
Abstract
Background
The prevalence of BRCA1 and BRCA2 mutations in Spain is heterogeneous and varies according to geographical origin of studied families. The contribution of these mutations to hereditary breast and ovarian cancer has not been previously investigated in Asturian populations (Northern Spain).
Methods
In the present work, 256 unrelated high-risk probands with breast and/or ovarian cancer from families living in Asturias were analyzed for the presence of a BRCA1 or BRCA2 gene mutation from October 2007 to May 2012. The entire coding sequences and each intron/exon boundaries of BRCA1/2 genes were screened both by direct sequencing and Multiplex Ligation-dependent Probe Amplification (MLPA).
Results
A total of 59 families (23%) were found to carry a pathogenic germ line mutation, 39 in BRCA1 and 20 in BRCA2. Twenty nine additional families (12%) carried an unknown significance variant. We detected 28 distinct pathogenic mutations (16 in BRCA1 and 12 in BRCA2), of which 3 mutations in BRCA1 (c.1674delA, c.1965C>A and c.2900_2901dupCT) and 5 in BRCA2 (c.262_263delCT, c.2095C>T, c.3263dupC, c.4030_4035delinsC, c.8042_8043delCA) had not been previously described.
The novel mutations c.2900_2901dupCT in BRCA1 and c.4030_4035delinsC in BRCA2 occurred in 8 and 6 families respectively and clustered in two separated small geographically isolated areas suggesting a founder effect. These 2 mutations, together with the Galician BRCA1 mutation c.211A>G (9 families), and the common BRCA1 mutation c.3331_3334delCAAG (6 families), account for approximately 50% of all affected families. By contrast, very frequent mutations in other Spanish series such as the BRCA1 Ashkenazi founder mutation c.68_69delAG, was found in only one family.
Conclusions
In this study we report the BRCA1 and BRCA2 spectrum of mutations and their geographical distribution in Asturias, which largely differ from other areas of Spain. Our findings may help design a first step recurrent mutation panel for screening high-risk breast and/or ovarian cancer families from this specific area.
Similar content being viewed by others


Background
Breast cancer is the most frequently diagnosed and the leading cause of cancer death among females, accounting for 23% of the total cases and 14% of the cancer deaths [1]. While most tumors are sporadic, about 5 to 10% are caused by germ line mutations in certain genes [2]. BRCA1 (MIM 113705) [3] and BRCA2 (MIM 600185) [4] genes are responsible for approximately 20-40% of inherited breast cancer [5, 6]. Women carrying germ line mutations in these genes have a high lifetime risk of developing both breast and ovarian cancer [7]. Testing these high-penetrant genes can make it feasible to identify individuals at risk as candidates for surveillance programs [8].
Prevalence of BRCA1 or BRCA2 germ line mutations varies considerably among ethnic groups, and in some countries, founder mutations are responsible for a significant proportion of breast cancer cases. Specific mutations have been described, for example, among Ashkenazi Jews, in Iceland, and in several other countries where isolated populations exists [9]. Mutational analysis of BRCA1 and BRCA2 in hereditary breast and ovarian cancer families have also been reported in several Spanish regions, showing a heterogeneous prevalence of recurrent mutations according to the geographical area [10]. However there are to date no comprehensive studies in the Asturian population, a geographically isolated area in the North of Spain.
In this article, we present mutation detection data corresponding to a set of 256 high-risk families living in Asturias, analyzed by direct sequencing and MLPA. Thirty-nine families were found to have a mutation in BRCA1 gene and 20 in BRCA2. We report the finding of 8 novel mutations and the clustered geographical distribution of two of them which may be founder mutations.
Methods
Study population
Patients were referred to the Familial Cancer Unit at Hospital Universitario Central de Asturias (HUCA) for genetic counseling by physicians between October 2007 and May 2012. Information about the number and type of cancers, age of diagnosis, age of death or current age and geographical origin were collected from each family. Genetic testing was offered to affected individuals from high-risk families meeting any of the following criteria:
-
A.
Three or more family members with breast and/or ovarian cancer.
-
A. Br: only breast cancer.
-
A. Ov: at least one ovarian cancer.
-
B.
Two family members with ovarian cancer.
-
C.
One family member with ovarian cancer and one with breast cancer.
-
D.
One family member with a male breast cancer and one or more with breast and/or ovarian cancer.
-
E.
Two family members with breast cancer, before the age of 50.
-
F.
One family member with bilateral breast cancer and one with breast cancer, at least one before the age of 50.
-
G.
Single affected individual with either:
-
Ga: bilateral breast cancer, first diagnosed before the age of 40, or
-
Gb: breast or ovarian cancer diagnosed before the age of 30, or
-
Gc: breast and ovarian cancer
-
H.
Affected probands from families close to fulfill any of the above criteria. We have included in this group affected probands with a history suggestive of hereditary breast and ovary cancer who do not fulfill completely any of the above inclusion criteria. This included, for instance, probands from small families with two affected relatives older than 50 years of age with triple negative breast cancer.
A total of 256 families fulfilled any of the selection criteria. The study was approved by the hospital ethical committee. All tested individuals provided a signed informed consent following appropriate genetic counseling. Blood samples were obtained from at least one affected member. DNA and RNA were obtained from peripheral blood from all studied members of the family. Index case was first analyzed for large genomic rearrangements and then for sequence mutations. Genomic DNA was obtained after red blood cell lyses using DNAzol method (MRC, USA). RNA was purified from leucocytes by using TriReagent solution (Ambion, Life Technologies).
Large genomic rearrangements of BRCA1 and BRCA2
BRCA1 and BRCA2 large genomic rearrangements were assayed by MLPA (multiplex ligation-dependent probe amplification) by using P002 and P087 kits for BRCA1 gene and P045 kit for BRCA2 gene (MRC-Holland), following essentially the instructions of the manufacturer. Amplified products were electrophoretically separated in an ABIPrism310 genetic analyzer and interpreted using the GeneMapper 4.0 software (Applied Biosystems). For normalizing the data, at least two genomic DNA samples obtained from peripheral blood cells of healthy donors were always run as controls in each analysis. Quantification of the results, measuring peak areas, was performed by using an Excel calculation sheet. The final allele dosage for each allele was obtained from the following formula:
(“n” peak area from sample / “n” peak area from control) divided by (∑ reference peak areas from sample / ∑ reference peak areas from control).
Normal values were considered when this ratio was between 0.8 and 1.2.
Mutation analysis of BRCA1 and BRCA2
Mutational screening of BRCA1 and BRCA2 genes was performed by direct sequencing of each exon and 20 bp exon-intron boundaries, by standard Sanger sequencing using BigDye™ terminator sequencing kits (Applied BioSystems). Sequence analyses for some index cases were performed at Sistemas Genómicos SL (Valencia, Spain) or at Imegen (Parque Científico, Universidad de Valencia, Spain).
Reference sequences used for BRCA1 and BRCA2 analyses were GenBank NM_007294.2 (BRCA1) and NM_000059.3 (BRCA2). Mutation nomenclature is described according to Human Genome Variation Society (v2.0) (http://www.hgvs.org) [11].
Results and discussion
Frequency of families with BRCA1 and BRCA2pathogenic mutations according to the inclusion criteria
We identified 59 families carrying a pathogenic mutation, 39 in BRCA1 and 20 in BRCA2, which represent 23% of all screened families (Table 1). The highest mutation detection rate in BRCA1 was observed in families with at least one ovarian cancer (groups A.Ov., B, C, and Gc.) and in families with a single case of bilateral breast cancer diagnosed before the age of 40 (group Ga). Regarding BRCA2, the highest detection rate was observed in families with one breast cancer and one ovarian cancer and in families with a male breast cancer (groups C and D), while mutations were not found in any of the groups with single cases (group G) or two ovarian cancers (group B). Three out of 11 families with a male breast cancer (group D) carried a pathogenic mutation, 1 in BRCA1 and 2 in BRCA2. These mutation detection rates are comparable to previously reported populations. No pathogenic mutations were found in 9 women with early onset breast cancer without family history (group Gb). Although this is an inclusion criterion in all clinical guides, other authors have also found very low mutation rate in this group [12, 13]. On the other hand, we identified a deleterious mutation in 2 out of 19 affected probands (10.5%) who did not fully meet our present inclusion criteria for testing. Within this group (H), a BRCA1 deleterious mutation was found in a family with two affected first degree relatives diagnosed with breast cancer over the age of 50, having one of them a “triple negative” phenotype in the tumor. The other proband was a 47-year-old woman, diagnosed with ovarian cancer and with no family history of cancer, in whom a BRCA2 pathogenic mutation was identified.
BRCA1deleterious mutations
Analysis of the BRCA1 gene revealed 16 distinct germ line mutations with predicted deleterious effects on the BRCA1 protein in 39 families (Table 2). Among these pathogenic mutations, 3 are novel and have not yet been reported in the Breast Cancer Information Core, BIC (http://research.nhgri.nih.gov/bic/). One of these novel BRCA1 mutations, c.2900_2901dupCT (p.Pro968Leufs), was shared by 8 apparently unrelated families and it accounted for 20% of BRCA1 mutated families. This mutation is responsible for 7 breast and 7 ovarian cancer cases. Mean age of diagnosis was 49 years (range 33–78) for breast and 53 years (range 41–82) for ovarian cancer. Clinical and pathological characteristics of these families are shown in Table 3. The two remaining BRCA1 novel mutations c.1674delA (p.Gly559Valfs) and c.1965C>A (p.Tyr655*) were found only in one family each.
Among previously described mutations in BRCA1, the most common was the mutation c.211A>G (p.Arg71Gly), which was present in 9 families in our series (23%). This mutation is a founder mutation in Galicia, a Spanish region located just alongside western Asturias, where it represents 50% of all mutations [14, 15], but it is almost absent in other Spanish populations [10]. All families found in our study to carry this mutation were either from Galicia or had Galician ancestors. Another common mutation, the c.3331_3334delCAAG (p.Gln1111Asnfs), was found in 6 families, thus representing 15% of BRCA1 mutated families. It was first described in 1996 in a Canadian family [16] and thereafter in populations from all over the word. More recently an haplotype analysis performed in Hispanic populations living in Colombia suggested that this mutation has arisen from a common ancestor and that could represent a founder effect of Spanish origin [17]. This common mutation has 40 records in the BIC database, and has been reported in other regions of Spain [18, 19]. The c.470_471delCT (p.Ser157*) mutation in exon 8 was detected in 3 families, being also reported in other Spanish [20, 21], Chinese [22] and in Portuguese populations [23].
Other previously reported mutations were less frequent in our study. Thus, the nonsense mutation 1687C>T (p.Gln563*) was found in one family originally from León, Spain. Although this mutation was first described in 1996 in a Swedish population where it was considered to have a founder effect [24], it has also been reported in the Spanish area where this family came from (Castilla-León) [21]. From the same region were also two families carrying the mutations c.791_794delGTTC (p.Ser264Metfs) and c.3689T>G (p.Leu1230*). Both mutations have been previously reported in Spanish populations [15, 20], as well as the mutation c.3770-3771delAG (p.Glu1257Glyfs), found in two families from our study.
Notably, the frame shift mutation c.4065_4068delTCAA (p.Asn1355Lysfs), which is one of the most frequently reported (133 entries in BIC) [25, 26], was only found in one of our families and has not been previously reported in Spain. The Ashkenazi founder mutations c.68_69delAG (p.Glu23Valfs) was found in only one family, while it is the most frequent BRCA1 alteration found in other Spanish series [20]. Finally, we found the missense mutation c.5117G>C (p.Gly1706Ala) in one family. Although this mutation is still annotated as one with unknown clinical importance in the BIC database, some studies support that it could be pathogenic [20, 27].
BRCA1large genomic deletion
An MLPA-based search for large genomic rearrangements was performed in all index cases. This approach revealed that three of them were positive for the presence of a large deletion in the BRCA1 gene. One family presented heterozygous deletions for all the probes specific for BRCA1, from exon 1 to exon 24, revealed with two different MLPA kits (P002B and p084). The index case was a 56 years old woman with ovarian cancer. Her mother had also ovarian cancer and a sister was affected of breast cancer. After a complete study of all available members of the family, we could confirm that the deletion segregated with the disease. BRCA1 complete deletions have only been previously reported in 3 families, being a de novo mutation in one of them [28–30].
A second case showed an MLPA profile suggesting heterozygous deletions in probes specific for exons 1 to 13. The pattern was displayed with probes from P002B kit and confirmed with P082 MLPA kit exons 1–13. The deletion was identified in a woman with ovarian cancer at the age of 64 who had had a bilateral breast cancer at ages 42 and 61 respectively. Her sister had also had an ovarian cancer at the age of 33. This mutation has also been described in another Spanish family [31] and in one from Finland [32], both with cases of ovarian cancer. Finally, we found an in frame deletion in exon 20 in a large family. More detailed data about this deletion will be reported elsewhere (manuscript in preparation).
Deleterious BRCA2mutations
Analysis of the BRCA2 gene revealed 12 distinct germ line mutations with predicted deleterious effects on the BRCA2 protein in 20 families (Table 4). Among these pathogenic mutations 5 have not yet been reported in the Breast Cancer Information Core, BIC. Thus, the novel truncating mutation c.4030_4035delinsC (p.Asn1344Hisfs), shared by 6 families, is BRCA2 most frequent mutation in our series and accounts for 30% of the BRCA2 mutations. In the eleven carriers identified, a total of ten breast cancers, three ovarian cancers and one squamous oesophagus cancer have been diagnosed. Median age at diagnosis was 48 for breast (range 32–74) and 52 for ovarian cancer (range 48–60). Clinic and pathological characteristics of tumors from individuals carrying this mutation are shown in Table 5.
The second most frequent novel mutation in BRCA2 was c.2095 C>T (p.Gln699*) identified in three Asturian families, while the following novel truncating mutations were found in one family each: c.262_263delCT (p.Leu88Alafs); c.3263dupC (p.Gln1089Serfs); and c.8042_8043delCA (p.Thr2681Serfs). Individuals with these 3 novel mutations come either from Andalucía (Southern Spain) or from Albacete (Eastern Spain) where, to our knowledge, a comprehensive mutational analysis has not been reported yet.
Eight families carried 7 previously reported BRCA2 mutations. Two families carried the mutation c.1813dupA (p.Ile605Asnfs), which is very common in Western Europe, mostly in Germany where a possible founder effect has been suggested [33]. The other 6 known mutations were found in one family each. The c.5116_5119delAATA (p.Asn1706Leufs) and c.9310_9311delAA (p.Lys3104Valfs) mutations, recurrent in Castilla-León, were found in two families originally from that area [34]. The pathogenic mutation c.5576_5579delTTAA (p.Ile1859Lysfs) was found in one family from the País Vasco (Northern Spain) and has been previously described in other Spanish populations [13, 35]. Another family carried the frame-shift mutation c.4631dupA (p.Asn1544Lysfs), which has also been described in Western European populations [36]. The frame-shift c.9026_9030delATCAT (p.Tyr3009Serfs), which is a founder mutation originating in the Northeast of Spain [37], was identified in one family from Cantabria (Northern Spain). Finally, an African American woman with early onset bilateral breast cancer carried the nonsense mutation c.2830A>T (p.Lys944*), which has 6 entries in the BIC database and has been found in a Swedish series [38] but has not been previously reported in the Spanish population.
In contrast to the large BRCA1 genomic deletions discussed above, no large genomic rearrangements were found to affect BRCA2 in any of the families included in our study.
Unclassified variants
Twenty two different variants of unknown significance were identified in a total of 29 affected index cases, 3 of which had not been previously reported, 2 in BRCA1 and 1 in BRCA2 (Table 6). The BRCA1 missense mutation c.287A>C (p.Asp96Ala) has no BIC records, although the mutation p.Asp96Asn in the same position is considered pathogenic by some authors due to the fact that this amino acid is fully conserved throughout evolution [39]. We could not perform co-segregation studies, as the only first degree relative with cancer was deceased at the time of the study. The second novel BRCA1 variant is the c.656A>T (p.Asp219Val) found in a family with 6 breast cancers. Two bioinformatics tools, Sorting Intolerant From Tolerant (SIFT) [40] and Polymorphism Phenotyping (PolyPhen) [41] indicate that this variant may alter the function of the protein. However, this variant does not segregate with the disease in this family and the affected residue is not conserved in mammals. A novel BRCA2 variant found in the present work is the missense mutation c.6847C>A (p.Pro2283Thr), affecting a conserved residue in chordates. According to the above bioinformatics tools, this variant could be also pathogenic but it did not segregate with the disease in a family with two ovarian cancers. Consequently, these two variants are probably non-pathogenic.
While the remaining variants have been previously reported, controversies persist in the literature regarding the pathogenicity of some of them. This is the case of missense mutation c.115T>A (p.Cys39Ser), which is registered as a variant of unknown significance in the BIC database, but alters the protein structure and is considered deleterious by some authors [39, 43]. We found it in a family with breast and ovarian cancer, but it did not segregate with the disease as it was not present in one of the sisters who developed breast cancer at the age of 33. On the other hand, two BRCA1 missense variants, c.5054C>T (p.Thr1685Ile) and c.5154G>T (p.Trp1718Cys), affecting highly conserved amino acid residues, have been predicted to be deleterious by a number of studies, including a multifactorial likelihood-ratio model [44], evolutionary conservation analyses and functional assays [39, 45, 46]. In our population, we could not study co-segregation of those variants, but the number of coincident results in favor of causality suggests that these variants can be deleterious.
We found the nonsense mutation c.9976A>T (p.Lys3326*) in two unrelated families. In spite that this mutation introduces a stop codon, causing premature termination of the protein, it is considered in BIC as with no clinical significance. In agreement with these observations, this mutation does not segregate with the disease in one of the families studied in this work. Some other variants are probably non-pathogenic, with odds of >100:1 in favor of neutrality according to multifactorial model by Easton et al. [44]. This can be the case of BRCA1 c.199G>T (p.Asp67Tyr) and BRCA2 c.6748A>G (p.Thr2250Ala) and c.8850G>T (p.Lys2950Asn).
Geographic distribution of two novel recurrent mutations
Asturias is a Northern Spain community of 1 million inhabitants, bordered by Galicia to the West, Cantabria to the East and Castilla-León to the South. It has been rather isolated from the rest of Spain by its high mountains and it was one of the few areas that never came under the Muslim control that lasted in most of the Iberian Peninsula for five centuries. Although modern society has made possible migration phenomena, small geographically isolated communities still remain within deep valleys in a rural environment.
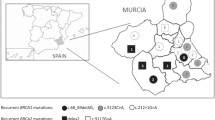
The origin of the eight families with the c.2900_2901dupCT (p.Pro968Leufs) BRCA1 novel mutation clustered along a region extending from the Asturias coastline to the mountains bordering the province of Leon to the South (Figure 1). An historical important roman pathway (Calzada de la Mesa) communicating these two regions is found in the vicinity. This is the area where Vaqueiros, a seasonal transhumance population were established. With different habits and their own dialect, they have been for centuries a distinct social group, suggesting a possible founder effect of this mutation.

Map of Asturias showing the geographical origin of the families with the two most frequent novel BRCA1 and BRCA2 mutations. Two main linguistic areas are separated by a solid line while dialect boundaries are separated by dotted lines (based in map created with data from dialectologists Zamora-Vicente and Fernandez-Rei). BRCA1 c.2900_2901dupCT mutation can be related to a transhumance population established in the vicinities of a Roman pathway of historical importance while c.4030_4035delinsC BRCA2 is closely associated to Eonavian dialect population.
Regarding the other recurrent mutation c.4030_4035delinsC (p.Asn1344Hisfs) in BRCA2, the six families with this mutations were from a small area situated on the western border of Asturias (Figure 1), geographically isolated by two deep river valleys (Eo and Navia rivers) running from South to North. Interestingly, the population living there has their own and distinct dialect called Eonavian confirming the isolation and thus suggesting a founder effect of this mutation.
Conclusions
In this study we conducted for the first time a comprehensive BRCA1/2 screening in a group of 256 high risk families living in Asturias. We found that 59 families carried a pathogenic mutation, 39 in BRCA1 and 20 in BRCA2, 8 of them being novel. Two mutations were found in families who cluster in two geographical and customary isolated areas, suggesting a founder effect, although future haplotype analysis would be necessary to confirm this hypothesis. Besides, all the families who carried the previously reported Galician founder mutations had Galician ancestors. In total, these three mutations plus a known recurrent mutation account for approximately 50% of all affected families, being 60% of BRCA1 and 30% of BRCA2 mutations recurrent. The results of the present study suggest that Asturias belongs to the group of geographical areas in which a small numbers of mutations account for a large proportion of BRCA1/2 mutations.
Consequently, testing a person from these areas for their respectively recurrent mutations before sequencing the complete genes could be a cost and time-efficient way to assess if the individual has BRCA1 or BRCA2 germ line mutations. Finally, we believe that conducting a specific mutational analysis on unselected cases of both breast and ovarian cancer for the Asturian women whose origin are these two small areas where the novel mutations seem to cluster, may be helpful and effective in cancer prevention terms.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D: Global cancer statistics. CA Cancer J Clin. 2011, 61 (2): 69-90. 10.3322/caac.20107.
Ripperger T, Gadzicki D, Meindl A, Schlegelberger B: Breast cancer susceptibility: current knowledge and implications for genetic counselling. Eur J Hum Genet. 2009, 17 (6): 722-731. 10.1038/ejhg.2008.212.
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W: A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994, 266 (5182): 66-71. 10.1126/science.7545954.
Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G: Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995, 378 (6559): 789-792. 10.1038/378789a0.
Wooster R, Weber BL: Breast and ovarian cancer. N Engl J Med. 2003, 348 (23): 2339-2347. 10.1056/NEJMra012284.
Fackenthal JD, Olopade OI: Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer. 2007, 7 (12): 937-948. 10.1038/nrc2054.
Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A: Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003, 72 (5): 1117-1130. 10.1086/375033.
Balmana J, Diez O, Rubio IT, Cardoso F: BRCA in breast cancer: ESMO Clinical Practice Guidelines. Ann Oncol. 2011, 22 (Suppl 6): vi31-34.
Ferla R, Calo V, Cascio S, Rinaldi G, Badalamenti G, Carreca I, Surmacz E, Colucci G, Bazan V, Russo A: Founder mutations in BRCA1 and BRCA2 genes. Ann Oncol. 2007, 18 (Suppl 6): vi93-98.
Diez O, Gutierrez-Enriquez S, Balmana J: Heterogeneous prevalence of recurrent BRCA1 and BRCA2 mutations in Spain according to the geographical area: implications for genetic testing. Fam Cancer. 2010, 9 (2): 187-191. 10.1007/s10689-009-9301-5.
den Dunnen JT, Antonarakis SE: Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000, 15 (1): 7-12. 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N.
Gonzalez-Hormazabal P, Gutierrez-Enriquez S, Gaete D, Reyes JM, Peralta O, Waugh E, Gomez F, Margarit S, Bravo T, Blanco R: Spectrum of BRCA1/2 point mutations and genomic rearrangements in high-risk breast/ovarian cancer Chilean families. Breast Cancer Res Treat. 2011, 126 (3): 705-716. 10.1007/s10549-010-1170-y.
Beristain E, Martinez-Bouzas C, Guerra I, Viguera N, Moreno J, Ibanez E, Diez J, Rodriguez F, Mallabiabarrena G, Lujan S: Differences in the frequency and distribution of BRCA1 and BRCA2 mutations in breast/ovarian cancer cases from the Basque country with respect to the Spanish population: implications for genetic counselling. Breast Cancer Res Treat. 2007, 106 (2): 255-262. 10.1007/s10549-006-9489-0.
Vega A, Campos B, Bressac-De-Paillerets B, Bond PM, Janin N, Douglas FS, Domenech M, Baena M, Pericay C, Alonso C: The R71G BRCA1 is a founder Spanish mutation and leads to aberrant splicing of the transcript. Hum Mutat. 2001, 17 (6): 520-521.
Vega A, Torres M, Martinez JI, Ruiz-Ponte C, Barros F, Carracedo A: Analysis of BRCA1 and BRCA2 in breast and breast/ovarian cancer families shows population substructure in the Iberian peninsula. Ann Hum Genet. 2002, 66 (Pt 1): 29-36.
Durocher F, Tonin P, Shattuck-Eidens D, Skolnick M, Narod SA, Simard J: Mutation analysis of the BRCA1 gene in 23 families with cases of cancer of the breast, ovary, and multiple other sites. J Med Genet. 1996, 33 (10): 814-819. 10.1136/jmg.33.10.814.
Torres D, Rashid MU, Gil F, Umana A, Ramelli G, Robledo JF, Tawil M, Torregrosa L, Briceno I, Hamann U: High proportion of BRCA1/2 founder mutations in Hispanic breast/ovarian cancer families from Colombia. Breast Cancer Res Treat. 2007, 103 (2): 225-232. 10.1007/s10549-006-9370-1.
Blesa JR, Garcia JA, Ochoa E: Frequency of germ-line BRCA1 mutations among Spanish families from a Mediterranean area. Hum Mutat. 2000, 15 (4): 381-382.
Esteban Cardenosa E, Bolufer Gilabert P, Palanca Suela S, Barragan Gonzalez E, Oltra Soler S, Chirivella Gonzalez I, Segura Huerta A, Guillen Ponce C, Martinez de Duenas E, Cuevas Cuerda D: [BRCA1 and BRCA2 mutations in families studied in the program of genetic counselling in cancer of the Valencian community (Spain)]. Med Clin (Barc). 2008, 130 (4): 121-126. 10.1157/13115767.
Diez O, Osorio A, Duran M, Martinez-Ferrandis JI, de la Hoya M, Salazar R, Vega A, Campos B, Rodriguez-Lopez R, Velasco E: Analysis of BRCA1 and BRCA2 genes in Spanish breast/ovarian cancer patients: a high proportion of mutations unique to Spain and evidence of founder effects. Hum Mutat. 2003, 22 (4): 301-312. 10.1002/humu.10260.
Salazar R, Cruz-Hernandez JJ, Sanchez-Valdivieso E, Rodriguez CA, Gomez-Bernal A, Barco E, Fonseca E, Portugal T, Gonzalez-Sarmiento R: BRCA1-2 mutations in breast cancer: identification of nine new variants of BRCA1-2 genes in a population from central Western Spain. Cancer Lett. 2006, 233 (1): 172-177. 10.1016/j.canlet.2005.03.006.
Kwong A, Wong LP, Wong HN, Law FB, Ng EK, Tang YH, Chan WK, Ho LS, Kwan KH, Poon M: A BRCA2 founder mutation and seven novel deleterious BRCA mutations in southern Chinese women with breast and ovarian cancer. Breast Cancer Res Treat. 2009, 117 (3): 683-686. 10.1007/s10549-009-0385-2.
Peixoto A, Salgueiro N, Santos C, Varzim G, Rocha P, Soares MJ, Pereira D, Rodrigues H, Bento MJ, Fraguas A: BRCA1 and BRCA2 germline mutational spectrum and evidence for genetic anticipation in Portuguese breast/ovarian cancer families. Fam Cancer. 2006, 5 (4): 379-387. 10.1007/s10689-006-0009-5.
Johannsson O, Ostermeyer EA, Hakansson S, Friedman LS, Johansson U, Sellberg G, Brondum-Nielsen K, Sele V, Olsson H, King MC: Founding BRCA1 mutations in hereditary breast and ovarian cancer in southern Sweden. Am J Hum Genet. 1996, 58 (3): 441-450.
Evans DG, Neuhausen SL, Bulman M, Young K, Gokhale D, Lalloo F: Haplotype and cancer risk analysis of two common mutations, BRCA1 4184del4 and BRCA2 2157delG, in high risk northwest England breast/ovarian families. J Med Genet. 2004, 41 (2): e21-10.1136/jmg.2003.012104.
Janavicius R: Founder BRCA1/2 mutations in the Europe: implications for hereditary breast-ovarian cancer prevention and control. EPMA J. 2010, 1 (3): 397-412. 10.1007/s13167-010-0037-y.
Osorio A, de la Hoya M, Rodriguez-Lopez R, Martinez-Ramirez A, Cazorla A, Granizo JJ, Esteller M, Rivas C, Caldes T, Benitez J: Loss of heterozygosity analysis at the BRCA loci in tumor samples from patients with familial breast cancer. Int J Cancer. 2002, 99 (2): 305-309. 10.1002/ijc.10337.
Konecny M, Zavodna K, Vranova V, Vizvaryova M, Weismanova E, Mlkva I, Kuglik P, Kausitz J, Bartosova Z: Identification of rare complete BRCA1 gene deletion using a combination of SNP haplotype analysis, MLPA and array-CGH techniques. Breast Cancer Res Treat. 2008, 109 (3): 581-583. 10.1007/s10549-007-9670-0.
de la Hoya M, Gutierrez-Enriquez S, Velasco E, Osorio A, Sanchez de Abajo A, Vega A, Salazar R, Esteban E, Llort G, Gonzalez-Sarmiento R: Genomic rearrangements at the BRCA1 locus in Spanish families with breast/ovarian cancer. Clin Chem. 2006, 52 (8): 1480-1485. 10.1373/clinchem.2006.070110.
Garcia-Casado Z, Romero I, Fernandez-Serra A, Rubio L, Llopis F, Garcia A, Llombart P, Lopez-Guerrero JA: A de novo complete BRCA1 gene deletion identified in a Spanish woman with early bilateral breast cancer. BMC Med Genet. 2011, 12: 134-10.1186/1471-2350-12-134.
del Valle J, Feliubadalo L, Nadal M, Teule A, Miro R, Cuesta R, Tornero E, Menendez M, Darder E, Brunet J: Identification and comprehensive characterization of large genomic rearrangements in the BRCA1 and BRCA2 genes. Breast Cancer Res Treat. 2010, 122 (3): 733-743. 10.1007/s10549-009-0613-9.
Pylkas K, Erkko H, Nikkila J, Solyom S, Winqvist R: Analysis of large deletions in BRCA1, BRCA2 and PALB2 genes in Finnish breast and ovarian cancer families. BMC Cancer. 2008, 8: 146-10.1186/1471-2407-8-146.
Meindl A: Comprehensive analysis of 989 patients with breast or ovarian cancer provides BRCA1 and BRCA2 mutation profiles and frequencies for the German population. Int J Cancer. 2002, 97 (4): 472-480.
Infante M, Duran M, Acedo A, Perez-Cabornero L, Sanz DJ, Garcia-Gonzalez M, Beristain E, Esteban-Cardenosa E, de la Hoya M, Teule A: BRCA1 5272-1G>A and BRCA2 5374delTATG are founder mutations of high relevance for genetic counselling in breast/ovarian cancer families of Spanish origin. Clin Genet. 2010, 77 (1): 60-69. 10.1111/j.1399-0004.2009.01272.x.
Miramar MD, Calvo MT, Rodriguez A, Anton A, Lorente F, Barrio E, Herrero A, Burriel J, Garcia de Jalon A: Genetic analysis of BRCA1 and BRCA2 in breast/ovarian cancer families from Aragon (Spain): two novel truncating mutations and a large genomic deletion in BRCA1. Breast Cancer Res Treat. 2008, 112 (2): 353-358. 10.1007/s10549-007-9868-1.
Al-Mulla F, Bland JM, Serratt D, Miller J, Chu C, Taylor GT: Age-dependent penetrance of different germline mutations in the BRCA1 gene. J Clin Pathol. 2009, 62 (4): 350-356. 10.1136/jcp.2008.062646.
Campos B, Diez O, Odefrey F, Domenech M, Moncoutier V, Martinez-Ferrandis JI, Osorio A, Balmana J, Barroso A, Armengod ME: Haplotype analysis of the BRCA2 9254delATCAT recurrent mutation in breast/ovarian cancer families from Spain. Hum Mutat. 2003, 21 (4): 452-10.1002/humu.9133.
Hakansson S, Johannsson O, Johansson U, Sellberg G, Loman N, Gerdes AM, Holmberg E, Dahl N, Pandis N, Kristoffersson U: Moderate frequency of BRCA1 and BRCA2 germ-line mutations in Scandinavian familial breast cancer. Am J Hum Genet. 1997, 60 (5): 1068-1078.
Abkevich V, Zharkikh A, Deffenbaugh AM, Frank D, Chen Y, Shattuck D, Skolnick MH, Gutin A, Tavtigian SV: Analysis of missense variation in human BRCA1 in the context of interspecific sequence variation. J Med Genet. 2004, 41 (7): 492-507. 10.1136/jmg.2003.015867.
Ng PC, Henikoff S: SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31 (13): 3812-3814. 10.1093/nar/gkg509.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR: A method and server for predicting damaging missense mutations. Nat Methods. 2010, 7 (4): 248-249. 10.1038/nmeth0410-248.
Campos B, Diez O, Domenech M, Baena M, Balmana J, Sanz J, Ramirez A, Alonso C, Baiget M: RNA analysis of eight BRCA1 and BRCA2 unclassified variants identified in breast/ovarian cancer families from Spain. Hum Mutat. 2003, 22 (4): 337-
Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D, Fukuda M, Ohta T, Klevit R: Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci U S A. 2003, 100 (10): 5646-5651. 10.1073/pnas.0836054100.
Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro AN, Iversen ES, Couch FJ: A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007, 81 (5): 873-883. 10.1086/521032.
Phelan CM, Dapic V, Tice B, Favis R, Kwan E, Barany F, Manoukian S, Radice P, van der Luijt RB, van Nesselrooij BP: Classification of BRCA1 missense variants of unknown clinical significance. J Med Genet. 2005, 42 (2): 138-146. 10.1136/jmg.2004.024711.
Mirkovic N, Marti-Renom MA, Weber BL, Sali A, Monteiro AN: Structure-based assessment of missense mutations in human BRCA1: implications for breast and ovarian cancer predisposition. Cancer Res. 2004, 64 (11): 3790-3797. 10.1158/0008-5472.CAN-03-3009.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/13/243/prepub
Acknowledgements
We thank the authorities at Hospital Universitario Central de Asturias-Servicio de Salud del Principado de Asturias (HUCA-SESPA) for developing and maintaining the regional Familial Cancer Program. We thank Fundación Caja Rural de Asturias for financial support to Laboratorio de Oncología Molecular (HUCA). Part of this work was also supported by SAF 2010–21165 (Ministerio de Ciencia e Innovación.). We thank Pilar F. Valdés and Mº Luisa Fernandez for excellent technical assistance. IUOPA is supported by Obra Social Cajastur.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PB, MB: designed the study, analyzed clinical and mutational data and drafted the manuscript. ISR, ASP, MGA, AL: performed mutational analyses and MLPA studies. ML, YF: revised clinical and pathological data. JMPF: revised the mutational data and helped to draft the manuscript. AP: completed data collection of families’ origins. All authors approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Blay, P., Santamaría, I., Pitiot, A.S. et al. Mutational analysis of BRCA1 and BRCA2 in hereditary breast and ovarian cancer families from Asturias (Northern Spain). BMC Cancer 13, 243 (2013). https://doi.org/10.1186/1471-2407-13-243
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-13-243