerko RM, Yan M, Platzer P, Guda K, Dotson A, Lawrence E, Dannenberg AJ, Lovgren AK, Luo G, et al: 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci USA. 2006, 103: 12098-12102. 10.1073/pnas.0603235103.
Wolf I, O'Kelly J, Rubinek T, Tong M, Nguyen A, Lin BT, Tai HH, Karlan BY, Koeffler HP: 15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of human breast cancer. Cancer Res. 2006, 66: 7818-7823. 10.1158/0008-5472.CAN-05-4368.
Yan M, Rerko RM, Platzer P, Dawson D, Willis J, Tong M, Lawrence E, Lutterbaugh J, Lu S, Willson JK, et al: 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-beta-induced suppressor of human gastrointestinal cancers. Proc Natl Acad Sci USA. 2004, 101: 17468-17473. 10.1073/pnas.0406142101.
Mann JR, Backlund MG, Buchanan FG, Daikoku T, Holla VR, Rosenberg DW, Dey SK, DuBois RN: Repression of prostaglandin dehydrogenase by epidermal growth factor and snail increases prostaglandin E2 and promotes cancer progression. Cancer Res. 2006, 66: 6649-6656. 10.1158/0008-5472.CAN-06-1787.
Acknowledgements
The assistance of John Timmer and Anthony Brown during manuscript preparation is gratefully acknowledged. The author apologizes for her inability to include many important primary literature citations due to space constraints. Funding for work in the author's laboratory is provided by the National Institutes of Health (R03 CA119273, R01 CA078480, R01 CA089578) and by the Breast Cancer Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Howe, L.R. Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res 9, 210 (2007). https://doi.org/10.1186/bcr1678
Published:
DOI: https://doi.org/10.1186/bcr1678