
Abstract
There is accumulating evidence that cysteine sulfenation (cys-SOH) in proteins plays an important role in cellular response to oxidative stress. The purpose of the present study was to identify mitochondrial proteins that undergo changes in cys-SOH during aging. Studies were conducted in rats when they were 5 or 30 months of age. Following blocking of free protein thiols with N-ethylmaleimide, protein sulfenic acids were reduced by arsenite to free thiol groups that were subsequently labeled with biotin-maleimide. Samples were then comparatively analyzed by two-dimensional Western blots, and proteins showing changes in sulfenation were selectively identified by mass spectrometry peptide sequencing. As a result, five proteins were identified. Proteins showing an age-related decrease in sulfenation include pyruvate carboxylase and pyruvate dehydrogenase; while those showing an age-related increase in sulfenation include aconitase, mitofilin, and tubulin (α-1). Results of the present study provide a general picture of mitochondrial protein sulfenation in brain oxidative stress and implicate the involvement of protein sulfenation in overall decline of mitochondrial function during brain aging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Brain aging is associated with a shift from cellular redox regulation to oxidative damage (Rebrin et al. 2007; Sohal and Forster 2007). This redox shift to a more oxidative status during aging is thought to be responsible for oxidative modification of reactive protein cysteine (cys) residues that are involved in redox signaling upon oxidative stress (Cai and Yan 2013; Ying et al. 2007). A major product of protein cys oxidation is a protein sulfenic acid (PSOH), a reversible oxidative modification of a protein thiol group (Heppner et al. 2017; Poole et al. 2004). Sulfenic acid is a central intermediate during cys oxidation and can be induced by hydrogen peroxide, alkyl hydroperoxides, and peroxynitrite (Poole et al. 2004). Although long considered highly reactive and unstable, accumulating evidence has indicated that stabilized sulfenic acids do exist and play a key redox regulatory role in a growing number of proteins (Boschi-Muller et al. 2008; Claiborne et al. 2001; Denu and Tanner 2002; Kaiserova et al. 2006; Shetty et al. 2007; Turell et al. 2008; Wood et al. 2003; Yeh et al. 1996). Moreover, the biological and pathological significance of PSOHs has been further demonstrated by reports that PSOH formation is required for T-cell activation (Michalek et al. 2007) and that many proteins in cancer cells contain sulfenic acids (Leonard et al. 2009; Seo and Carroll 2009).
The purpose of the present study was to identify mitochondrial proteins that undergo changes in cys sulfenation (cys-SOH) during brain aging and oxidative stress. Mitochondria were used in this study because mitochondrial oxidative stress has been associated with brain aging (Sohal and Forster 2007) and a number of neurodegenerative disorders including Parkinson’s and Alzheimer’s disease (Keating 2008). In this study, rats aged at 5 or 30 months were used. Following mitochondria isolation and blocking of free protein thiols with N-ethylmaleimide (NEM), sulfenic acids in proteins were captured by an arsenite-specific reduction/biotin switch method (Saurin et al. 2004), whereby a sulfenic acid was reduced back to a free thiol group that was subsequently labeled with biotin-maleimide (Fig. 1). Samples were then analyzed by comparative two-dimensional (2D) Western blots and sulfenated proteins were identified by mass spectrometry (MS) peptide sequencing. Results of the present study suggest that mitochondrial PSOHs play a role in brain aging.

Scheme outlining the procedure of arsenite reduction/biotin switch used for detection and identification of proteins that underwent sulfenation in rat brain mitochondria. All available free protein thiol groups were blocked with N-ethylmaleimide (NEM) in the presence of 1% SDS that facilitates the access of free thiols by the alkylating reagent. Excessive NEM was removed by gel filtration using PD-10 columns. The resulting protein solution was treated with sodium arsenite in the presence of biotin-maleimide. Following precipitation with 10% TCA and washing with ethyl acetate/ethanol, protein pellet was dissolved in corresponding buffers for 2D Western blot detection or for affinity purification
RESULTS
Specificity of the arsenite reduction/biotin switch method in conjunction with reducing gel electrophoresis
The method of arsenite reduction/biotin switch (as shown in Fig. 1) was originally applied under non-reducing electrophoretic conditions (Saurin et al. 2004). However, given the fact that the resolution of a reducing gel is always better than that of a non-reducing gel, we first performed one-dimensional (1D) reducing SDS-PAGE (polyacrylamide gel electrophoresis) and Western blot to test whether our procedure of trichloroacetic acid (TCA) precipitation and organic solvent washing following arsenite reduction/biotin labeling could completely remove any unincorporated biotin-maleimide that would otherwise interfere with reducing gel electrophoresis and Western blot detection. Mitochondria were oxidatively stressed in vitro in the presence of succinate and antimycin A (Yan et al. 2008) and the results are shown in Fig. 2. When the sample was treated with biotin-maleimide in the absence of sodium arsenite, no biotin signals were detected (lane 2). When the sample was treated with dimedone that specifically reacts with sulfenic acids (Saurin et al. 2004) before arsenite reduction and biotin labeling, no biotin signals were detected either (lane 3), indicating a complete blocking of PSOHs by dimedone. Biotin signals were only detected in the samples treated with sodium arsenite and biotin-maleimide (lane 4). Therefore, these results demonstrate that the steps of TCA precipitation and organic solvent washing used in our procedure completely removed unincorporated biotin-maleimide, and that samples treated in this way were compatible with reducing gel electrophoresis and Western blot detection of PSOHs. Nevertheless, as shown in Fig. 3, 1D Western blot analysis was not applicable in detecting age-related changes in protein sulfenation because the 1D approach yielded a very low resolution that could not produce well-defined and distinguished bands for protein identification.

A representative 1D Western blot demonstrating a complete removal of biotin-maleimide by the procedure of TCA precipitation and organic solvent washing. Lane 1: biotin-conjugated protein markers (Bio-Rad); Lane 2: biotin labeling of the protein mixture in the absence of arsenite; Lane 3: biotin labeling of the protein mixture in the presence of arsenite following dimedone treatment; Lane 4: biotin labeling of the protein mixture in the presence of sodium arsenite. Samples (20 μg/lane) loaded onto the gel were derived from mitochondria that underwent in vitro oxidative stress in the presence of succinate and antimycin A as previously described (Yan et al. 2008)

One-dimensional Western blot detection of mitochondrial protein sulfenation during brain aging. Mitochondrial proteins were treated according to the procedures presented in Fig. 1. Biotin-conjugated proteins from Bio-Rad were used as immunoblot markers
Identification of mitochondrial proteins showing changes in cysteine sulfenation
As 1D Western blot analysis didn’t work for our purpose (Fig. 3), we then took a comparative 2D Western blot approach so that comparison of sulfenation signal intensities could be made between corresponding gel spots. While there were numerous proteins on Coomassie blue-stained 2D gels (Fig. 4A), there was a less number of gel spots showing age-related changes in sulfenation when probed with HRP–streptavidin (Fig. 4B). Based on changes of the selected spot intensities between the two age groups (5 and 30 months), spots 1 and 5 showed a decrease in sulfenation; spots 2–4 showed an increase in sulfenation. These selected gel spots were then excised for MS peptide sequencing and the identified proteins are listed in Table 1, which includes pyruvate carboxylase (spot 1), aconitase (spot 3), pyruvate dehydrogenase (spot 5), mitofilin (spot 2), and tubulin α-1 (spot 4). It should be noted that no age-related changes in individual protein content could be observed between 5 and 30 months of age (Fig. 4A). Hence, any changes in sulfenation for the selected proteins were indeed due to sulfenic acid formation, rather than to changes in the levels of protein expression.

Two-dimensional gel analysis of sulfenated brain mitochondrial proteins. A Representative 2D maps comparing brain mitochondrial protein expression levels between 5- and 30-month-old rats. Brain mitochondria were prepared and analyzed as described in the text. Gels were stained by Coomassie blue G-250. B Representative 2D Western blots of age-related protein sulfenation in rat brain mitochondria. Shown are 2D gel maps of protein sulfenation in brain mitochondria isolated from 5- to 30-month-old rats (40 μg protein/gel). For these experiments, fresh mitochondria fractions were prepared followed by immediate analysis of protein sulfenic acids. Each age group contained brain mitochondria isolated from three rats
In vitro oxidative stress, protein sulfenation, and aconitase activity
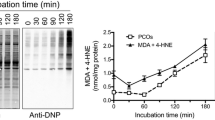
To investigate whether there is a relationship between overall protein sulfenation and changes in enzyme activity, we used an in vitro system whereby HT22 cells were treated by CoCl2 that is known to induce hypoxia–reperfusion oxidative stress (Sun et al. 2015; Wang et al. 2016) and then measured protein sulfenation and aconitase activity. Here PSOHs were labeled by a recently developed probe DCP-Bio1 (Klomsiri et al. 2010) followed by Western blot analysis. Results in Fig. 5 indicate that protein sulfenation showed an overall increase in sulfenic acid content that is reperfusion time-dependent (Fig. 5A) while aconitase activity showed a steady decrease in a reperfusion time-dependent manner (Fig. 5B). These results indicate that there was a time-dependent loss in aconitase activity in this in vitro oxidative stress system. While aconitase activity was inversely related to Western blot protein sulfenation profile, whether the loss of its activity is due to sulfenation to a specific cys residue on aconitase remains to be investigated. It is possible that the loss of aconitase activity could also be due to the formation of sulfinic or sulfonic acid (Han et al. 2005). It is also worth noting that the DCP-Bio1 method is much better and simpler than the arsenite reduction/biotin switch method as the former does not involve any washing and precipitation steps and saves time and samples. DCP-Bio1 is commercially available but is kind of expensive. For those who cannot afford to purchase this probe, the arsenite reduction/biotin switch should still be the method of choice.

Western blot analysis of HT22 cell protein sulfenation after CoCl2-induced hypoxia–reperfusion. A Reperfusion time-dependent increase in protein sulfenation measured by DCP-Bio1 labeling and Western blot. B Reperfusion time-dependent decrease in aconitase activity
DISCUSSION
In the present study, we mainly used the arsenite-specific reduction/biotin switch method (Saurin et al. 2004) to capture PSOHs formed during brain aging. The biotin-tagged protein samples were then comparatively analyzed by 2D Western blots probed with HRP–streptavidin. Five gel spots showing age-related changes in sulfenic acid formation were excised for protein identification. Results of the present study indicate a differential nature of protein sulfenation and suggest that mitochondrial PSOHs play a role in brain oxidative stress and aging.
Given the fact that PSOHs are key intermediates during protein thiol oxidation (Poole et al. 2004), a decrease in a protein’s sulfenation during aging should not be surprising. When the sulfenic acids on target proteins are not stable and can undergo further oxidation, the level of protein sulfenation detected by this method would show a decrease because all those further cys oxidation products (Poole et al. 2004), formed via sulfenic acids, are resistant to arsenite reduction, which leads to less biotin labeling and hence a lower level of PSOH content. This is in contrast to that of protein carbonylation, an irreversible oxidative modification that occurs on multiple amino acid residues and rarely shows a decrease during aging (Yan et al. 1997; Yan and Sohal 1998).
Aconitase is an enzyme of the citric acid cycle. Its possession of an iron–sulfur cluster makes it not only a target susceptible to oxidative modification (Yan et al. 1997; Yarian et al. 2006), but also a sensitive marker for evaluating the magnitude of oxidative stress (Gardner 2002; Zourlidou et al. 2007). Aconitase not only undergoes irreversible oxidation such as protein carbonylation (Yan et al. 1997) and sulfenation of the cys residue that binds to the Fe–S cluster (Han et al. 2005), but can also experience reversible oxidative modifications such as sulfenation (this study and Charles et al. 2007; Saurin et al. 2004), S-glutathionylation, and S-nitrosylation (Han et al. 2005; Tsou et al. 2007). All these findings indicate that both irreversible and reversible modifications to aconitase can occur. Furthermore, given the findings that rat heart aconitase incurs a reversible redox inhibition upon cardiac ischemia–reperfusion (Bulteau et al. 2005), it’s probable that aconitase sulfenation in brain mitochondria plays a role in age-related occurrence of neurodegeneration. It should be noted that in vitro studies using HT22 cell treated by CoCl2 to mimic hypoxia–reperfusion indicate that aconitase activity showed a reperfusion time-dependent decrease (Fig. 5B) while the overall mitochondrial protein sulfenation showed a reperfusion time-dependent increase. It is likely that the decrease in aconitase activity under these experimental conditions also involves other types of protein oxidation such as carbonylation and nitrosylation (Han et al. 2005; Yan et al. 1997).
Mitofilin is an inner membrane protein that regulates mitochondrial cristae morphogenesis (John et al. 2005). Down-regulation of mitofilin by siRNA leads to decreased cellular proliferation and increased apoptosis due to a disorganized inner mitochondrial membrane (John et al. 2005). In senescence-accelerated mouse, mitofilin is implicated in cognitive disorder associated with mitochondrial dysfunction (Wang et al. 2008). Moreover, relevant to the pathogenesis of Parkinson’s disease, in vitro exposure of isolated rat brain mitochondria to reactive dopamine quinone induced a rapid loss of mitofilin protein content (van Laar et al. 2008), suggesting that sulfenation of mitofilin, altering the protein’s function, could be implicated in age-related neurodegenerative disorders.
Regarding sulfenation of pyruvate dehydrogenase and pyruvate carboxylase, it is expected that sulfenation of these proteins would impair mitochondrial function. Pyruvate dehydrogenase is the first component of pyruvate dehydrogenase complex that catalyzes the conversion of pyruvate to acetyl-CoA. It serves as a key link between glycolysis and the Krebs cycle. Inactivation of this protein would switch cells from aerobic to anaerobic metabolism, producing lactate instead of acetyl-CoA (Kim et al. 2006). Likewise, pyruvate carboxylase catalyzes the conversion of pyruvate to oxaloacetate that also feeds into the Krebs cycle (Marin-Valencia et al. 2010). Hence, this protein is also an important enzyme in pyruvate metabolism. On the other hand, tubulin is known to undergo cys oxidation (McDonagh and Sheehan 2007) as well as reversible glutathionylation that is most likely derived from sulfenic acid (Britto et al. 2005). This protein interacts with voltage-dependent anion channel (Carre et al. 2002; Rostovtseva et al. 2008) that is a part of mitochondrial permeability transition pore structure (Yan et al. 2002). As all these identified proteins play critical roles in mitochondrial bioenergetics, their sulfenation may collectively contribute to the overall compromise of mitochondrial respiration and oxidative phosphorylation that occur during brain aging (Vancova et al. 2010; Yao et al. 2009).
Finally, it should be pointed out that protein sulfenation during aging could also provide beneficial effects on an organism. This is because there has been increasing evidence that oxidative stress can play a positive role in the adaption to non-injurious stress (Ristow and Schmeisser 2011; Yan 2014) such as caloric restriction (Kwon et al. 2017) and preconditioning or postconditioning (Cohen and Downey 2011; Dost et al. 2008). Indeed, it has been reported that protein sulfenation can be protective against oxidative stress (Fetherolf et al. 2017; Mateos et al. 2017). Therefore, identification of protein sulfenation that is beneficial to the retardation of the aging process may provide insights into the mechanisms of beneficial redox signaling processes in fighting aging and diseases.
In summary, using comparative 2D Western blot analysis in conjunction with MS peptide sequencing, we have identified mitochondrial proteins that underwent sulfenation changes during brain aging. The alterations in the identified proteins’ sulfenic acid intensities highlight the nature of mitochondrial protein sulfenation, which can go either up or down and is likely dictated not only by the stability of the sulfenic acids in target proteins, but also by the intensity and the duration of the oxidative stress that occurs during aging. Results of this study suggest that mitochondrial protein sulfenation plays a role in mitochondrial function and/or dysfunction during brain aging. Finally, it should be alerted that sulfenation of certain cys residues in some of the identified proteins may not have any functional consequences. Hence, future studies need to identify the specific cys residues that have impacts on protein functions upon sulfenation.
EXPERIMENTAL PROCEDURES
Animal and chemicals
Male Sprague–Dawley rats were obtained from Harlan (Indianapolis, IN). Experiments were conducted in adherence with the NIH Guidelines for the Care and Use of Laboratory Animals and were approved by the University of North Texas Health Science Center Animal Care and Use Committee. All chemicals were purchased from Sigma (St. Louis, MO) unless otherwise indicated.
Preparation of brain mitochondria
The isolation of whole brain mitochondria was carried out using Percoll gradient centrifugation as previously described (Yan et al. 2007). Brains were removed rapidly and homogenized in 15 ml of ice-cold, mitochondrial isolation buffer containing 0.32 mol/L sucrose, 1 mmol/L EDTA, and 10 mmol/L Tris–HCl, pH 7.1. The homogenate was centrifuged at 1330 g for 10 min and the supernatant was saved. The pellet was resuspended in 0.5 (7.5 ml) volume of the original isolation buffer and centrifuged again under the same conditions. The two supernatants were combined and centrifuged further at 21,200 g for 10 min. The resulting crude mitochondrial pellet was resuspended in 12% Percoll solution that was prepared in mitochondrial isolation buffer and centrifuged at 6900 g for 10 min. The resulting supernatant was then carefully removed by vacuum. The obtained soft pellet was resuspended in 10 ml of the mitochondrial isolation buffer and centrifuged again at 6900 g for 10 min. All of the mitochondrial pellets obtained after centrifugation were either used immediately or frozen at −80 °C until analysis. Protein concentrations were determined by bicinchoninic acid assay (Smith et al. 1985).
In vitro protein sulfenation by mitochondria-generated ROS
Protein sulfenation in isolated brain mitochondria was induced by incubating mitochondria with succinate and antimycin A, a condition that is known to enhance mitochondrial reactive oxygen species (ROS) generation and mitochondrial oxidative stress (Turrens 1997; Turrens et al. 1985). Mitochondrial incubation was carried out as previously described (Schonfeld and Reiser 2006; Yan et al. 2008). Briefly, mitochondria (0.25 mg/mL) were incubated at 25 °C for 60 min in incubation buffer (110 mmol/L mannitol, 10 mmol/L KH2PO4, 60 mmol/L Tris, 60 mmol/L KCl, and 0.5 mmol/L EGTA, pH 7.4) in the presence of 50 μmol/L antimycin A. The mixture was then supplemented with succinate (10 mmol/L). Control samples were incubated under the same conditions in the absence of antimycin A and succinate. At the end of the incubation, mitochondria were pelleted by centrifugation at 8000 g for 10 min followed by labeling of PSOHs as described below.
Labeling of protein sulfenic acids by arsenite reduction/biotin switch assay
The procedure used to label PSOHs, shown in Fig. 1, was performed as previously described (Saurin et al. 2004) with modifications. Mitochondrial pellet, immediately following isolation or in vitro oxidative stress challenge, was solubilized in a thiol-group blocking buffer containing 100 mmol/L sodium acetate (pH 7.0), 20 mmol/L NaCl, 1% SDS, and 100 mmol/L NEM. The protein mixture was incubated on a rotator at room temperature for 2 h followed by clarification of the mixture by centrifugation at 13,000 g for 10 min. Excess NEM in the supernatant was removed by gel filtration using PD-10 columns. This was followed by addition of 0.1 mmol/L biotin-maleimide and 20 mmol/L sodium arsenite (both final concentrations) to the eluate. The sample was further incubated on a rotator at room temperature for 30 min. Proteins were then precipitated by 10% TCA (final concentration) on ice for 10 min followed by centrifugation on a bench top centrifuge at 1000 g for 5 min. The pellet was washed three times with ethyl acetate:ethanol (1:1, v/v). Protein pellet after the third wash was used for either affinity capture or Western blot probing of the biotinylated (sulfenated) proteins.
Two-dimensional Western blot detection of sulfenated proteins
2D Western blot was performed as previously described (Yan 2009) with modifications. Following biotinylation of the sulfenated cys residues, mitochondrial pellets were resuspended in 2D rehydration buffer (8 mol/L urea, 4% CHAPS, 0.2% ampholytes (pH 3–10), and 100 mmol/L DTT). First-dimensional protein separation was performed with Bio-Rad Protean IEF Cell. Samples (40 μg/IPG strip) were applied to immobilized pH gradient strips (7-cm, non-linear pH 3–10, Bio-Rad) for 1 h at room temperature. The strips were then covered with mineral oil overnight, and isoelectric focusing was performed using the preset rapid voltage ramping method. For the second dimension, the immobilized pH gradient strips were equilibrated in room temperature for 25 min in equilibration buffer (6 mol/L urea, 2% SDS, 0.05 mmol/L Tris–HCl, 20% glycerol) to which 2% DTT was added before use. An additional 25 min equilibration period was then used with the same equilibration buffer to which 2.5% iodoacetamide, instead of 2% DTT, was added. The strips were then embedded in 0.7% agarose on the top of 7.5% Laemmli polyacrylamide slab gels (no stacking gel) and run by Tricine–SDS/PAGE running buffer (Khalkhali-Ellis 1995). One of the resulting 2D gels was stained with Coomassie colloidal blue as previously described (Yan and Forster 2009), and the other gel underwent electrophoretic transfer to PVDF membrane followed by immunoblotting with HRP–streptavidin. Signals on the PVDF membrane were visualized with an enhanced chemiluminescence kit. Biotin-conjugated protein marker was used for the purpose of both molecular weight ladders and positive controls. For 1D Western blot analysis, gel electrophoresis under reducing conditions was conducted. All images were scanned by an Epson Perfection 1670 scanner.
Affinity capture of sulfenated proteins
For affinity capture of sulfenated proteins that were converted to biotinylated proteins, the pellet after TCA precipitation and organic solvent washing was dissolved in 10 ml of 50 mmol/L phosphate buffer (pH 7.0) containing 150 mmol/L NaCl, 0.1% SDS, 1% Triton X-100, and 0.5% sodium deoxycholate (all final concentrations). The sample was then clarified at 13,000 g for 10 min and the supernatant was brought to 50 ml using the above pellet-dissolving phosphate buffer. One milliliter of high-capacity streptavidin agarose beads (Pierce, Inc.) was then added. The beads-containing sample was then rotated end-to-end for 1 h at room temperature, followed by centrifugation at 1500 g for 5 min. The agarose beads were then transferred to a small column, washed with 200 ml of 50 mmol/L phosphate buffer (pH 7.0) containing 1 mol/L NaCl, 0.1% SDS, 1% Triton X-100, and 0.5% sodium deoxycholate (all final concentration). The washed beads were then emptied out of the column, boiled in 0.5 ml elution buffer containing 62.5 mmol/L Tris–HCl (pH 6.8) and 1% SDS. The supernatant was then collected for analysis by Nano-liquid chromatography–mass spectrometry/mass spectrometry (LC–MS/MS).
Protein identification by mass spectrometry
Protein identification was performed at ProtTech (Norristown, PA) by the LC–MS/MS peptide sequencing technology. For gel-based identification, each 2D gel spot was destained, cleaned, and in-gel digested with sequencing grade trypsin. For solution-based identification, the sample was first reduced by DTT (10 mmol/L, final concentration) and then alkylated by iodoacetamide (20 mmol/L, final concentration). Proteins were denatured by 8 mol/L urea, followed by dilution to 2 mol/L urea with 100 mmol/L ammonium bicarbonate, pH 8.5. Following trypsin digestion, the resulting peptide mixture was cleaned and analyzed by LC–MS/MS sequencing. The mass spectrometric data collected were used to search the most recent non-redundant protein database using ProtTech’s proprietary software suite; and the relative abundance of a protein in a given gel spot was determined by the corresponding spectral count number (redundant peptides and non-redundant peptides) as previously described (Liu et al. 2004; Roth et al. 2006; Vogel and Marcotte 2008). Only the proteins identified in a given gel spot that were also identified by affinity capture were reported in this study.
Cell culture and in vitro hypoxia/reperfusion treatment
HT22 cells were cultured in DMEM (Hyclone, USA) with 10% fetal bovine serum (Hyclone, USA) and were incubated in a humidified incubator with 5% CO2 at 37 °C, and were seeded on 100-mm culture dishes at 100,000 cells/dish. Cell density was maintained at 80% or less confluency to attenuate excessive growth. CoCl2, a chemical hypoxia inducer (Ardyanto et al. 2006; Naves et al. 2013), was added into HT22 cells to develop the hypoxia model. In the hypoxia and reperfusion injury experiments, HT22 cells were incubated with 500 µmol/L CoCl2 for 16 h. After the removal of the culture medium, DMEM with 10% fetal bovine serum was added and maintained for 0, 6, 12, and 24 h, respectively. Control cell groups were treated with DMEM with 10% fetal bovine serum in the absence of CoCl2 and were maintained under the same condition. Mitochondria from these cultured cells were isolated by the same method as described above. For HT22 cellular mitochondrial sulfenic acid labeling, a DCP-Bio1 probe purchased from Kerafast (Boston, MA) was used to specifically label PSOHs (Klomsiri et al. 2010). Mitochondrial aconitase activities in these cells after varying treatment conditions were measured using a kit from BioAssay System (Hayward, CA) and aconitase activity was expressed at mU/mg protein as previously described (Yan et al. 1997).
Abbreviations
- 2D-PAGE:
-
Two-dimensional polyacrylamide gel electrophoresis
- Cys:
-
Cysteine
- LC–MS:
-
Liquid chromatography–mass spectrometry
- NEM:
-
N-ethylmaleimide
- ROS:
-
Reactive oxygen species
- TCA:
-
Trichloroacetic acid
References
Ardyanto TD, Osaki M, Tokuyasu N, Nagahama Y, Ito H (2006) CoCl2-induced HIF-1alpha expression correlates with proliferation and apoptosis in MKN-1 cells: a possible role for the PI3K/Akt pathway. Int J Oncol 29:549–555
Boschi-Muller S, Gand A, Branlant G (2008) The methionine sulfoxide reductases: catalysis and substrate specificities. Arch Biochem Biophys 474(2):266–273
Britto PJ, Knipling L, McPhie P, Wolff J (2005) Thiol-disulphide interchange in tubulin: kinetics and the effect on polymerization. Biochem J 389:549–558
Bulteau AL, Lundberg KC, Ikeda-Saito M, Isaya G, Szweda LI (2005) Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc Natl Acad Sci USA 102:5987–5991
Cai Z, Yan LJ (2013) Protein oxidative modifications: beneficial roles in disease and health. J Biochem Pharmacol Res 1:15–26
Carre M, Andre N, Carles G, Borghi H, Brichese L, Briand C, Braguer D (2002) Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J Biol Chem 277:33664–36669
Charles RL, Schroder E, May G, Free P, Gaffney PR, Wait R, Begum S, Heads RJ, Eaton P (2007) Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics 6:1473–1484
Claiborne A, Mallett TC, Yeh JI, Luba J, Parsonage D (2001) Structural, redox, and mechanistic parameters for cysteine-sulfenic acid function in catalysis and regulation. Adv Protein Chem 58:215–276
Cohen MV, Downey JM (2011) Ischemic postconditioning: from receptor to end-effector. Antioxid Redox Signal 14:821–831
Denu JM, Tanner KG (2002) Redox regulation of protein tyrosine phosphatases by hydrogen peroxide: detecting sulfenic acid intermediates and examining reversible inactivation. Methods Enzymol 348:297–305
Dost T, Cohen MV, Downey JM (2008) Redox signaling triggers protection during the reperfusion rather than the ischemic phase of preconditioning. Basic Res Cardiol 103:378–384
Fetherolf MM, Boyd SD, Taylor AB, Kim HJ, Wohlschlegel JA, Blackburn NJ, Hart PJ, Winge DR, Winkler DD (2017) Copper–zinc superoxide dismutase is activated through a sulfenic acid intermediate at a copper ion entry site. J Biol Chem 292:12025–12040
Gardner PR (2002) Aconitase: sensitive target and measure of superoxide. Methods Enzymol 349:9–23
Han D, Canali R, Garcia J, Aguilera R, Gallaher TK, Cadenas E (2005) Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry 44:11986–11996
Heppner DE, Janssen-Heininger YM, van der Vliet A (2017) The role of sulfenic acids in cellular redox signaling: reconciling chemical kinetics and molecular detection strategies. Arch Biochem Biophys 616:40–46
John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ, Zha J (2005) The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell 16:1543–1554
Kaiserova K, Srivastava S, Hoetker JD, Awe SO, Tang XL, Cai J, Bhatnagar A (2006) Redox activation of aldose reductase in the ischemic heart. J Biol Chem 281:15110–15120
Keating DJ (2008) Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J Neurochem 104:298–305
Khalkhali-Ellis Z (1995) An improved SDS-polyacrylamide gel electrophoresis for resolution of peptides in the range of 3.5–200 kDa. Prep Biochem 25:1–9
Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177–185
Klomsiri C, Nelson KJ, Bechtold E, Soito L, Johnson LC, Lowther WT, Ryu SE, King SB, Furdui CM, Poole LB (2010) Use of dimedone-based chemical probes for sulfenic acid detection evaluation of conditions affecting probe incorporation into redox-sensitive proteins. Methods Enzymol 473:77–94
Kwon YY, Lee SK, Lee CK (2017) Caloric restriction-induced extension of chronological lifespan requires intact respiration in budding yeast. Mol Cells 40:307–313
Leonard SE, Reddie KG, Carroll KS (2009) Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol 4:783–799
Liu H, Sadygov RG, Yates JR III (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem 76:4193–4201
Marin-Valencia I, Roe CR, Pascual JM (2010) Pyruvate carboxylase deficiency: mechanisms, mimics and anaplerosis. Mol Genet Metab 101:9–17
Mateos LM, Villadangos AF, de la Rubia AG, Mourenza A, Marcos-Pascual L, Letek M, Pedre B, Messens J, Gil JA (2017) The arsenic detoxification system in corynebacteria: basis and application for bioremediation and redox control. Adv Appl Microbiol 99:103–137
McDonagh B, Sheehan D (2007) Effect of oxidative stress on protein thiols in the blue mussel Mytilus edulis: proteomic identification of target proteins. Proteomics 7:3395–3403
Michalek RD, Nelson KJ, Holbrook BC, Yi JS, Stridiron D, Daniel LW, Fetrow JS, King SB, Poole LB, Grayson JM (2007) The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J Immunol 179:6456–6467
Naves T, Jawhari S, Jauberteau MO, Ratinaud MH, Verdier M (2013) Autophagy takes place in mutated p53 neuroblastoma cells in response to hypoxia mimetic CoCl(2). Biochem Pharmacol 85:1153–1161
Poole LB, Karplus PA, Claiborne A (2004) Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol 44:325–347
Rebrin I, Forster MJ, Sohal RS (2007) Effects of age and caloric intake on glutathione redox state in different brain regions of C57BL/6 and DBA/2 mice. Brain Res 1127:10–18
Ristow M, Schmeisser S (2011) Extending life span by increasing oxidative stress. Free Radic Biol Med 51:327–336
Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, Sackett DL (2008) Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci USA 105:18746–18751
Roth AF, Wan J, Green WN, Yates JR, Davis NG (2006) Proteomic identification of palmitoylated proteins. Methods 40:135–142
Saurin AT, Neubert H, Brennan JP, Eaton P (2004) Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci USA 101:17982–17987
Schonfeld P, Reiser G (2006) Rotenone-like action of the branched-chain phytanic acid induces oxidative stress in mitochondria. J Biol Chem 281:7136–7142
Seo YH, Carroll KS (2009) Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci USA 106:16163–16168
Shetty V, Spellman DS, Neubert TA (2007) Characterization by tandem mass spectrometry of stable cysteine sulfenic acid in a cysteine switch peptide of matrix metalloproteinases. J Am Soc Mass Spectrom 18:1544–1551
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85
Sohal RS, Forster MJ (2007) Coenzyme Q, oxidative stress and aging. Mitochondrion 7(Suppl):S103–S111
Sun Z, Mohamed MA, Park SY, Yi TH (2015) Fucosterol protects cobalt chloride induced inflammation by the inhibition of hypoxia-inducible factor through PI3K/Akt pathway. Int Immunopharmacol 29:642–647
Tsou CC, Lu TT, Liaw WF (2007) EPR, UV–Vis, IR, and X-ray demonstration of the anionic dimeric dinitrosyl iron complex [(NO)(2)Fe(micro-S(t)Bu)(2)Fe(NO)(2)](−): relevance to the products of nitrosylation of cytosolic and mitochondrial aconitases, and high-potential iron proteins. J Am Chem Soc 129:12626–12627
Turell L, Botti H, Carballal S, Ferrer-Sueta G, Souza JM, Duran R, Freeman BA, Radi R, Alvarez B (2008) Reactivity of sulfenic acid in human serum albumin. Biochemistry 47:358–367
Turrens JF (1997) Superoxide production by the mitochondrial respiratory chain. Biosci Rep 17:3–8
Turrens JF, Alexandre A, Lehninger AL (1985) Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 237:408–414
Van Laar VS, Dukes AA, Cascio M, Hastings TG (2008) Proteomic analysis of rat brain mitochondria following exposure to dopamine quinone: implications for Parkinson disease. Neurobiol Dis 29:477–489
Vancova O, Baciak L, Kasparova S, Kucharska J, Palacios HH, Horecky J, Aliev G (2010) In vivo and in vitro assessment of brain bioenergetics in aging rats. J Cell Mol Med 14:2667–2674
Vogel C, Marcotte EM (2008) Calculating absolute and relative protein abundance from mass spectrometry-based protein expression data. Nat Protoc 3:1444–1451
Wang Q, Liu Y, Zou X, An M, Guan X, He J, Tong Y, Ji J (2008) The hippocampal proteomic analysis of senescence-accelerated mouse: implications of Uchl3 and mitofilin in cognitive disorder and mitochondria dysfunction in SAMP8. Neurochem Res 33(9):1776–1782
Wang G, Cui J, Guo Y, Wang Y, Kang L, Liu L (2016) Cyclosporin A protects H9c2 cells against chemical hypoxia-induced injury via inhibition of MAPK signaling pathway. Int Heart J 57:483–489
Wood ZA, Poole LB, Karplus PA (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300:650–653
Yan LJ (2009) Analysis of oxidative modification of proteins. Curr Protoc Protein Sci Chapter 14(Unit14):4
Yan LJ (2014) Positive oxidative stress in aging and aging-related disease tolerance. Redox Biol 2:165–169
Yan LJ, Forster MJ (2009) Resolving mitochondrial protein complexes using nongradient blue native polyacrylamide gel electrophoresis. Anal Biochem 389:143–149
Yan LJ, Sohal RS (1998) Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc Natl Acad Sci USA 95:12896–12901
Yan LJ, Levine RL, Sohal RS (1997) Oxidative damage during aging targets mitochondrial aconitase. Proc Natl Acad Sci USA 94:11168–11172
Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ (2002) Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J 21:5164–5172
Yan LJ, Yang SH, Shu H, Prokai L, Forster MJ (2007) Histochemical staining and quantification of dihydrolipoamide dehydrogenase diaphorase activity using blue native PAGE. Electrophoresis 28:1036–1045
Yan LJ, Thangthaeng N, Forster MJ (2008) Changes in dihydrolipoamide dehydrogenase expression and activity during postnatal development and aging in the rat brain. Mech Ageing Dev 129:282–290
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 106:14670–14675
Yarian CS, Toroser D, Sohal RS (2006) Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech Ageing Dev 127:79–84
Yeh JI, Claiborne A, Hol WG (1996) Structure of the native cysteine-sulfenic acid redox center of enterococcal NADH peroxidase refined at 2.8 Å resolution. Biochemistry 35:9951–9957
Ying J, Clavreul N, Sethuraman M, Adachi T, Cohen RA (2007) Thiol oxidation in signaling and response to stress: detection and quantification of physiological and pathophysiological thiol modifications. Free Radic Biol Med 43:1099–1108
Zourlidou A, Gidalevitz T, Kristiansen M, Landles C, Woodman B, Wells DJ, Latchman DS, de Belleroche J, Tabrizi SJ, Morimoto RI, Bates GP (2007) Hsp27 overexpression in the R6/2 mouse model of Huntington’s disease: chronic neurodegeneration does not induce Hsp27 activation. Hum Mol Genet 16:1078–1090
Acknowledgements
This work was supported in part by the National Institutes of Health (Grant Number R01NS079792). The authors thank Dr. Drake Zhang and Ms. Kerri Jin at ProtTech for their assistance in mass spectrometry peptide sequencing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Xiaorong Yang, Jinzi Wu, Siqun Jing, Michael J. Forster, and Liang-Jun Yan declare that they have no conflict of interest.
Human and animal rights and informed consent
This article involves the use of rats. Experiments were conducted in adherence with the NIH Guidelines for the Care and Use of Laboratory Animals and were approved by the University of North Texas Health Science Center Animal Care and Use Committee. All institutional and national guidelines for the care and use of laboratory animals were followed.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yang, X., Wu, J., Jing, S. et al. Mitochondrial protein sulfenation during aging in the rat brain. Biophys Rep 4, 104–113 (2018). https://doi.org/10.1007/s41048-018-0053-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41048-018-0053-3