
Abstract
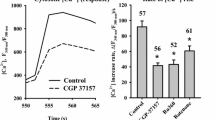
Data obtained in studies of the nature of the correlation which we have previously observed [10, 17] between mitochondrial depolarization and the level of disruption of Ca2+ homeostasis in cultivated brain neurons are summarized. Experiments were performed on cultured cerebellar granule cells loaded with Fura-2-AM or rhodamine 123 to measure changes in cytoplasmic Ca2+ and mitochondrial potential during pathogenic treatments of the cells. Prolonged exposure to 100 μM glutamate induced a reversible increase in [Ca2+]i, which was accompanied by only a small degree of mitochondrial depolarization. A sharp increase in this mitochondrial depolarization, induced by addition of 3 mM NaCN or 300 μM dinitrophenol (DNP) to the glutamate-containing solution, resulted in further increase in [Ca2+]i, due to blockade of electrophoretic mitochondrial Ca2+ uptake. Prolonged exposure to CN– or DNP in the post-glutamate period maintained [Ca2+]i at a high level until the metabolic inhibitors were removed. In most cells, this plateau was characterized by low sensitivity to removal of external Ca2+, demonstrating that the mechanisms of Ca2+ release from neurons were disrupted. Addition of oligomycin, a blocker of mitochondrial ATP synthase/ATPase, to the solution containing glutamate and CN– or DNP eliminated the post-glutamate plateau. Parallel experiments with direct measurements of intracellular ATP levels ([ATP]) showed that profound mitochondrial depolarization induced by CN– or DNP sharply enhanced the drop in ATP due to glutamate, while oligomycin significantly weakened this effect of the metabolic inhibitors. Analysis of these data led to the conclusion that blockade of mitochondrial Ca2+ uptake and inhibition of ATP synthesis resulted from mitochondrial depolarization and plays a key role in the mechanism disrupting [Ca2+]i homeostasis after toxic exposure to glutamate.
Similar content being viewed by others


REFERENCES
A. Bogachev, L. Bykova, B. Khodorov, N. Andreeva, L. Khaspekov, V. Pinelis, and I. Victorov, “Stable decrease in ATP contents in cultured cerebellar and hippocampal neurons during and after toxic glutamate treatment,” Biol. Membranes, 9, 1057–1059 (1992).
F. Buttgereit and M. Brand, “A hierarchy of ATP-consuming processes in mammalian cells,” Biochem. J., 312, 163–167 (1995).
R. DiPolo and L. Beauge, “Ca2+ transport in nerve fibers,” Biochem. Biophys. Acta, 947, 549–569 (1988).
M. R. Duchen, “Topical review: contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signaling and cell death,” J. Physiol. (London), 516, 1–17 (1999).
B. I. Khodorov, “Mechanisms of destabilization of Ca2+ homeostasis of brain neurons caused by toxic glutamate challenge,” Membrane Cell Biol., 14, 149–162 (2000).
B. I. Khodorov, D. A. Fayuk, S. G. Koshelev, O. V. Vergun, V. G. Pinelis, N. P. Vinskaya, T. P. Storozhevikh, E. N. Arsen'eva, L. G. Khaspekov, A. P. Lyzhin, I. V. Victorov, and J. M. Dubinsky, “Effect of a prolonged glutamate challenge on plasmalemmal calcium permeability in mammalian central neurons. Mn2+ as a tool to study calcium influx pathways,” Int. J. Neurosci., 88, 215–241 (1996).
B. Khodorov, P. Pinelis, T. Storozhevikh, and A. Juriavichus, “Blockade of mitochondrial Ca2+ uptake by mitochondrial inhibitors amplifies the glutamate-induced calcium response in cultured cerebellar granule cells,” FEBS Lett., 458, 162–166 (1999).
B. Khodorov, V. Pinelis, T. Storozhevikh, O. Vergun, and N. Vinskaya, “Dominant role of mitochondria in protection against a delayed neuronal Ca2+ overload induced by endogenous excitatory amino acids following a glutamate pulse,” FEBS Lett., 393, 135–138 (1996).
B. Khodorov, V. Pinelis, T. Storozhevykh, A. Jurevicius, A. Borodin, A. Surin, N. Vinskaya, and L. Khaspecov, “Mitochondrial calcium uptake following prolonged glutamate challenge is required for nerve cell recovery from calcium load,” J. Physiol., 525, 69 (2000).
B. Khodorov, V. Pinelis, O. Vergun, T. Storozhevykh, and N. Vinskaya, “Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge,” FEBS Lett., 397, 230–234 (1996).
G. Marcaida, M. Minana, S. Grisolia, and V. Felipo, “Lack of correlation between glutamate-induced depletion of ATP and neuronal death in primary cultures of cerebellum,” Brain Res., 695, 146–150 (1995).
D. Nicholls and S. Budd, “Mitochondria and neuronal survival,” Physiol. Rev., 80, 315–360 (2000).
V. G. Pinelis, L. P. Bykova, A. P. Bogachev, N. K. Isaev, I. V. Vickorov, and B. I. Khodorov, “Toxic glutamate treatment of cerebellar granule cells decreases the intracellular ATP content. Role of Ca2+ ions,” Bulleten Eksperimentalnoy Biologii i Medicini, 123, 162–164 (1997).
A. F. Schinder, E. C. Olson, N. C. Spitzer, and M. Montal, “Mitochondrial dysfunction is a primary event in glutamate neurotoxicity,” J. Neurosci., 16, 6125–6133 (1996).
A. Surin, T. Storozhevykh, A. Turavichus, E. Sorokina, E. Arsen'eva, N. Vinskaya, A. Borodin, V. Pinelis, and B. Khodorov, “Contribution of plasmalemmal Ca2+ pump to clearance of glutamate-imposed load in cerebellar granule cells,” J. Physiol.(in press) (2000).
D. M. Turetsky, J. E. Huettner, D. I. Gottlieb, M. P. Goldberg, and D. W. Choi, “Glutamate receptor-mediated currents and toxicity in embryonal carcinoma cells,” J. Neurobiology, 24, 1157–1169 (1993).
O. Vergun, J. Keelan, B. I. Khodorov, and M. R. Duchen, “Glutamate-induced mitochondrial depolarization and perturbation of calcium homeostasis in cultured rat hippocampal neurones,” J. Physiol., 519, 451–466 (1999).
R. J. White and I. J. Reynolds, “Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxic exposure,” J. Neurosci., 16, 5688–5697 (1996).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Khodorov, B.I., Storozhevykh, T.P., Surin, A.M. et al. The Leading Role of Mitochondrial Depolarization in the Mechanism of Glutamate-Induced Disruptions in Ca2+ Homeostasis. Neurosci Behav Physiol 32, 541–547 (2002). https://doi.org/10.1023/A:1019819925257
Issue Date:
DOI: https://doi.org/10.1023/A:1019819925257