
Abstract
Maintenance of pluripotency, lineage commitment and differentiation of mammalian embryonic stem cells into all somatic cell types involves differential regulation of different subsets of genes, as does reprogramming of somatic cells back into a pluripotent state. It is now understood that the three-dimensional organization of the human genome asserts a key role in these processes in two ways. First, by providing a largely invariable scaffold onto which dynamic changes in chromatin may manifest; second, by allowing the spatial clustering of genes contributing to the same functional pathways. In this review, we discuss the rapidly growing volume of literature on the structure-to-function relationship of mammalian genomes as regards key developmental transitions of stem cell populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lineage commitment and differentiation of embryonic stem (ES) cells into all somatic cell types involves upregulation and downregulation of different subsets of genes [1]. This cell type-specific regulation of gene expression occurs predominantly at the level of transcription, and this was exemplified by a seminal experiment where mammalian fibroblasts were reprogrammed into an induced pluripotent cell state via the overexpression of only four transcription factors, Oct-4, Myc, Klf4, and Sox2 [2, 3]. Similarly, the key pluripotency factors, Oct-4, Sox2, and Nanog, have been shown to maintain ESCs in an undifferentiated state by driving the expression of genes associated with cell identity, including those encoding these TFs themselves [1]. The maintenance of pluripotency also requires the repression of lineage-specific genes that can stimulate various differentiation pathways; this is mediated by Polycomb group proteins that bind these genes and their cognate cis-regulatory elements, thus introducing an epigenetic environment preventing the loss of ESC identity. Given the diverse, sometimes overlapping [4] binding patterns of these factors, it has become evident that control of transcription programs is manifested via organizational features that extend beyond the mere linear order of primary genomic sequence.
These organizational features of the genome stem from the need to fit mammalian chromosomes within the confined dimensions of cell nuclei. As a result, DNA molecules are wrapped around histones and the resulting chromatin fibers are compacted into interphase chromosomes, which in turn assume a non-random arrangement and form spatially distinct territories [5]. Hence, even at this large-scale view of the nucleus, physical proximity of regions that are otherwise non-adjacent on the linear fiber (or even between regions lying on different chromosomes) becomes apparent. Most of the aforementioned knowledge comes from cell biology approaches, which—however powerful at the single-cell level—suffer from limitations in resolution and throughput [6]. But, as such, the need to study regulatory interactions between defined, sub-resolution, genomic regions called for the development of novel technologies able to capture these at the molecular level. In this review, we will discuss how new insight into the three-dimensional (3D) organization of mammalian genomes reshapes our understanding of pluripotency, cell differentiation, and organismal aging.
A New Tool to Study Chromatin Organization at Sub-Gene Resolution
Already in 1993, Cullen and colleagues described the elegant idea of “proximity ligation” by which one could detect short-range chromatin looping [7]. But, it was not until several years later that the introduction of the chromosome conformation capture (3C) technology revolutionized the study of chromosome folding at high resolution [8]. Within the last decade, a number of 3C variants have emerged, like circularized 3C (4C), carbon copy 3C (5C), or chromatin interaction analysis coupled to paired-end tagging (ChIA-PET) that exploit latest sequencing technologies to analyze events of chromatin looping at an increasingly higher genome coverage [9]. The key principles of 3C-based methods involve formaldehyde fixation to preserve chromatin interactions, digestion of DNA using a restriction enzyme, and cross-linked complexes undergo ligation under conditions that favor fusions also between fragments originally not in close proximity on the linear fiber; finally, identification of these ligation junctions, and of the frequency by which these occur, provides a snapshot of the in vivo genomic architecture.
At the whole-genome level, development and application of Hi-C [10] has verified older findings and at the same time uncovered novel organizational principles of mammalian genomes. For example, the preferential co-associations between euchromatic and heterochromatic regions—termed compartment “A” and “B,” respectively—were not unforeseen, but were extensively charted by Hi-C [10, 11, 12•]. At the sub-compartment scale, a striking new feature was that of topologically associating domains (TADs); these represent spatial neighborhoods that harbor high-frequency interactions and are insulated from adjacent TADs by sharp boundaries of low interaction capacity [12•, 13, 14••]. Finally, at the sub-TAD level, the dynamics of promoter-promoter and enhancer-promoter crosstalk are the highlights of 3D architecture and change considerably upon establishment of lineage-specific transcriptional programs [15, 16•]. Under the light of a multi-level hierarchical genome organization, the control of cell fate can be studied in all four dimensions, in 3D space and over time [17–19], with the capacity to investigate variations down to a ∼1-kbp resolution [20] or even at the single-cell level [21]. Notably, the recent emergence of “targeted” 3C variants will allow lower cost, single-restriction-fragment resolution analysis of selected loci involved in cellular pathways of particular interest [16•, 22, 23].
Large-Scale Reorganization of Higher-Order Chromatin Structure During Differentiation
A more careful look into the pioneering Hi-C studies reveals that genomic regions correlating with both genetic features (e.g., gene density, GC content) and epigenetic indicators of transcriptional status (e.g., chromatin accessibility, transcriptional co-regulation, “activating” or “repressive” histone marks, early or late replication timing) preferentially interact with regions exhibiting similar characteristics [10, 24, 25]. This is consistent with numerous studies linking transcriptional co-regulation to spatial clustering of the relevant genes and their cis-regulatory elements [26–32] around the nucleoplasmic supra-molecular entities that harbor most nuclear transcription—around transcription factories [19].
Advances in sequencing capacity, accuracy, and read length allowed Hi-C studies to achieve resolution down to ∼10 [33] and then ∼1 kbp [20]. In a recent analysis, comparison of high-resolution Hi-C data generated using human ES cells and four ES-derived cell types revealed extensive A/B compartment rearrangements. Transition from the ES cell state to lung fibroblasts or mesenchymal stem cells coincides with a marked expansion of the B compartment [12•]; this is in agreement with the previously documented spread of heterochromatic modifications upon differentiation [34]. As a result, changes in expression correlate with genes switching from compartment A to B (or the converse); however, these are in average small, indicative of a contributory rather than deterministic role of compartments during lineage commitment [12•].
The Role of TADs in the Regulatory Landscape
At a 0.1–1-Mbp resolution, TADs represent a prominent module of 3D genome organization. Dixon and colleagues identified a partitioning of the genome in ∼2000 such TADs, covering more than 90 % of its length [11]. More recently, Rao and colleagues, pushing resolution to the 1-kbp limit, claim a reduced number of TADs with a median loop size of 185 kbp [20]. Either way, cell types of endodermal, mesodermal, and ectodermal origin share between 55 and 75 % of such loops, while ∼45 % of TADs called in mouse are also seen in man [20]. In addition, TADs are restricted to interphase chromosomes, when transcription is widespread, suggestive of their influence in regulation of gene expression [35]. As a result, transitions between the A/B compartments during cell differentiation, which rearrange TAD boundaries, are accompanied by the gain or loss of interactions within these TADs. This occurs in parallel with concomitant changes in replication timing, histone mark profiles, and association to the nuclear lamina [12•, 14••, 36•]. Especially, the latter lead to the repositioning of TADs within the typically repressive context of the lamina and often harbor genes silenced along the course of differentiation [37].
Nevertheless, as the majority of TADs and their boundaries remain largely invariable, an overarching architecture of the genome can be imagined that acts as an evolutionarily selected scaffold onto which finer-scale changes allow for lineage commitment. It is worth noting here that both inflammatory and hormonal signaling only marginally remodel (<10 %) TAD boundaries, as well as that pro-inflammatory, pluripotency-specific, and key developmental enhancers have been seen pre-looped onto the gene promoters they control before these become activated [33, 38, 39••, 40, 41, 42••]. Collectively, these beg the question of how this overall “stable” overarching architecture allows for the gene expression changes seen along the different differentiation paths [1].
A View of Gene Expression Regulation at the Sub-TAD Scale
A key functional feature of TADs is the insulation of intra-domain interactions from “leaking” into neighboring domains, and the disruption of TAD boundaries was shown to cause gene expression misregulation [42••]. For example, in the well-studied HoxD gene cluster, early and late developmental genes are differentially induced via a switch between the two TADs that span the locus and act to direct promoter-enhancer crosstalk [43].
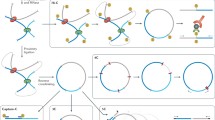
This crosstalk has been globally investigated using ChIA-PET and a targeted Hi-C approach. ChIA-PET connectivity maps have charted ∼40,000 interactions amongst mouse enhancers and promoters; the majority of which involve enhancers contacting promoters that are located beyond their nearest active gene but are contained within the same TAD. Again, genes pivotal for stem cell identity are found in physical proximity with one another, indicative of co-regulation at the surface of “specialized” transcription factories [15]; this non-random 3D clustering was verified in a comparison of the interactomes of >22,000 mouse promoters in ES versus fetal liver cells using Capture Hi-C, its most emphatic manifestation seen in genes controlling key developmental transitions [16•]. It is worth noting however that a large number of associations persist throughout differentiation, pointing to a robust spatial network underlying cellular homeostasis, which —nonetheless—is characterized by the preferential clustering of genes involved in the same biological processes or pathways [15, 16•]. Similarly, the application of 5C at six developmentally regulated genes in ES and neural progenitor cells (NPCs) revealed additional sub-domains within a single TAD, indicating reorganization at the sub-Mbp scale during differentiation [44]. Owing to the single-fragment resolution achieved here, these “sub-TAD” chromatin loops can be classified into three groups. First, constitutive looping interactions seen in both cell types; second, enhancer-promoter loops specific to the pluripotent state in ESCs; third, lineage-specific looping occurring only upon differentiation. Taken together, changes in chromatin architecture interplay with gene expression during the process of cell-type commitment, but the question whether 3D genome topology is a functional cause or a result of transcription is still unresolved.
Finally, using the more focused 4C approach, where all interactions of a single genomic “viewpoint” are recorded [45], the promoters of a number of key pluripotency genes have been studied in both mouse and human ES cells. Again, long-range looping appears mostly confined within TADs, and co-associations, completely absent from differentiated cells, preferentially involve sites bound by the Oct-4, Sox2, and Nanog (OSN) transcription factors [38, 46]. Critically, Oct-4 or Nanog knock-down diminishes contact frequencies between OSN-bound regions [38, 39••], and insertion of a Nanog-binding array in the ESC genome led to the nucleation of endogenous OSN-bound loci around this ectopic array [46]. Not surprisingly, reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) by OSN overexpression recapitulates the pluripotency-specific genome configuration [38, 47]. Taken together, this data convincingly demonstrate that the three key pluripotency factors drive spatial organization via a specialized network of interactions as proposed [19] (Fig. 1a).

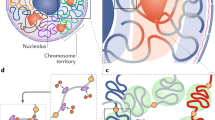
Hierarchical principles of 3D genome organization and cellular differentiation. a Multi-scale 3D organization. Interphase chromosomes in the human nucleus occupy distinct territories, which can intermingle at the edges. Chromosome 7 (yellow) is shown, and three exemplary TADs (grey, orange, purple; 0.1–1 Mbp in size) are sketched. For one, chromatin loops (10–250 kbp in size) forming via association with two transcription factories (orange spheres; ∼90 nm in diameter) are depicted. Upon differentiation signaling towards a specific cell type, some of these loops reshuffle, owing to the specialization of some factories (green spheres) for transcribing a cell type-specific gene subset. b Chromatin interaction changes within and between TADs. Hi-C interaction data (from ref. 11; 40-kbp resolution) along a 10-Mbp region (positions 70 to 80 Mbp; hg19) on human chromosome 12 for embryonic stem (ES) cells (middle; mirrored graphs) and differentiated lymphoblasts (GM12878; top) and fibroblasts (IMR-90; bottom). Gain and loss of ES-specific interactions (orange shading) are shown (inter-TAD ones; green; intra-TAD ones; yellow)
Genome organization does not solely owe to the “active” compartment of chromatin. The “inactive” ES cell compartment contains, amongst others, genes that are to be activated later in development. As it has been demonstrated that some of these associate with a subset of developmentally important “poised” promoters also marked by H3K27me3 [4] and that this Polycomb-instated histone mark is a key structural feature of the Drosophila genome [48], Denholtz and colleagues investigated Polycomb contribution to stem cell chromatin folding. A segregation between OSN-occupied and Polycomb-bound and H3K27me3-marked loci was revealed, and this feature is largely specific to the ES cell state. Moreover, loss of a central component of the Polycomb complex partially disassembles clusters formed around such H3K27me3-marked loci [49•]. Hence, changes in both the transcriptionally permissive and non-permissive parts of the ES cell genome contribute to the spatial arrangement and facilitate the state of pluripotency (Fig. 1b).
Protein-Mediated 3D Organization of the Pluripotent Genome
All the aforementioned novel insight into genome 3D organization has prompted researchers to look for proteins other than transcription factors that may contribute to looping, both on its “static” and on its dynamic aspects. These proteins, usually referred to as “architectural co-factors”, include the insulator factor CTCF [20, 42••, 50] and the cohesin [42••, 51, 52] and condensin II complexes [53], and have been implicated in the folding of genomes of various cell types across species [54].
Genome-wide analyses of TAD boundaries showed an enrichment of specific features at these sites, predominated by the presence of tRNA and house-keeping genes and of binding sites for the protein CTCF [11]. Later redefinition of TADs increased the correlation to CTCF [55], and ultra-resolution Hi-C studies implicated consecutive convergent CTCF sites in the formation of key structural loops [20]. Loss of CTCF in ES cells does not result in the complete disintegration of TAD boundaries but does induce partial fusion of adjacent TADs [42••].
Similarly, cohesin complexes have been implicated in chromatin 3D organization but mainly in the maintenance of intra-TAD looping, as its depletion does not affect TAD-boundary integrity but leads to misregulation of a considerable number of genes in both ES and differentiated cells [44, 56–58]. Most interestingly, analysis of 5C data revealed that >80 % of charted interactions in ESCs associated with different combinations of CTCF, cohesion, and the co-activator complex of Mediator, by contrast to ∼40 % interactions anchored via OSN; the majority of these interactions involved enhancers and “super-enhancers” [38, 42••, 44]. Comparison of the interaction profiles between ES and NP cells shows that the vast majority of sites occupied by both CTCF and cohesin in ES cells remained bound also upon differentiation, while the few CTCF-cohesin-Mediator-bridged interactions are endemic to the pluripotent state and thus lost [44]. Consistent with these data, depletion of cohesin or of a Mediator subunit spontaneously induces differentiation of ES cells and impairs somatic cell reprogramming [15, 38, 47].
Finally, the condensin II complex—mainly known for its involvement in cell division—was shown to be associated with “active” euchromatic sites, often alongside cohesin. This feature appears to be unique for ES cells, displays enrichment for enhancer elements, and is expected to also contribute to local chromatin folding [53].
Conclusions and Outlook
Lineage commitment of ES cells is accompanied by significant changes in both gene expression and epigenetic distribution of euchromatic and heterochromatic histone marks. As a result, the discovery of a largely invariable 3D genome organization between ES and differentiated cells came as a surprise. Nonetheless, differences in chromatin folding seen when comparing the pluripotent to any differentiated cell are markedly larger than when comparing any two differentiated cell types. Given the now-documented heterogeneity of genome organization at the single-cell level [21], the well-accepted heterogeneity within the various stem cell compartments in vivo [59], and the fact that deterioration of adult stem cells (in terms of population size and “functional quality”) accounts for much of aging-associated tissue defects [60], it will be interesting to delineate the structure of the genome in single stem cells of progressively older age. Such an analysis might uncover principles of dis-/re-organization that are linked with a decline in regenerative capacity, as recently documented in cell ensembles upon senescence [61].
Lastly, the emerging discussion on formaldehyde crosslinking biases imposed onto 3C approaches to study nuclear organization [62], and contrasting views on chromatin folding obtained using independent methods [63] highlight the need for the development of novel tools for interrogating the in vivo architecture of the genome in the absence of crosslinking and from increasingly lower cell counts.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Hochedlinger K, Plath K. Epigenetic reprogramming and induced pluripotency. Development. 2009;136:509–23.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72.
Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–83.
Cremer T, Cremer M. Chromosome territories. Cold Spring Harb Perspect Biol. 2010;2:a003889.
Buckley MS, Lis JT. Imaging RNA Polymerase II transcription sites in living cells. Curr Opin Genet Dev. 2014;25:126–30.
Cullen KE, Kladde MP, Seyfred MA. Interaction between transcription regulatory regions of prolactin chromatin. Science. 1993;261:203–6.
Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–11.
de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012;26:11–24.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93.
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80.
Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–6. This study describes changes in 3D organization during stem cell differentiation genome-wide.
Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–13.
Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–5. This study, together with ref. 11, defines for the first time TADs at high resolution.
Kieffer-Kwon KR, Tang Z, Mathe E, Qian J, Sung MH, Li G, et al. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell. 2013;155:1507–20.
Schoenfelder S, Furlan-Magaril M, Mifsud B, Tavares-Cadete F, Sugar R, Javierre BM, et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 2015. doi:10.1101/gr.185272.114. This study, together with ref. 15, describes high resolution of enhancer-promoter crosstalk during differentiation.
Gibcus JH, Dekker J. The hierarchy of the 3D genome. Mol Cell. 2013;49:773–82.
Krijger PH, de Laat W. Identical cells with different 3D genomes; cause and consequences? Curr Opin Genet Dev. 2013;23:191–6.
Papantonis A, Cook PR. Transcription factories: genome organization and gene regulation. Chem Rev. 2013;113:8683–705.
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80.
Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64.
Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat Genet. 2014;46:205–12.
Kolovos P, van de Werken HJ, Kepper N, Zuin J, Brouwer RW, Kockx CE, et al. Targeted chromatin capture (T2C): a novel high resolution high throughput method to detect genomic interactions and regulatory elements. Epigenetics Chromatin. 2014;7:10.
Dong X, Li C, Chen Y, Ding G, Li Y. Human transcriptional interactome of chromatin contribute to gene co-expression. BMC Genomics. 2010;11:704.
Liu L, Zhang Y, Feng J, Zheng N, Yin J, Zhang Y. GeSICA: genome segmentation from intra-chromosomal associations. BMC Genomics. 2012;13:164.
Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet. 2010;42:53–61.
Papantonis A, Kohro T, Baboo S, Larkin JD, Deng B, Short P, et al. TNFα signals through specialized factories where responsive coding and miRNA genes are transcribed. EMBO J. 2012;31:4404–14.
Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98.
Crepaldi L, Policarpi C, Coatti A, Sherlock WT, Jongbloets BC, Down TA, et al. Binding of TFIIIC to sine elements controls the relocation of activity-dependent neuronal genes to transcription factories. PLoS Genet. 2013;9:e1003699.
Li HB, Ohno K, Gui H, Pirrotta V. Insulators target active genes to transcription factories and polycomb-repressed genes to polycomb bodies. PLoS Genet. 2013;9:e1003436.
Park SK, Xiang Y, Feng X, Garrard WT. Pronounced cohabitation of active immunoglobulin genes from three different chromosomes in transcription factories during maximal antibody synthesis. Genes Dev. 2014;28:1159–64.
Rieder D, Ploner C, Krogsdam AM, Stocker G, Fischer M, Scheideler M, et al. Co-expressed genes prepositioned in spatial neighborhoods stochastically associate with SC35 speckles and RNA polymerase II factories. Cell Mol Life Sci. 2014;71:1741–59.
Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–4.
Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. 2014;14:68–80.
Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA, et al. Organization of the mitotic chromosome. Science. 2013;342:948–53.
Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515:402–5. In this study, a causal link between replication timing and 3D topology is described.
Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010;38:603–13.
Apostolou E, Ferrari F, Walsh RM, Bar-Nur O, Stadtfeld M, Cheloufi S, et al. Genome wide chromatin interactions of the Nanog locus in pluripotency, differentiation, and reprogramming. Cell Stem Cell. 2013;12:699–712.
Wei Z, Gao F, Kim S, Yang H, Lyu J, An W, et al. Klf4 organizes long-range chromosomal interactions with the oct4 locus in reprogramming and pluripotency. Cell Stem Cell. 2013;13:36–47. This study, together with refs. 38 and 46, describes the formation-coordinated spatial clusters where key pluripotency genes are transcribed, driven by the Oct-4, Sox2, and Nanog TFs.
Le Dily F, Baù D, Pohl A, Vicent GP, Serra F, Soronellas D, et al. Distinct structural transitions of chromatin topological domains correlate with coordinated hormone-induced gene regulation. Genes Dev. 2014;28:2151–62.
Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, et al. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 2014;512:96–100.
Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–87. This work demonstrates how CTCF/cohesin insulation at TAD boundaries allows for the efficient regulation of stem cell identity genes.
Andrey G, Montavon T, Mascrez B, Gonzalez F, Noordermeer D, Leleu M, et al. A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science. 2013;340:1234167.
Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–95.
Stadhouders R, Kolovos P, Brouwer R, Zuin J, van den Heuvel A, Kockx C, et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat Protoc. 2013;8:509–24.
de Wit E, Bouwman BA, Zhu Y, Klous P, Splinter E, Verstegen MJ, et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 2013;501:227–31.
Zhang H, Jiao W, Sun L, Fan J, Chen M, Wang H, et al. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming. Cell Stem Cell. 2013;13:30–5.
Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–72.
Denholtz M, Bonora G, Chronis C, Splinter E, de Laat W, Ernst J, et al. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 2013;13:602–16. This work highlights the importance of polycomb-mediated interactions for stem cell identity.
Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet. 2011;43:630–8.
DeMare LE, Leng J, Cotney J, Reilly SK, Yin J, Sarro R, et al. The genomic landscape of cohesin-associated chromatin interactions. Genome Res. 2013;23:1224–34.
Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–5.
Dowen JM, Bilodeau S, Orlando DA, Hübner MR, Abraham BJ, Spector DL, et al. Multiple structural maintenance of chromosome complexes at transcriptional regulatory elements. Stem Cell Rep. 2013;1:371–8.
Vietri Rudan M, Barrington C, Henderson S, Ernst C, Odom DT, Tanay A, et al. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015;10:1297–309.
Filippova D, Patro R, Duggal G, Kingsford C. Identification of alternative topological domains in chromatin. Algoritm Mol Biol. 2014;9:14.
Zuin J, Dixon JR, van der Reijden MI, Ye Z, Kolovos P, Brouwer RW, et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A. 2014;111:996–1001.
Seitan VC, Faure AJ, Zhan Y, McCord RP, Lajoie BR, Ing-Simmons E, et al. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013;23:2066–77.
Sofueva S, Yaffe E, Chan WC, Georgopoulou D, Vietri Rudan M, Mira-Bontenbal H, et al. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J. 2013;32:3119–29.
Motohashi N, Asakura A. Muscle satellite cell heterogeneity and self-renewal. Front Cell Dev Biol. 2014;2:1.
Parker MH. The altered fate of aging satellite cells is determined by signaling and epigenetic changes. Front Genet. 2015;6:59.
Chandra T, Ewels PA, Schoenfelder S, Furlan-Magaril M, Wingett SW, Kirschner K, et al. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015; pii: S2211-1247(14)01122-X.
Gavrilov A, Razin SV, Cavalli G. In vivo formaldehyde cross-linking: it is time for black box analysis. Brief Funct Genomics. 2015;14:163–5.
Williamson I, Berlivet S, Eskeland R, Boyle S, Illingworth RS, Paquette D, et al. Spatial genome organization: contrasting views from chromosome conformation capture and fluorescence in situ hybridization. Genes Dev. 2014;28:2778–91.
A cknowledgments
We would like to thank the members of our lab for discussions, and the UoC Advanced Researcher Grant (via the DFG Excellence Initiative) and CMMC intramural funding for the support.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Lilija Brant and Argyris Papantonis declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Age-related Stem Cell Modifiers
Rights and permissions
About this article

Cite this article
Brant, L., Papantonis, A. Contribution of 3D Chromatin Architecture to the Maintenance of Pluripotency. Curr Stem Cell Rep 1, 170–175 (2015). https://doi.org/10.1007/s40778-015-0016-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-015-0016-2