

Abstract
Nonalcoholic fatty liver disease (NAFLD) and its more severe form, nonalcoholic steatohepatitis (NASH), represent a growing worldwide epidemic and a high unmet medical need, as no licensed drugs have been approved thus far. Currently, histopathological assessment of liver biopsies is mandatory as a primary endpoint for conditional drug approval. This requirement represents one of the main challenges in the field, as there is substantial variability in this invasive histopathological assessment, which leads to dramatically high screen-failure rates in clinical trials. Over the past decades, several non-invasive tests have been developed to correlate with liver histology and, eventually, outcomes to assess disease severity and longitudinal changes non-invasively. However, further data are needed to ensure their endorsement by regulatory authorities as alternatives to histological endpoints in phase 3 trials. This Review describes the challenges of drug development in NAFLD–NASH trials and potential mitigating strategies to move the field forward.
Similar content being viewed by others


Main
NAFLD is a worldwide health care concern and a growing epidemic1,2 with an estimated global prevalence of 25% (ref. 1). Its more severe form, NASH, has become the most common indication for liver transplantation in women and the second most common indication in men in the United States3.
NAFLD is a spectrum that ranges from isolated steatosis (buildup of fat in the liver) with a relatively benign non-progressive clinical course to the serious condition of NASH, characterized by a state of hepatocellular injury, inflammation and fibrosis, with a progressive course that may lead to cirrhosis and its complications (including hepatocellular carcinoma)4,5,6. The central feature of NASH pathogenesis is a dysfunctional stress response to an excess supply of nutrient substrates to the liver, promoting fibrosis and ultimately leading to cirrhosis (stage 4 fibrosis)7,8,9,10 (Box 1). Additionally, genomic instability increases the carcinogenic risk leading to hepatocellular carcinoma11.
Despite a high unmet medical need to prevent, stop or reverse NASH, there are currently no approved licensed drugs, and developing them has been challenging12. NASH drug candidates must demonstrate the ability to prevent or delay disease progression, as measured by a composite endpoint that includes progression to cirrhosis, liver-related outcome events (ascites, hepatic encephalopathy, upper gastrointestinal bleeding) and all-cause death. However, considering the low incidence rate of these outcomes13, regulatory authorities have recommended the use of liver histopathology as a surrogate endpoint of outcomes to accelerate conditional drug approval14, while clinical outcome data from trials are awaited. However, there are many challenges with identifying the correct patients to qualify for these paired liver biopsy trials, as more than 70% of screened patients are failing to meet the eligibility criteria. Additional challenges include the highly variable placebo response rates15,16 and the need for correct trial design with appropriate endpoints and a duration of treatment that will permit accurate assessment of drug efficacy.
In this review, we provide an overview of the NASH trial landscape; we then review in detail the biggest challenges associated with trial design and endpoints, and their analysis and interpretation. Finally, we discuss new and emerging approaches to mitigating these challenges, including innovative trial designs, non-invasive tests (NITs) and biomarkers.
Overview of the NASH trial landscape
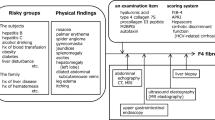
Since the recognition of NASH as a major unmet medical need, clinical trials have undergone major changes that have led to modifications and improvements in primary endpoints and interpretation of results12,17. Eligibility criteria defined in NASH clinical trials are usually sequentially assessed following multiple steps including laboratories, imaging and liver biopsy. Patients can be excluded at any of these steps18 (Fig. 1). The surrogate (liver histology) endpoints defined by the Food and Drug Administration (FDA) are used both at the screening stage to define patients’ eligibility criteria and as efficacy endpoints (change in liver histology from baseline to end of treatment). Histology assessment includes evaluation of the NAFLD activity score (NAS), developed by the NASH Clinical Research Network in 2005 (ref. 19), and the fibrosis stage. The NAS is a numeric semi-quantitative score distinguishing three lesions: steatosis (scored 0–3), lobular inflammation (scored 0–3) and ballooning (scored 0–2) (Fig. 2a); their sum reflects the disease grade (total score, 0–8). The score also assesses the severity of fibrosis (scored 0–4) and enables assessment of the disease stage (Fig. 2b). This score has allowed analysis of histologic changes for comparative and correlative studies in therapeutic intervention trials. The NAS was adopted in clinical trials soon after its inception; one of its most notable initial contributions was the readout of the PIVENS trial in 2010, in which vitamin E and pioglitazone were tested against placebo20. The PIVENS trial, together with additional landmark studies21,22, led to recommendation of vitamin E for patients with NASH who did not have diabetes and pioglitazone for patients with NASH with or without diabetes in the 2012 and 2018 American Association Study of Liver Disease guidance on NASH23,24.

Typical rate of screen failures (SFs) at each step of the usual NASH clinical trials. The liver biopsy (step 3) represents the main hurdle in the screening process. DXA, dual-energy X-ray absorptiometry; eGFR, estimated glomerular filtration rate; HbA1c, glycated hemoglobin.

Liver biopsy slides using the two usual staining approaches for NASH assessment. a, Hematoxylin and eosin staining reveals histologic features of NASH: steatosis (yellow arrows), ballooning recognized as swollen hepatocytes (black arrows) and inflammation recognized as a mixed inflammatory infiltrate (black circles). b, Masson trichrome staining (measuring collagen content; collagen is blue): peri-sinusoidal fibrosis (black arrows). Blue-stained collagen fibers outline the sinusoids surrounding the central vein (CV). Images courtesy of P. Bedossa, Liverpat, Paris, France.
Primary efficacy endpoints and the inclusion criteria for NASH clinical trials have undergone major changes since the PIVENS trial. The first major change was the emergence of data showing that patients with NASH who have stage 2 fibrosis or higher are at increased risk for morbidity (decompensation) and mortality from the disease25,26,27,28. This observation led to adoption of the term ‘at risk NASH’ (NASH with F2 and higher fibrosis) as a key inclusion criterion, especially for phase 3 trials (excluding patients with cirrhosis). However, this artificial classification of disease severity leaves many patients with advanced disease outside the inclusion criteria for clinical trials. As an example, a patient with stage 3 fibrosis but no ballooning on histology will be deemed as not having active disease required for inclusion in a trial, despite the evidence that this is a high-risk patient who may progress to cirrhosis in a relatively short period of time.
The second major change in the field followed a white paper published by the FDA in 2011, detailing the roadmap of drug approval, which stated that one of two histological endpoints should be achieved as surrogate endpoints: resolution of NASH (defined as an inflammation score of 0 or 1 and a ballooning score of 0) without worsening of fibrosis or improvement in fibrosis by one stage or more, without worsening of NASH29. Liver biopsy was then considered the gold standard for the assessment of disease severity, and biopsy endpoints became the main criteria for conditional drug approval in phase 3 trials. This policy allows for accelerated approval of a drug (given the unmet need of the disease) while outcome data for final approval are pending. These two surrogate endpoints have been widely applied in many large phase 3 clinical trials but remain the main divergent point between the FDA and the European Medicines Agency (EMA). The FDA accepts as primary endpoint either resolution of NASH or improvement in fibrosis, while the EMA requires that co-primary endpoints are met (including both resolution of NASH and improvement in fibrosis)14.
The initial implementation of these histological endpoints, especially fibrosis improvement, was widely accepted, given the correlation between the degree of fibrosis and disease outcomes in patients with NASH25. On the other hand, robust data correlating NASH resolution with hard clinical outcomes have been lacking, but NASH resolution has been accepted as steatohepatitis is the disease driver that leads to fibrosis. In addition, recent data have shown that NASH resolution correlates with fibrosis improvement30. The NAS remains the standard for histologic assessment in phase 2b and 3 trials; however, due to widely recognized limitations such as inter- and intra-reader variability, the EMA and the FDA allow for alternative validated scoring systems such as the Steatosis, Activity and Fibrosis (SAF) score developed by Bedossa et al.31. The SAF score has three components: steatosis (scored 0–3), activity (addition of ballooning, scored 0–2 and lobular inflammation, scored 0–2) and fibrosis (scored 0–4)31.
In addition to efficacy endpoints, drug-safety assessment is a key element for drug approval and conditional approval. The goal of drug-safety assessment is to protect the population from rare and severe adverse reactions. Although the primary purpose of preclinical and clinical drug development is to balance risk against the expected clinical benefit, post-marketing surveillance remains the most common pathway for safety signal detection. Indeed, in addition to being short term, with small sample size and often excluding high-risk populations32, randomized clinical trials cannot be fully powered to assess unexpected or unknown adverse reactions32.
Although considered as a rare complication in the general population (up to 19 of 100,000 persons)33, drug-induced liver injury (DILI) is a leading cause of acute liver failure associated with high mortality34 and has been the main cause of marketed drug withdrawals (broadly speaking) over the past decades (for example, troglitazone35, bromfenac36) and a common cause for halting drug development (for example, tasosartan37). The identification of DILI has been a challenge across drug development for both sponsors and regulatory authorities, which have jointly led efforts to attempt to address the challenge38. In that context, the FDA has developed guidelines and a software tool, eDISH (evaluation of drug-induced serious hepatotoxicity), which uses alanine transaminase (ALT) and total bilirubin for assisting assessment of potential hepatotoxicity of pre-marketing drugs39,40. This tool allows for identification of patients matching the Hy’s law criteria (ALT of 3× upper limit of normal or more and total bilirubin of 2× upper limit of normal or more)41 and provides time courses of liver enzymes for each patient. It has been widely adopted by sponsors42, and some of them have withdrawn their drugs before regulatory consideration based on these assessments. However, considerable limitations in specificity for DILI require that histologic assessment remains necessary for definitive diagnosis of hepatotoxicity, especially in light of known fluctuations in liver enzymes in NAFLD and NASH. Potential modifications of thresholds for the NAFLD–NASH population might be helpful to improve the use of eDISH in NASH clinical trials.
Challenges in NASH clinical trials
Limitations of standard histological assessment
The need for liver biopsy to identify the appropriate individuals for enrollment and as efficacy endpoints in clinical trials has several important limitations43,44. First, the invasive nature of the procedure creates hesitancy for patients to participate in trials and is associated with potential for serious complications45. Second, there is concern for sampling variability given the heterogeneous nature of the disease and the small size of the histologic sample. Finally, and most importantly, there are major issues related to suboptimal intra- and inter-reader reliability. In a study that investigated 51 patients with NAFLD who underwent liver biopsy with two samples collected simultaneously, histological lesions of NASH were found unevenly distributed throughout the two samples, illustrating that sampling error can lead to misdiagnosis and inaccuracy in reading46. Furthermore, multiple studies have exposed the inconsistencies in histological reading, where the inter- and intra-reader agreement varied widely43,47. In the most recent and largest study that examined inter- and intra-reader variabilities, agreement among three pathologists on eligibility criteria was found in only 53.7% of patients, demonstrating the lack of reliability43.
One of the most debated histological features is hepatocellular ballooning. It is thought to represent a form of hepatocyte injury that is not seen in non-progressive disease48. However, ballooning (using the current definition) is too subjective. A recent study, performed on digitalized slides evaluated independently by nine internationally recognized expert liver pathologists, demonstrated a substantial divergence with regard to identification of ballooned hepatocytes among the experts49,50. The results of this study challenge the applicability of the current scoring system for assessing hepatocellular ballooning in clinical trials. Fortunately, new artificial intelligence (AI) and machine learning methods have emerged and show promising results51,52. Recent data have shown high concordance of AI with consensus pathologist reading and high reproducibility when applied to a selection of patients for clinical trials53. However, these algorithms will not address issues related to the invasive nature of biopsy and the heterogeneous nature of the disease that is not captured by a limited-size biopsy. In addition, while incorporating AI and/or including two or more pathologists for consensus on reading can potentially mitigate the inter- and intra-reader variability, the number of ongoing and future clinical trials and patients needed to complete phase 3 studies make this solution impractical for the long term.
In addition to the substantial variability of these histologic endpoints, there is a lack of guidelines on the biopsy-reading process. This leads to the lack of standardization across clinical trials on several issues such as the number of readers, consensus between readers (on every variable or only the gestalt reading for NASH resolution or fibrosis improvement), number of slides to be read, digital versus glass slide reads and the possibility of re-reading baseline slides. Further refinement with regulatory authorities is warranted and urgently needed.
Limitations of imaging endpoints
Hepatic steatosis can be assessed and quantified by several imaging modalities. Controlled attenuation parameter (CAP) via FibroScan (Echosens) provides rapid point-of-care estimation of steatosis, with a value of ≥302 dB m−1 corresponding to having 5% or more steatosis on a liver biopsy54. Magnetic resonance imaging (MRI)-derived proton density fat fraction (MRI-PDFF) has emerged as one of the most accurate methods to quantify liver steatosis at baseline for clinical trials55, with most studies requiring a proton density fat fraction value of ≥8–10% for inclusion in the trial. In that context, MRI-PDFF has become one of the main primary endpoints for phase 2a studies, especially with drugs that target metabolic pathways. A reduction of ≥30% of fat fraction in MRI-PDFF has been used as an efficacy endpoint, together with the average relative and absolute change in liver fat content56. MRI fat reductions of ≥50% or ≥70% and even complete resolution of liver fat content (in ‘super responders’) have been introduced as other measurements to assess efficacy of highly potent drugs56. Nevertheless, although the change in fat in MRI-PDFF correlates with histological improvement in steatohepatitis57, even a large relative decrease of 70% predicts NASH resolution in only less than 50% of cases58. Thus, the formula for conversion to a meaningful histological NASH resolution is unknown; as such, power calculation might be challenging in the design of future phase 3 trials.
Carefully generated and assessed data on NITs other than MRI-PDFF are needed. Additional NITs, including blood-based ALT or enhanced liver fibrosis (ELF) tests, and ultrasound-based vibration-controlled transient elastography (VCTE; using the FibroScan device), along with MRI-PDFF will aid in interpreting changes in phase 2 studies and the design of future phase 3 studies that use histology as a primary endpoint. In fact, a recent study demonstrated that a combination of MRI-PDFF and ALT response predicted higher likelihood of histologic improvement than either MRI-PDFF or ALT response alone59. Nevertheless, the area under the curve (a measure of diagnostic accuracy) for the combination of ALT and MRI-PDFF in predicting histological response was low, signaling that further studies are needed to investigate which combination of NITs will better assess histological response.
Trial duration
Another issue with study design is determining the appropriate trial duration, which involves many considerations: mechanism of drug action, magnitude of effect in phase 2 trials and severity of disease (F2–F3 versus cirrhosis). Failure to correctly interpret all these factors can lead to studies with durations that could be too short to demonstrate efficacy60,61. This might be especially problematic in NASH cirrhosis trials, in which the histological endpoint of fibrosis regression by one stage without worsening of NASH may be more challenging to achieve in 48–72 weeks (the standard duration for phase 2b trials). Although regression of cirrhosis is associated with a reduction in liver-related events as early as 24 months, as seen in the simtuzumab and STELLAR-4 clinical trials62, longer-duration studies with adequate sample size to achieve this endpoint are costly. Soon, the use of complementary NITs, such as liver-stiffness measurement (LSM) by magnetic resonance elastography (MRE), alone or in combination with other NITs, may be used, as measurement on a continuous scale (rather than categorical fibrosis stages by histology) may provide an opportunity for earlier detection of drug response.
Impact of comorbidities
In addition to liver efficacy endpoints, multiple factors should be assessed carefully when evaluating a new drug in development for treatment of NASH. Cardiovascular disease is the major cause of death in patients with non-cirrhotic NASH63,64. Analysis from the National Health and Nutrition Examination Survey data from 1988 to 1994 demonstrated that cardiovascular risk factors such as blood pressure and glycemic control were strongly associated with cardiovascular and overall mortality in patients with NAFLD65. Other studies have shown that mortality in patients with NAFLD is mainly driven by cardiovascular risk factors and that an increased number of metabolic syndrome components was associated with lower survival66,67.
The impact of a NASH drug on cardiovascular risk factors such as dyslipidemia, insulin resistance and obesity is therefore highly important when determining the net benefit of that drug. In fact, certain new drugs for NASH that are associated with weight loss and a positive effect on dyslipidemia and insulin sensitivity may improve long-term clinical outcomes beyond their hepatic benefits by providing cardioprotective benefits. On the other hand, drugs that induce weight gain or worsening in dyslipidemia as adverse events will need to demonstrate significant improvement in liver efficacy endpoints to justify their use in this susceptible patient population. These drugs will also require careful monitoring for cardiovascular outcomes and the potential need to use additional drugs to mitigate negative adverse events (for example, adding a statin drug to mitigate increases in low-density lipoprotein cholesterol).
The presence of baseline comorbidities in clinical trials may also affect the therapeutic response rate in clinical trials, as some drugs may be more or less efficacious in a certain NAFLD–NASH phenotype than in another one. The best example is the influence of type 2 diabetes in NASH trials; several drugs (initially developed for type 2 diabetes) have demonstrated stronger effects in patients with type 2 diabetes versus non-diabetic patients20,21,68. Similarly, in the FLINT trial, the baseline triglyceride level was predictive of histological response69. Additional efforts should be considered to collect exhaustive information on those comorbidities and associated treatments as well as lifestyle during the conduct of the trials. This should be eased by the rapid growth of digital health and internet-connected monitoring devices. Stratification on these major confounding factors might be essential to improve the reliability of clinical trials.
Drawbacks of the placebo response
In NASH clinical trials, an unexpectedly high and variable placebo response has been a chronic and vexing problem. The placebo response has varied over the years throughout changes in the selection of primary endpoints and the targeted population. One of the first failures was the GOLDEN-505 trial, which did not meet its primary endpoint due to an unexpected placebo response of more than 57% (ref. 70). Post hoc analyses demonstrated that focusing on more advanced disease (NAS of 4 or higher, rather than the predefined eligibility criterion of a NAS of 3 or higher) would have reduced the placebo response and resulted in the primary endpoint being met70. A meta-analysis derived from a NASH study of 956 patients found that 25% of patients given placebo had an improvement in NAS by 2 or more points and 21% of patients had an improvement in fibrosis score15,71. Univariate and multivariate meta-regression showed that trials enrolling patients with a higher baseline NAS, trials conducted in South America and trials in which patients had a decrease in body mass index were associated with greater improvements in NAS among patients given placebo. Nevertheless, with the shift in endpoints from a two-point improvement in NAS to NASH resolution without fibrosis improvement or fibrosis improvement without worsening of NASH, there was a lower placebo response in the STELLAR-3–4 and REGENERATE trials (~10–16% for fibrosis improvement and 4–9% for NASH resolution), both of which included large numbers of patients60,72.
Newer trials have found that the placebo response rate can still be high; indeed, it reached 33% for fibrosis improvement without worsening of NASH and 17% for NASH resolution without fibrosis worsening in the phase 2 trial of semaglutide in patients with NASH73. Importantly, when both endpoints were combined (fibrosis improvement without worsening of NASH and NASH resolution without fibrosis worsening), the placebo improvement rate seemed to decrease significantly. A recent meta-analysis including 43 randomized controlled trials and 2,649 placebo-treated patients showed a pooled rate of 11.65% (95% confidence interval, 7.98–16.71) for NASH resolution without worsening of fibrosis and 18.82% (95% confidence interval, 15.65–22.47) for at least one-stage reduction in fibrosis16. These placebo changes are mainly studied in the F2–F3 population, whereas they are less known in patients with NASH and cirrhosis.
Multiple strategies have been proposed or applied to mitigate the placebo effect. Among these are weight stabilization for 3–6 months before liver biopsy is performed, verification of the amount of alcohol intake via the AUDIT questionnaire or with blood tests, histologic assessment by AI methods and randomization based on comorbidities and their severity (for example, type 2 diabetes)74. Alcohol consumption, even at low or moderate levels, has been raised as one of the major confounding factors to the placebo effect, and development of blood biomarkers to assess alcohol consumption (such as phosphatidylethanol75) has been suggested, but appropriate cutoffs remain debated. Another proposed strategy includes a study lead-in phase, in which patients can be followed for a time to assess their diet and weight changes and standardize their lifestyle changes74. These proposed approaches, however, could lead to further delays in the screening period or confounding from strict monitoring of dietary recommendations that result in substantial weight loss.
One of the promising approaches to mitigate the placebo effect is the application of AI and machine learning to read liver histology51. For instance, in a recent study from the ATLAS trial that examined the combination of cilofexor and firsocostat in patients with NASH, the authors developed a machine learning-based score to quantify changes in fibrosis between baseline and the end of treatment52. By using this score, they showed a statistically significant change in fibrosis improvement with the drug combination in comparison to the placebo arm after 48 weeks of therapy, whereas the conventional pathology reading failed to show this difference52. Notably, this difference seems to be mostly driven by lower-magnitude changes in fibrosis between baseline and the end of therapy in the placebo arm52.
Finally, given the heterogeneity of liver biopsies and lower inter–intra-observer reading, it is thought that using NITs may reduce the placebo effect, as an initial report found smaller placebo changes with NITs than with histologic grading15, but more data are needed to confirm this observation.
Strategies to advance drug development
Innovations in clinical trial design and endpoints
Proper design of clinical trials is one of the fundamental requirements in proving that a drug is efficacious and achieving its approval via the acceptable regulatory pathways76.
Adaptive study design, allowing modifications of the study after its initiation without affecting validity and integrity, has gained interest among sponsors due to the potential to improve trial efficiency (for example, smaller sample size required for achieving the same level of statistical power) and their attractiveness for patients (for example, closure of ineffective arm(s) and replacement by more promising arm(s)). However, there are also some important considerations and challenges associated with adaptive design. For example, outcomes used in the interim analysis should allow for detection of differences between treatment arms in a relatively short timeframe and before all patients are enrolled, to enable modifications to the trial design, and the interim endpoint must be highly predictive of the final primary endpoint. Moreover, the rate of enrollment must be compatible with that of the interim analysis.
In 2008, the FDA issued guidance requiring additional long-term safety trials for antidiabetic drugs (type 2 diabetes) to collect cardiovascular outcomes. This was largely due to the recognition of the growing impact of these outcomes in this population77 as well as increasing concerns about potentially higher cardiovascular risks associated with certain antidiabetic drugs (for example, rosiglitazone)77. Considering the important entanglement of NAFLD–NASH and type 2 diabetes and the increased recognition of cardiovascular outcomes as a growing burden in the NAFLD–NASH population, the inclusion of major adverse cardiovascular events in trials is an important factor to consider in NASH drug development. The experience gained from a decade of cardiovascular outcomes in type 2 diabetes drug trials should be considered and applied to NASH drug development.
Given the multifaceted mechanisms of NASH, adaptive design trials may benefit sponsors by allowing for better selection of a target population; for example, interim analyses could demonstrate better treatment response rates in a subset of patients defined by comorbidities, lifestyle and/or genetics. This would ultimately lead to restriction of the drug label but would increase the likelihood of drug approval based on a specific mechanism of action (MOA), opening the market to combination therapy strategies.
The emergence of NITs as tools to correlate with liver histology and, eventually, as outcomes to assess disease severity and longitudinal changes non-invasively is an important advance in the NASH trial landscape78. These biomarkers evaluate for different features of the disease that correspond to histologic findings, namely, liver steatosis, disease activity (ballooning and inflammation) and the stage of fibrosis79.
Several NITs are available to quantify liver fibrosis such as MRE and VCTE, which estimate stiffness by inducing a shear wave through the liver tissue54,80,81. Serologic biomarkers of extracellular matrix turnover have been developed and validated to determine fibrosis severity, such as the ELF score (a proprietary algorithm that uses three serum biomarkers: hyaluronic acid, procollagen III amino-terminal peptide (PIIINP) and tissue inhibitor of metalloproteinase 1 (TIMP-1)) (https://www.siemens-healthineers.com/en-us/press-room/press-releases/elftest.html).
The non-invasive diagnosis of NASH remains limited by lower accuracy of biomarkers. Several serologic biomarkers are available, such as serum ALT, cleaved cytokeratin 18 fragments82,83,84 and NIS4 (a proprietary blood-based biomarker panel that uses four biomarkers: miR-34a-5p, α2-macroglobulin, YKL-40 and glycated hemoglobin)85. Multiparametric MRI to estimate iron-corrected T1 (cT1) values can quantify extracellular water content, which rises with liver inflammation and fibrosis86. A study that included 264 patients with biopsy-proven NAFLD showed that cT1 correlated with all features of the NAS including ballooning and inflammation87. A combination of imaging parameters obtained from MRI and MRE can diagnose NASH and regression after weight loss88,89. An algorithm combining automated measurements of MRI-PDFF and liver stiffness by MRE detected NASH with an area under the receiver operating characteristic curve of 0.87 in a small study90.
Recently, the combination of serologic and imaging biomarkers in one score was proposed as a strategy to enhance the accuracy of predicting patients with fibrotic NASH. As an example, the FibroScan and aspartate transaminase (FAST) score combines VCTE as a biomarker for fibrosis, CAP as a biomarker for steatosis and aspartate transaminase (AST) as a biomarker of activity into one score that ranges from 0 to 1.00, with values over 0.67 having high positive predictive value for fibrotic NASH91. Similarly, the MRI and AST (MAST) score was developed using MRI-PDFF to quantify liver fat, MRE for fibrosis and AST for activity to identify fibrotic NASH with good accuracy92. The MEFIB index, which combines MRE with FIB-4 (fibrosis 4 score, which includes age, AST, ALT and platelets), also demonstrated good diagnostic accuracy to identify patients with fibrotic NASH93.
Several NITs that are used to identify patients with fibrotic NASH have been employed to decrease the screen-failure rate in clinical trials at the liver biopsy stage94. These NITs can be used at two levels: the study level (as part of the protocol developed by sponsors) or at the clinical site level (in prescreening strategies). In the phase 2b FASCINATE-2 trial of denifanstat in patients with NASH, after amending the protocol to add AST > 20 U l−1 and FibroScan CAP ≥ 280 dB m−1 as eligibility criteria, the overall screen-failure rate decreased from 96% to 80% (ref. 95). The screening and baseline data of FASCINATE-2 trial participants also showed moderate-to-good correlations between simple NITs (including AST, FAST, FIB-4) and fibrosis quantification by AI-based technology (HistoIndex)95. The correlation between liver fat content assessed by MRI-PDFF and steatosis quantification by AI-based technology was also excellent in this study95. In the phase 2b SYNERGY trial of tirzepatide for NASH, after a protocol amendment adding FAST ≥ 0.35 and AST > 23 U l−1 as eligibility criteria, the proportion of patients failing at the biopsy stage decreased from 72% to 66% among patients who underwent a ‘per protocol’ biopsy96. The addition of these eligibility criteria had a stronger effect on clinical sites where the specialty of the principal investigator was neither gastroenterology nor endocrinology, with a screen-failure-rate decrease from 80% to 62% at the ‘per protocol’ liver biopsy stage96.
The published FAST thresholds of ≤0.35 for ruling out the diagnosis of NASH and ≥0.67 for ruling in the diagnosis are not adapted for clinical trials. Indeed, the use of the rule-out threshold did not substantially decrease the screen-failure rate at biopsy in the SYNERGY trial, and, although the use of the rule-in threshold would efficiently reduce the screen-failure rate at biopsy, it would also reduce the screening volume and a substantial proportion of potentially eligible patients would be missed. A FAST threshold of 0.50 has been proposed as a prescreening or screening criteria in the setting of NASH drug development18. Research to define the best NIT thresholds for clinical trial purposes are underway and will facilitate an optimal balance between screening volume and screen-failure rate18.
It is of utmost importance to note that several of the above-mentioned biomarkers such as ELF, VCTE, MRE and cT1 have their own prognostic value in predicting liver-related outcomes; therefore, they should not be thought of solely as surrogates for predicting NAFLD histologic severity but also as direct surrogates for clinical outcomes97,98,99,100. Use of NITs for establishing primary endpoints in NASH trials has been supported recently by correlation of such scores with long-term outcomes. For instance, score changes on MRE, FIB-4, NFS (NAFLD fibrosis score), ELF and liver stiffness on VCTE have all correlated with worse clinical liver-related outcomes98, 100,101,102,103 (Table 1). While further data are needed to firmly establish the results of NIT scores with histological changes and proof that improvement in NIT scores leads to improved disease clinical outcomes, it is plausible that disease experts and regulators can soon reach consensus and endorse these NITs (likely in combination) as alternatives to the histological endpoints currently used in phase 3 trials.
On the way to personalized medicine
In addition to the challenges related to participants and clinical trial design described above, several additional challenges directly relate to the therapeutic agents. Most investigational agents have been developed to target a specific mechanism in the pathogenesis of NASH, from energy intake and disposal to liver metabolism and the response to lipotoxic liver injury resulting in inflammation and fibrosis10 (Fig. 3). It is becoming increasingly evident that the principal driving mechanism in NASH pathogenesis may not be replicable in all individuals, and the hepatic dysfunctional stress response to substrate overload varies with sex, race, ethnicity, comorbidities, genetic background, epigenetics and other characteristics yet to be elucidated.

NASH pathogenesis is complex and involves several pathways. The three most common pathways to hit NASH are to act on metabolism, fibrosis and inflammation. Adapted from ref. 131. DNL, de novo lipogenesis; ER, endoplasmic reticulum; FASN, fatty acid synthetase; FFA, free fatty acid; FGF, fibroblast growth factor; GLP1-RA, GLP1 receptor agonist; IL-6, interleukin 6; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharides; NLRP3, NLR family pyrin domain-containing 3; PPAR, peroxisome proliferator-activator receptor; ROS, reactive oxygen species; SEFA, structurally engineered fatty acids; SHP, small heterodimer partner; SREPB-1, sterol regulatory element-binding protein 1; TGF-β, transforming growth factor β; TGR5, Takeda G protein-coupled receptor 5; THR-β, thyroid hormone receptor β; TNF, tumor necrosis factor; UPR, unfolded protein response.
Although several agents have pleiotropic effects, the suboptimal efficacy demonstrated in most clinical trials thus far suggests that a ‘one size fits all’ approach is unlikely to lead to a groundbreaking treatment for NASH. Therefore, there is a great need to understand more about individualized treatment using agents with the right MOA for the right patient. One approach is development of predictive biomarkers of response to a specific MOA as investigational agents are tested. Current examples include the genetic polymorphisms associated with response to pioglitazone104 and obeticholic acid105, but the field is ripe for exploration. The application of unsupervised machine learning methods to large, heterogeneous cohorts exposed to investigational agents with various MOA represents an untapped opportunity in the development of the precision medicine drug-discovery pipeline106. New therapeutic assets can be screened with single-cell omics to evaluate the on- and off-target effects, the immunophenotype of different cell populations and the potential toxicology107. Another exciting opportunity is the utilization of patient-derived induced pluripotent stem cells to form organoids to test a personalized response to a specific drug108.
There is an emerging role for genetics in particular, as single-nucleotide polymorphisms in PNPLA3, GCKR, TM6SF2 and HSD17B13 have all been associated with NASH development or progression109. The expression of pathogenic gene variants can be modulated using antisense oligonucleotides or double-stranded short interfering RNA110 (Table 2). These strategies may be used for therapeutic intervention in patients with NASH to restore lipid droplets, secretion of hepatic very-low-density lipoprotein (VLDL) and de novo lipogenesis. The underlying mechanisms connecting the common patatin-like phospholipase domain-containing 3 (PNPLA3)I148M variant with NAFLD are the best-studied to date. PNPLA3I148M has been associated with liver steatosis, steatohepatitis, increased liver enzyme levels, hepatic fibrosis and cirrhosis111,112,113. The PNPLA3I148M protein is a trans‐repressor of hepatocyte lipid droplet lipase activity, which can lead to lipid buildup in hepatocytes. PNPLA3I148M has also been found to diminish retinol production via hepatic stellate cells (retinol is involved in lipid metabolism, and steatosis and has been found to be lower in patients with NAFLD). Thus, targeting this genetic pathway might mitigate hepatic steatosis, steatohepatitis and fibrosis. Interestingly, lifestyle adjustment and bariatric surgery have been more effective in improving hepatic steatosis in PNPLA3I148M carriers than in noncarriers114,115. By contrast, omega‐3 fatty acid may be less efficacious in decreasing liver fat levels in PNPLA3I148M carriers than in noncarriers115,116. Although it is plausible that patients with NASH and PNPLA3I148M lose more liver fat than noncarriers after an effective therapy, they are also likely to begin from a worse condition at baseline. Therefore, risk stratification based on PNPLA3I148M status may lead to a balanced approach to ensure that the number of patients with or without this mutation in treatment arms does not confound interpretation of results. This is particularly important in light of recent studies showing an association between PNPLA3I148M and liver‐related and all‐cause mortality in a large US population117.
The recently discovered hydroxysteroid 17β dehydrogenase 13 (HSD17B13) variant (rs6834314 and rs72613567:TA) has been recognized as a liver lipid droplet‐associated protein with retinol dehydrogenase activity. This variant has been shown to protect against NAFLD via loss of this enzymatic activity118,119. Although the underlying mechanism of HSD17B13 protection is not well understood, evidence points toward the effect of the hydroxysteroid 17β dehydrogenase family on steroid and fatty acid metabolism118. The rs72613567:TA loss‐of‐function variant of HSD17B13 has been found to be associated with a reduced risk of NASH in human livers and decreased liver injury in those predisposed to liver steatosis by PNPLA3I148M and was also associated with lower PNPLA3 messenger RNA (mRNA) expression118.This observation calls for personalized medicine, where people with functional variants of HSD17B13 and carriers of PNPLA3I148M could benefit most from therapeutic intervention targeting the activity or expression of HSD17B13 (ref. 118). Furthermore, studies evaluating therapies that target HSD17B13 should stratify treatment arms according to the presence or absence of the PNPLA3I148M variant.
Other genetic variants in genes such as TM6SF2 (ref. 120) (involved in the pathway for hepatic VLDL secretion), GCKR121 (encoding a glucokinase involved in hepatic de novo lipogenesis) and MBOAT7 (ref. 122) (the product of which plays a role in remodeling endomembrane phospholipid acyl chains) are being investigated as potential therapeutic targets in NASH. However, it is unlikely that all of them will prove to be an attractive therapeutic choice. For instance, approaches to decrease transmembrane 6 superfamily member 2 (TM6SF2) activity in patients with NASH may increase hepatic VLDL secretion while diminishing triglyceride levels in the liver123. This outcome may increase the risk of undesirable cardiovascular events, which might make TM6SF2E167K a less attractive therapeutic target in NASH, especially as the odds ratios of the association between TM6SF2E167K and NASH are modest123.
Combination therapy approaches
The NASH therapeutic landscape includes agents that target one or more of the main pathogenic pathways, including those regulating energy imbalance (to limit the influx of excess nutrients), pathways regulating the fate of excess metabolic substrates in the liver or inflammatory and fibrotic components of the wound-repair response124. Given the complex NASH pathophysiology, it is conceivable that any single therapeutic agent will lack adequate potency to effectively reverse the disease. The rationale behind combination therapies is based on improved efficacy by means of complementary or synergistic MOA and improved tolerability by allowing for lower doses of individual compounds. Finding the right combination among the endless possible permutations of drugs remains a key challenge. The most appealing combinations involve drugs from different classes targeting multiple steps in NASH pathogenesis, such as energy balance, liver metabolism, hepatocyte stress and fibrogenesis. Given the pleiotropic effects of farnesoid X receptor (FXR) agonists, they have been used in most trials thus far in combination with the 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor atorvastatin125 (CONTROL trial: mitigation of low-density lipoprotein cholesterol increase induced by obeticholic acid through the addition of atorvastatin), apoptosis signal-regulating kinase 1 (ASK-1) inhibitor plus acetyl-CoA carboxylase (ACC) inhibitor126 (primary endpoint not met), C–C motif chemokine receptor (CCR)2–CCR5 inhibitor cenicriviroc (TANDEM NCT03517540: primary endpoint not met), solute carrier family 5 member (SGLT)1–SGLT2 inhibitor licogliflozin (ELIVATE NCT04065841, ongoing), leukotriene A4 (LTA4) hydrolase inhibitor (NEXSCOT NCT04147195: terminated early by sponsor) and diacylglycerol O-acyltransferase 2 (DGAT2) inhibitor plus ACC inhibitor127 (potential to address limitations of ACC inhibition alone such as triglyceride increase). In the largest completed trial to date (the ATLAS study), cilofexor combination therapy did not meet the primary endpoint of fibrosis improvement by one stage or more with no worsening of NASH in participants with F3 or F4 at week 48 (ref. 126).
Future combination therapies should include drugs with proven benefit in terms of metabolic drivers and comorbidities in addition to liver-directed therapy. Examples include weight-loss medications such as glucagon-like peptide 1 (GLP1) receptor agonists128 and SGLT2 inhibitors with additional benefits in management of diabetes and cardiovascular disease prevention. The potential benefit of combination therapy must be carefully weighed with the challenges that come with the enrollment of a larger sample size for multiple arms and the potential for more types of side effects.
Currently, the optimal length of treatment for NASH remains unclear. Phase 3–4 trials plan for 5–8 years of longitudinal follow-up to assess whether the histologic response leads to improvement in clinical endpoints. Nevertheless, in a slowly progressive disease such as NASH, in which progression to cirrhosis or complications can span over decades13, this timeframe may be trivial for a large proportion of individuals129. Akin to diabetes mellitus, it is conceivable that lifelong treatment might be necessary, unless the therapy is supplemented by sustained weight loss. Alternatively, a model of ‘induction’ therapy with a potent combination of drugs with complementary MOA, followed by de-escalation to ‘maintenance’ therapy using drugs that target underlying metabolic abnormalities to prevent disease recurrence, could be considered130, similar to the autoimmune hepatitis-treatment paradigm.
It is also important for sponsors to consider developing companion diagnostics (non-invasive biomarker tests to predict response to therapy) for their respective drugs, in parallel to the regular drug-development process. Indeed, this is an essential tool to allow personalized medicine by targeting patients who are more likely to benefit from a specific therapy.
Regardless of the model, the cost of a lifelong NASH treatment could add a substantial health care burden, hopefully balanced by a decrease in costs associated with management of complications of end-stage liver disease. Cost-effectiveness will be enhanced by applying individualized therapies based on predictors of response for the optimal length of time, guided by high-performing non-invasive biomarkers of disease severity and response to treatment.
Conclusion
The major challenge in NASH drug development is the substantial variability in the assessment of the two primary histology endpoints used for conditional drug approval. Despite several attempts to improve the interpretation of liver histology, including AI or multiple-reader consensus, the mitigating actions will never be sufficient to overcome the limitations of these endpoints. The highest priority in the NASH field is the further development of NITs. All stakeholders should collectively work on refining the existing NITs and demonstrating that one, or a combination of them, strongly correlate with major adverse liver outcomes and treatment response. The ultimate goal is to improve the reliability of endpoints in NASH trials and to enable treatment decision making and follow-up in clinical practice.
References
Younossi, Z. et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 69, 2672–2682 (2019).
Younossi, Z. M. et al. Global epidemiology of nonalcoholic fatty liver disease—meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016).
Noureddin, M. et al. NASH leading cause of liver transplant in women: updated analysis of indications for liver transplant and ethnic and gender variances. Am. J. Gastroenterol. 113, 1649–1659 (2018).
Ascha, M. S. et al. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 51, 1972–1978 (2010).
Kabbany, M. N. et al. Prevalence of nonalcoholic steatohepatitis-associated cirrhosis in the United States: an analysis of national health and nutrition examination survey data. Am. J. Gastroenterol. 112, 581–587 (2017).
Noureddin, M. & Rinella, M. E. Nonalcoholic fatty liver disease, diabetes, obesity, and hepatocellular carcinoma. Clin. Liver Dis. 19, 361–379 (2015).
Donnelly, K. L. et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 (2005).
Hirsova, P., Ibrabim, S. H., Gores, G. J. & Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J. Lipid Res. 57, 1758–1770 (2016).
Marra, F. & Svegliati-Baroni, G. Lipotoxicity and the gut–liver axis in NASH pathogenesis. J. Hepatol. 68, 280–295 (2018).
Neuschwander-Tetri, B. A. Therapeutic landscape for NAFLD in 2020. Gastroenterology 158, 1984–1998 (2020).
Cornell, L. et al. DNA-PK—a candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin. Cancer Res. 21, 925–933 (2015).
Vuppalanchi, R., Noureddin, M., Alkhouri, N. & Sanyal, A. J. Therapeutic pipeline in nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 18, 373–392 (2021).
Sanyal, A. J. et al. Prospective study of outcomes in adults with nonalcoholic fatty liver disease. N. Engl. J. Med. 385, 1559–1569 (2021).
Anania, F. A., Dimick-Santos, L., Mehta, R., Toerner, J. & Beitz, J. Nonalcoholic steatohepatitis: current thinking from the Division of Hepatology and Nutrition at the Food and Drug Administration. Hepatology 73, 2023–2027 (2021).
Han, M. A. T. et al. Rates of and factors associated with placebo response in trials of pharmacotherapies for nonalcoholic steatohepatitis: systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 17, 616–629 (2019).
Ng, C. H. et al. Placebo effect on progression and regression in NASH: evidence from a meta-analysis. Hepatology 75, 1647–1661 (2022).
Sanyal, A. J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 16, 377–386 (2019).
Harrison, S. Mitigating Screen Failures: Finding the Right Patients for Trial Recruitment (EASL, 2022).
Kleiner, D. E. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005).
Sanyal, A. J. et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 362, 1675–1685 (2010).
Cusi, K. et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann. Intern. Med. 165, 305–315 (2016).
Belfort, R. et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 355, 2297–2307 (2006).
Chalasani, N. et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357 (2018).
Chalasani, N. et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 142, 1592–1609 (2012).
Dulai, P. S. et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta-analysis. Hepatology 65, 1557–1565 (2017).
Angulo, P. et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149, 389–397 (2015).
Ekstedt, M. et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 61, 1547–1554 (2015).
Younossi, Z. M. et al. Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver-related mortality. Hepatology 53, 1874–1882 (2011).
Sanyal, A. J. et al. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 54, 344–353 (2011).
Kleiner, D. E. et al. Association of histologic disease activity with progression of nonalcoholic fatty liver disease. JAMA Netw. Open 2, e1912565 (2019).
Bedossa, P. et al. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology 56, 1751–1759 (2012).
Singh, S. & Loke, Y. K. Drug safety assessment in clinical trials: methodological challenges and opportunities. Trials 13, 138 (2012).
Björnsson, E. S. Incidence and outcomes of DILI in Western patients. Clin. Liver Dis. 4, 9–11 (2014).
Reuben, A., Koch, D. G. & Lee, W. M. Drug-induced acute liver failure: results of a U.S. multicenter, prospective study. Hepatology 52, 2065–2076 (2010).
LiverTox: Clinical and Research Information on Drug-Induced Liver Injury (National Institute of Diabetes and Digestive and Kidney Diseases, 2012).
Goldkind, L. & Laine, L. A systematic review of NSAIDs withdrawn from the market due to hepatotoxicity: lessons learned from the bromfenac experience. Pharmacoepidemiol. Drug Saf. 15, 213–220 (2006).
Grudzinskas, C. Design of clinical development programs. In Principles of Clinical Pharmacology (Second Edition) (eds. Atkinson, A. Jr. et al.) 501–517 (Academic Press, 2007).
Regev, A. et al. Consensus: guidelines: best practices for detection, assessment and management of suspected acute drug-induced liver injury during clinical trials in patients with nonalcoholic steatohepatitis. Aliment. Pharm. Ther. 49, 702–713 (2019).
Guo, T. A tool to help you decide (detect potentially serious liver injury). In Annual Conference of the American Association for the Study of Liver Diseases (AASLD, 2008).
Guo, T. How a SAS/IntrNet tool was created at the FDA for the detection of potential drug-induced liver injury using data with CDISC standard. In Western Users of SAS Software Annual Conference (WUSS, 2009).
Temple, R. Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol. Drug Saf. 15, 241–243 (2006).
Watkins, P. B. et al. Evaluation of drug-induced serious hepatotoxicity (eDISH): application of this data organization approach to phase III clinical trials of rivaroxaban after total hip or knee replacement surgery. Drug Saf. 34, 243–252 (2011).
Davison, B. A. et al. Suboptimal reliability of liver biopsy evaluation has implications for randomized clinical trials. J. Hepatol. 73, 1322–1332 (2020).
Sumida, Y., Nakajima, A. & Itoh, Y. Limitations of liver biopsy and non-invasive diagnostic tests for the diagnosis of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 20, 475–485 (2014).
Tian, G., Kong, D., Jiang, T. & Li, L. Complications after percutaneous ultrasound-guided liver biopsy: a systematic review and meta-analysis of a population of more than 12,000 patients from 51 cohort studies. J. Ultrasound Med. 39, 1355–1365 (2020).
Ratziu, V. et al. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology 128, 1898–1906 (2005).
Gawrieh, S., Knoedler, D. M., Saeian, K., Wallace, J. R. & Komorowski, R. A. Effects of interventions on intra- and interobserver agreement on interpretation of nonalcoholic fatty liver disease histology. Ann. Diagn. Pathol. 15, 19–24 (2011).
Gramlich, T. et al. Pathologic features associated with fibrosis in nonalcoholic fatty liver disease. Hum. Pathol. 35, 196–199 (2004).
Brunt, E. M. Liver biopsy reliability in clinical trials: thoughts from a liver pathologist. J. Hepatol. 73, 1310–1312 (2020).
Brunt, E. M. et al. Complexity of ballooned hepatocyte feature recognition: defining a training atlas for artificial intelligence-based imaging in NAFLD. J. Hepatol. 76, 1030–1041 (2022).
Noureddin, M. Artificial intelligence in NASH histology: human teaches a machine for the machine to help humans. Hepatology 74, 9–11 (2021).
Taylor-Weiner, A. et al. A machine learning approach enables quantitative measurement of liver histology and disease monitoring in NASH. Hepatology 74, 133–147 (2021).
Pokkalla, H. et al. Machine learning models accurately interpret liver histology in patients with nonalcoholic steatohepatitis (NASH). Hepatology 70, 121A–122A (2019).
Eddowes, P. J. et al. Accuracy of FibroScan controlled attenuation parameter and liver stiffness measurement in assessing steatosis and fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 156, 1717–1730 (2019).
Noureddin, M. et al. Utility of magnetic resonance imaging versus histology for quantifying changes in liver fat in nonalcoholic fatty liver disease trials. Hepatology 58, 1930–1940 (2013).
Loomba, R. MRI-proton density fat fraction treatment response criteria in nonalcoholic steatohepatitis. Hepatology 73, 881–883 (2021).
Stine, J. G. et al. Change in MRI-PDFF and histologic response in patients with nonalcoholic steatohepatitis: a systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 19, 2274–2283 (2020).
Bril, F., Barb, D., Lomonaco, R., Lai, J. & Cusi, K. Change in hepatic fat content measured by MRI does not predict treatment-induced histological improvement of steatohepatitis. J. Hepatol. 72, 401–410 (2020).
Huang, D. Q. et al. Clinical utility of combined MRI-PDFF and ALT response in predicting histologic response in nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. https://doi.org/10.1016/j.cgh.2022.08.036 (2022).
Younossi, Z. M. et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 394, 2184–2196 (2019).
Harrison, S. A. et al. Efficacy and safety of aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology 160, 219–231 (2021).
Sanyal, A. J. et al. Cirrhosis regression is associated with improved clinical outcomes in patients with nonalcoholic steatohepatitis. Hepatology 75, 1235–1246 (2022).
Vilar-Gomez, E. et al. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: a multi-national cohort study. Gastroenterology 155, 443–457 (2018).
Adams, L. A. et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 129, 113–121 (2005).
Paik, J. M. et al. The impact of modifiable risk factors on the long-term outcomes of non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 51, 291–304 (2019).
Younossi, Z. M., Otgonsuren, M., Venkatesan, C. & Mishra, A. In patients with non-alcoholic fatty liver disease, metabolically abnormal individuals are at a higher risk for mortality while metabolically normal individuals are not. Metabolism 62, 352–360 (2013).
Golabi, P. et al. Components of metabolic syndrome increase the risk of mortality in nonalcoholic fatty liver disease (NAFLD). Medicine 97, e0214 (2018).
Armstrong, M. J. et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 387, 679–690 (2016).
Loomba, R. et al. Factors associated with histologic response in adult patients with nonalcoholic steatohepatitis. Gastroenterology 156, 88–95 (2019).
Ratziu, V. et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150, 1147–1159 (2016).
Altayar, O., Noureddin, N., Thanda Han, M. A., Murad, M. H. & Noureddin, M. Fibrosis changes in the placebo arm of NASH clinical trials. Clin. Gastroenterol. Hepatol. 17, 2387 (2019).
Harrison, S. A. et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: results from randomized phase III STELLAR trials. J. Hepatol. 73, 26–39 (2020).
Newsome, P. N. et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N. Engl. J. Med. 384, 1113–1124 (2021).
Glass, O. et al. Standardisation of diet and exercise in clinical trials of NAFLD–NASH: recommendations from the Liver Forum. J. Hepatol. 73, 680–693 (2020).
Neumann, J., Beck, O. & Böttcher, M. Phosphatidylethanol, ethyl glucuronide and ethanol in blood as complementary biomarkers for alcohol consumption. J. Mass Spectrom. Adv. Clin. Lab 22, 3–7 (2021).
Pocock, S. J. & Stone, G. W. The primary outcome is positive—is that good enough? N. Engl. J. Med. 375, 971–979 (2016).
Cefalu, W. T. et al. Cardiovascular outcomes trials in type 2 diabetes: where do we go from here? Reflections from a Diabetes Care Editors’ Expert Forum. Diabetes Care 41, 14–31 (2018).
Noureddin, N., Schattenberg, J. M., Alkhouri, N. & Noureddin, M. Noninvasive testing using magnetic resonance imaging techniques as outcomes in nonalcoholic steatohepatitis clinical trials: how full is the glass? Hepatol. Commun. 4, 141–144 (2020).
Castera, L., Friedrich-Rust, M. & Loomba, R. Noninvasive assessment of liver disease in patients with nonalcoholic fatty liver disease. Gastroenterology 156, 1264–1281 (2019).
Loomba, R. et al. Magnetic resonance elastography predicts advanced fibrosis in patients with nonalcoholic fatty liver disease: a prospective study. Hepatology 60, 1920–1928 (2014).
Selvaraj, E. A. et al. Diagnostic accuracy of elastography and magnetic resonance imaging in patients with NAFLD: a systematic review and meta-analysis. J. Hepatol. 75, 770–785 (2021).
Feldstein, A. E. et al. Serum cytokeratin-18 fragment levels are useful biomarkers for nonalcoholic steatohepatitis in children. Am. J. Gastroenterol. 108, 1526–1531 (2013).
Feldstein, A. E. et al. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology 50, 1072–1078 (2009).
Lee, J. et al. Accuracy of cytokeratin 18 (M30 and M65) in detecting non-alcoholic steatohepatitis and fibrosis: a systematic review and meta-analysis. PLoS ONE 15, e0238717 (2020).
Harrison, S. A. et al. A blood-based biomarker panel (NIS4) for non-invasive diagnosis of non-alcoholic steatohepatitis and liver fibrosis: a prospective derivation and global validation study. Lancet Gastroenterol. Hepatol. 5, 970–985 (2020).
Dennis, A. et al. A composite biomarker using multiparametric magnetic resonance imaging and blood analytes accurately identifies patients with non-alcoholic steatohepatitis and significant fibrosis. Sci. Rep. 10, 15308 (2020).
Dennis, A. et al. Correlations between MRI biomarkers PDFF and cT1 with histopathological features of non-alcoholic steatohepatitis. Front. Endocrinol. 11, 575843 (2020).
Allen, A. M. et al. The role of three-dimensional magnetic resonance elastography in the diagnosis of nonalcoholic steatohepatitis in obese patients undergoing bariatric surgery. Hepatology 71, 510–521 (2020).
Allen, A. M. et al. Multiparametric magnetic resonance elastography improves the detection of NASH regression following bariatric surgery. Hepatol. Commun. 4, 185–192 (2020).
Dzyubak, B. et al. Automated analysis of multiparametric magnetic resonance imaging/magnetic resonance elastography exams for prediction of nonalcoholic steatohepatitis. J. Magn. Reson. Imaging 54, 122–131 (2021).
Aggarwal, P. et al. Identifying patients who require pharmacotherapy for nonalcoholic steatohepatitis (NASH) using the FAST score. Hepatology 72, 922A–923A (2020).
Noureddin, M. et al. MRI-based (MAST) score accurately identifies patients with NASH and significant fibrosis. J. Hepatol. 76, 781–787 (2022).
Kim, B. K. et al. Head-to-head comparison between MEFIB, MAST, and FAST for detecting stage 2 fibrosis or higher among patients with NAFLD. J. Hepatol. 77, 1482–1490 (2022).
Alkhouri, N. & McCullough, A. Noninvasive diagnosis of NASH and liver fibrosis within the spectrum of NAFLD. Gastroenterol. Hepatol. 8, 661–668 (2012).
Harrison, S. et al. Correlation between AI-based digital pathology and non-invasive tests (NITs) of NASH fibrosis stage: baseline data from the FASCINATE-2 phase 2b clinical study of denifanstat in patients with F2/F3 NASH. In Americam Association for the Study of Liver Diseases—the Liver Meeting (AASLD, 2022).
Hartman, M. L., Tang, Y., Robins, D. B. & Vuppalanchi, R. FibroScan-AST (FAST) score improves qualification for at-risk nonalcoholic steatohepatitis (NASH) with different effects by specialty of the primary investigator in the phase 2b SYNERGY-NASH trial. In American Association for the Study of Liver Diseases—the Liver Meeting (AASLD, 2022).
Pavlides, M. et al. Multiparametric magnetic resonance imaging predicts clinical outcomes in patients with chronic liver disease. J. Hepatol. 64, 308–315 (2016).
Are, V. S., Vuppalanchi, R., Vilar-Gomez, E. & Chalasani, N. Enhanced liver fibrosis score can be used to predict liver-related events in patients with nonalcoholic steatohepatitis and compensated cirrhosis. Clin. Gastroenterol. Hepatol. 19, 1292–1293 (2021).
Boursier, J. et al. Diagnostic accuracy and prognostic significance of blood fibrosis tests and liver stiffness measurement by FibroScan in non-alcoholic fatty liver disease. J. Hepatol. 65, 570–578 (2016).
Gidener, T. et al. Liver stiffness by magnetic resonance elastography predicts future cirrhosis, decompensation, and death in NAFLD. Clin. Gastroenterol. Hepatol. 19, 1915–1924 (2020).
Han, M. A. T. et al. MR elastography-based liver fibrosis correlates with liver events in nonalcoholic fatty liver patients: a multicenter study. Liver Int. 40, 2242–2251 (2020).
Petta, S. et al. Monitoring occurrence of liver-related events and survival by transient elastography in patients with nonalcoholic fatty liver disease and compensated advanced chronic liver disease. Clin. Gastroenterol. Hepatol. 19, 806–815 (2021).
Younes, R. et al. Long-term outcomes and predictive ability of non-invasive scoring systems in patients with non-alcoholic fatty liver disease. J. Hepatol. 75, 786–794 (2021).
Kawaguchi-Suzuki, M. et al. A genetic score associates with pioglitazone response in patients with non-alcoholic steatohepatitis. Front. Pharmacol. 9, 752 (2018).
Gawrieh, S. et al. A pilot genome-wide analysis study identifies loci associated with response to obeticholic acid in patients with NASH. Hepatol. Commun. 3, 1571–1584 (2019).
Dinani, A. M., Kowdley, K. V. & Noureddin, M. Application of artificial intelligence for diagnosis and risk stratification in NAFLD and NASH: the state of the art. Hepatology 74, 2233–2240 (2021).
Nassar, S. F., Raddassi, K. & Wu, T. Single-cell multiomics analysis for drug discovery. Metabolites 11, 729 (2021).
Kuret, T., Sodin-Šemrl, S., Leskošek, B. & Ferk, P. Single cell RNA sequencing in autoimmune inflammatory rheumatic diseases: current applications, challenges and a step toward precision medicine. Front. Med. 8, 822804 (2021).
Carlsson, B. et al. Review article: the emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 51, 1305–1320 (2020).
Alkhouri, N. & Gawrieh, S. A perspective on RNA interference-based therapeutics for metabolic liver diseases. Expert Opin. Investig. Drugs 30, 237–244 (2021).
Burza, M. A. et al. PNPLA3 I148M (rs738409) genetic variant is associated with hepatocellular carcinoma in obese individuals. Dig. Liver Dis. 44, 1037–1041 (2012).
Grimaudo, S. et al. Association between PNPLA3 rs738409 C>G variant and liver-related outcomes in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 18, 935–944 (2020).
Pennisi, G. et al. PNPLA3 rs738409 C>G variant predicts fibrosis progression by noninvasive tools in nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 19, 1979–1981 (2021).
Romeo, S. et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int. J. Obes. 34, 190–194 (2010).
Krawczyk, M. et al. PNPLA3 p.I148M variant is associated with greater reduction of liver fat content after bariatric surgery. Surg. Obes. Relat. Dis. 12, 1838–1846 (2016).
Oscarsson, J. et al. Effects of free omega-3 carboxylic acids and fenofibrate on liver fat content in patients with hypertriglyceridemia and non-alcoholic fatty liver disease: a double-blind, randomized, placebo-controlled study. J. Clin. Lipidol. 12, 1390–1403 (2018).
Unalp-Arida, A. & Ruhl, C. E. Patatin-like phospholipase domain-containing protein 3 I148M and liver fat and fibrosis scores predict liver disease mortality in the U.S. population. Hepatology 71, 820–834 (2020).
Abul-Husn, N. S. et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N. Engl. J. Med. 378, 1096–1106 (2018).
Ma, Y. et al. 17-β hydroxysteroid dehydrogenase 13 is a hepatic retinol dehydrogenase associated with histological features of nonalcoholic fatty liver disease. Hepatology 69, 1504–1519 (2019).
Kozlitina, J. et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 46, 352–356 (2014).
Tan, H. L. et al. Association of glucokinase regulatory gene polymorphisms with risk and severity of non-alcoholic fatty liver disease: an interaction study with adiponutrin gene. J. Gastroenterol. 49, 1056–1064 (2014).
Mancina, R. M. et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 150, 1219–1230 (2016).
Eslam, M., Valenti, L. & Romeo, S. Genetics and epigenetics of NAFLD and NASH: clinical impact. J. Hepatol. 68, 268–279 (2018).
Noureddin, M., Muthiah, M. D. & Sanyal, A. J. Drug discovery and treatment paradigms in nonalcoholic steatohepatitis. Endocrinol. Diabetes Metab. 3, e00105 (2020).
Pockros, P. J. et al. CONTROL: a randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 39, 2082–2093 (2019).
Loomba, R. et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology 73, 625–643 (2021).
Calle, R. A. et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 27, 1836–1848 (2021).
Alkhouri, N. et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: a randomised, open-label phase II trial. J. Hepatol. 77, 607–618 (2022).
Allen, A. M. et al. Clinical course of non-alcoholic fatty liver disease and the implications for clinical trial design. J. Hepatol. 77, 1237–1245 (2022).
Alkhouri, N., Tincopa, M., Loomba, R. & Harrison, S. A. What does the future hold for patients with nonalcoholic steatohepatitis: diagnostic strategies and treatment options in 2021 and beyond? Hepatol. Commun. 5, 1810–1823 (2021).
Konerman, M. A., Jones, J. C. & Harrison, S. A. Pharmacotherapy for NASH: current and emerging. J. Hepatol. 68, 362–375 (2018).
Fu, S., Watkins, S. M. & Hotamisligil, G. S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 15, 623–634 (2012).
Ertunc, M. E. & Hotamisligil, G. S. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 57, 2099–2114 (2016).
Loomba, R., Friedman, S. L. & Shulman, G. I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564 (2021).
Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
S.A.H.: grant support for research (Akero Therapeutics, Altimmune, Axcella Health, Cirius Therapeutics, CiVi Biopharma, CymaBay Therapeutics, Enyo Pharma, Galectin Therapeutics, Galmed Research and Development, Genfit, Gilead Sciences, Hepion Pharmaceuticals, HighTide Therapeutics, Intercept Pharmaceuticals, Madrigal Pharmaceuticals, Metacrine, NGM Biopharmaceuticals, NorthSea Therapeutics, Novartis Pharmaceuticals, Novo Nordisk, Poxel, Sagimet Biosciences, Terns Pharmaceuticals, Viking Therapeutics, 89Bio), advisory board and/or consultant (Akero Therapeutics, Alentis Therapeutics, Alimentiv, Altimmune, Arrowhead, Axcella Health, Boston Pharmaceuticals, B Riley FBR, ChronWell, Corcept Therapeutics, Echosens North America, ENYO Pharma, Foresite Labs, Galectin Therapeutics, Genfit, Gilead Sciences, GNS, GSK, Hepion Pharmaceuticals, HighTide Therapeutics, HistoIndex, Intercept Pharmaceuticals, Ionis, Kowa Research Institute, Madrigal Pharmaceuticals, Medpace, Merck, Metacrine, NeuroBo, NGM Biopharmaceuticals, NorthSea Therapeutics, Novo Nordisk, Nutrasource, Perspectum Diagnostics, Piper Sandler, Poxel, Sagimet Biosciences, Sonic Incytes Medical, Terns, Viking Therapeutics), stock and/or options (self-managed) (Akero Therapeutics, ChronWell, Cirius Therapeutics, Galectin Therapeutics, Genfit, Hepion Pharmaceuticals, HistoIndex, Metacrine, NGM Biopharmaceuticals, NorthSea Therapeutics). J.D.: shareholder in Poxel. A.M.A.: research support to her institution from Novo Nordisk, Pfizer and Target Pharma and is a consultant for Novo Nordisk and Pfizer. M.N.: advisory board and/or consultant for 89Bio, Altimmune, BI, Gilead, CohBar, CytoDyn, Pfizer, GSK, Novo Nordisk, Echosens, Madrigal, NorthSea, Perspectum, Terns, Takeda, Sami-Sabinsa Group, Siemens and Roche Diagnostics; M.N. has received research support from Allergan, Akero, BMS, Gilead, Galmed, Galectin, Genfit, Conatus, Enanta, Madrigal, Novartis, Pfizer, Shire, Viking and Zydus; M.N. is a shareholder or has stocks in Anaetos, ChronWell, CytoDyn, Ciema, Rivus Pharma and Viking. N.A.: research funding (89Bio, Akero, AbbVie–Allergan, Better Therapeutics, Boehringer Ingelheim, Bristol Myers Squibb, Corcept, DSM, Galectin, Genentech, Genfit, Gilead, Hepagene, Healio, Intercept, Inventiva, Ionis, Madrigal, Merck, NGM, Noom, NorthSea, Novo Nordisk, Perspectum, Pfizer, Poxel, Viking and Zydus), speaker bureau (AbbVie–Allergan, Alexion, Echosens, Eisai, Exelixis, Gilead, Intercept, Perspectum, Salix and Theratechnologies), consultant (AbbVie–Allergan, Echosens, Fibronostics, Gilead, Intercept, Madrigal, Novo Nordisk, Perspectum, Pfizer and Zydus).
Peer review
Peer review information
Nature Medicine thanks Raj Vuppalanchi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Karen O’Leary, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Harrison, S.A., Allen, A.M., Dubourg, J. et al. Challenges and opportunities in NASH drug development. Nat Med 29, 562–573 (2023). https://doi.org/10.1038/s41591-023-02242-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-023-02242-6
- Springer Nature America, Inc.
This article is cited by
-
Updated mechanisms of MASLD pathogenesis
Lipids in Health and Disease (2024)
-
Role of steatotic liver disease in prediction and prevention of cardiometabolic diseases
Nature Reviews Gastroenterology & Hepatology (2024)
-
Opportunities and challenges following approval of resmetirom for MASH liver disease
Nature Medicine (2024)
-
Knockout of STE20-type kinase TAOK3 does not attenuate diet-induced NAFLD development in mice
Molecular Medicine (2023)
-
Friend or foe? The elusive role of hepatic stellate cells in liver cancer
Nature Reviews Gastroenterology & Hepatology (2023)