

Abstract
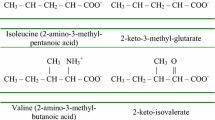
RECENTLY, a new metabolic defect, which was believed to arise from congenital lysine intolerance, was described in a 3-month-old baby girl1. An enzymatic impairment in the pathway for the degradation of lysine seemed the most probable explanation of the main finding, that is, high plasma lysine levels. Because the enzymes responsible for the metabolism of lysine in human beings had not previously been investigated, efforts were made to find an enzyme system capable of degrading lysine. This report presents evidence for the existence of a hitherto unknown enzyme, L-lysine: nicotinamide-adenine dinucleotide (NAD) - oxidoreductase (deaminating) (trivial name: L-lysine dehydrogenase) in human liver homogenates.
Similar content being viewed by others

References
Colombo, J. P., Richterich, R., Donath, A., Spahr, A., and Rossie, E., Lancet, 1014 (1964).
Meister, A., The Biochemistry of Amino Acids (Academic Press, Inc., New York, 1957).
Büngi, W., and Richterich, R., Helv. Physiol. Acta, 23, 183 (1965).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
BÜRGI, W., RICHTERICH, R. & COLOMBO, J. L-Lysine Dehydrogenase Deficiency in a Patient with Congenital Lysine Intolerance. Nature 211, 854–855 (1966). https://doi.org/10.1038/211854a0
Issue Date:
DOI: https://doi.org/10.1038/211854a0
- Springer Nature Limited
This article is cited by
-
Hyperdibasicaminoaciduria in a Turkish infant without evident protein intolerance
European Journal of Pediatrics (1979)